In Vitro and In Vivo Models for the Investigation of Potential Drugs Against Schizophrenia


1
Department of Synthesis and Chemical Technology of Pharmaceutical Substances, Faculty of Pharmacy, Medical University of Lublin, 4A Chodźki St., PL-20093 Lublin, Poland
2
Department of Biopharmacy, Faculty of Pharmacy, Medical University of Lublin, 4A Chodźki St., PL-20093 Lublin, Poland
3
Department of Pharmacology and Pharmacodynamics, Faculty of Pharmacy, Medical University of Lublin, 4A Chodźki St., PL-20093 Lublin, Poland
4
School of Pharmacy, University of Eastern Finland, P.O. Box 1627, FI-70211 Kuopio, Finland
*
Author to whom correspondence should be addressed.
Biomolecules 2020, 10(1), 160; https://0-doi-org.brum.beds.ac.uk/10.3390/biom10010160
Submission received: 4 December 2019
/
Revised: 12 January 2020
/
Accepted: 13 January 2020
/
Published: 19 January 2020
(This article belongs to the Section Molecular Medicine)
Abstract
:Schizophrenia (SZ) is a complex psychiatric disorder characterized by positive, negative, and cognitive symptoms, and is not satisfactorily treated by current antipsychotics. Progress in understanding the basic pathomechanism of the disease has been hampered by the lack of appropriate models. In order to develop modern drugs against SZ, efficient methods to study them in in vitro and in vivo models of this disease are required. In this review a short presentation of current hypotheses and concepts of SZ is followed by a description of current progress in the field of SZ experimental models. A critical discussion of advantages and limitations of in vitro models and pharmacological, genetic, and neurodevelopmental in vivo models for positive, negative, and cognitive symptoms of the disease is provided. In particular, this review concerns the important issue of how cellular and animal systems can help to meet the challenges of modeling the disease, which fully manifests only in humans, as experimental studies of SZ in humans are limited. Next, it is emphasized that novel clinical candidates should be evaluated in animal models for treatment-resistant SZ. In conclusion, the plurality of available in vitro and in vivo models is a consequence of the complex nature of SZ, and there are extensive possibilities for their integration. Future development of more efficient antipsychotics reflecting the pleiotropy of symptoms in SZ requires the incorporation of various models into one uniting model of the multifactorial disorder and use of this model for the evaluation of new drugs.
1. Introduction
Schizophrenia (SZ) is a severe mental disorder with a complex pathomechanism. The symptoms of SZ are organized in three classes: positive (e.g. hallucinations, delusions, and thought disorders), negative (e.g. social withdrawal, flattened affect, and anhedonia), and cognitive (e.g. deficits in working memory, poverty of speech, difficulties in attention) [1]. The first concept of SZ was proposed by Emil Kraepelin in 1919 under the name "dementia praecox", who claimed that mental disorders are associated with neuropathology, physiology, and biological chemistry of the brain [2]. According to Kraepelin, different disease syndromes can be associated with different areas of the brain that are more susceptible to pathological stimuli [3]. Eugen Bleuler modified the Kraepelin concept and coined the new name “schizophrenia” for the disease in 1950. He thought that SZ was a heterogenous group of diseases and introduced the concept of “group of schizophrenias” [4,5]. Bleuler elaborated two dichotomous classifications of SZ symptoms: i.e., basic and accessory and primary and secondary symptoms [6]. Basic symptoms are necessarily present in all types of SZ while accessory symptoms may or may not occur. The dichotomy between primary and secondary symptoms is based both on etiology and pathogenesis. Primary symptoms are derived directly from neurobiological abnormalities whereas secondary symptoms are rather psychological reactions that can result from primary symptoms. Moreover, Bleuler considered the alteration of associations as the only symptom which is both basic and primary [6]. Kraepelin and Bleuler speculated that brain dysfunction is responsible for SZ symptoms, in particular that frontal and/or temporal lobe dysfunctions are linked with cognitive abnormalities [7]. However, they were not able to prove this hypothesis with the tools of their era. In 1972, Plum stated that “schizophrenia is a graveyard for neuroanatomists” as no single pathological lesion could be found. On the other hand, Mirsky concluded in 1969 that there was evidence of brain dysfunction in SZ from cognitive, neurological, and electroencephalographic examinations [7]. This was further confirmed by Seidman in 1983 who searched for the neurobiological basis of SZ, in an era when computer tomography (CT) and positron emission tomography (PET) began to be applied to patients with SZ. The SZ concept has changed significantly over the last 30 years, as SZ has been increasingly considered as a neurodevelopmental disease [7].
In spite of numerous attempts to provide reliable diagnostic tools of SZ, it remains defined by the identification of behavioral disorders, in particular psychotic symptoms. Several putative biological markers associated with this disorder have been identified [8], including metabolic biomarkers, neurocognitive dysfunction, brain dysmorphology, and neurochemical abnormalities. However, none of these markers have the appropriate sensitivity and specificity in diagnostic tests. Genetic and association studies targeting multiple loci and genes have also failed to fully demonstrate that any particular gene variant or combination of genes underlies the formation of SZ [4]. Genetic factors are rather thought to predispose to the development of this disease when accompanied by unfavorable environmental factors such as long-lasting stress or multiple stressors simultaneously.
There are significant limitations of current antipsychotics used to treat SZ, including classical first-generation drugs such as chlorpromazine but also second- and third-generation drugs, olanzapine, risperidone, clozapine, aripiprazole, brexpiprazole, cariprazine, and many others. First of all, they are mainly efficient against positive symptoms. Secondly, epidemiologic studies have demonstrated that approximately 50% of the patients with chronic SZ do respond to treatment and have a favorable outcome in terms of functionality measures [9,10]. Finally, current antipsychotics exert severe side effects such as Parkinson-like extrapyramidal symptoms (in particular first-generation antipsychotics), and metabolic side effects including obesity, diabetes etc. (in particular second-generation antipsychotics). The side effects of drugs used to treat SZ reduce patient compliance with the drug regimen, which often leads to recurrence of the disease. There is no doubt that novel antipsychotics are urgently needed and the methods to investigate them in the early, preclinical, and clinical phase of development have to be improved.
In the light of above, the aim of this review is to summarize shortly current concepts and hypotheses of SZ and to discuss current in vitro and in vivo models to study SZ and antipsychotics. Special attention is given to advantages and limitations of the presented approaches as well as their applicability to model the disease which fully manifests only in humans and which is often treatment-resistant with available drugs.
2. Hypotheses and Concepts of SZ
2.1. Neuroanatomy of SZ
Over the last 30 years technologic achievements in functional brain imaging have supplied exciting and informative insights into the functional neuroanatomy and neurochemistry of SZ [11]. Modern imaging methods also enable the study of abnormalities on the receptor level. PET and single photon emission computed tomography (SPECT) have been applied to measure dopamine-related parameters using radioligands which bind to receptors, transporters, or other target molecules [12], and to trace a metabolic pathway [13].
In schizophrenic patients, dilation of the brain cavities, volume changes of some nuclei basales, and changes in the hippocampus were observed. SZ patients often have an enlarged ventricular system, third ventricle enlargement, temporal anomalies of the upper comers [14], and irregularities within the frontal and temporal lobe structures [15]. One approach to understanding of the lesions is assessment of changes occurring in healthy siblings, used as so-called endophenotype [14]. Most often the changes in healthy siblings are less intensive than in suffering ones but still more visible than in healthy individuals.
According to many preclinical studies, anatomical and physiological changes are frequently observed in the hippocampus in patients with SZ. A structural decrease in hippocampus volume was observed in post-mortem and imaging studies, in particular in patients with the first episode. In addition, the greater reduction of hippocampal volume was observed in chronic patients vs early illness patients [16]. In addition to anatomic changes, people with SZ also have differences in the activity of the hippocampus. This structure plays a major role in declarative memory. which leads to disability during memory tasks in patients with SZ. Moreover, numerous cellular and molecular changes have been observed in people suffering from this disorder. People with SZ have reduced stem cell proliferation in the dentate gyrus. Next, SZ patients demonstrate a reduced number of synapses between the hippocampal mossy fibers and pyramidal neurons, which is associated with changes in synaptic plasticity markers [17,18]. All above findings confirm the dysfunction of the hippocampus structure in SZ.
The prefrontal cortex is also involved in the pathology of the disorder. The region called the dorsolateral prefrontal cortex is responsible for the working memory, which is disturbed in the disease. Therefore, it is considered that abnormal prefrontal cortex activity contributes to cognitive dysfunction observed in patients with SZ. Interestingly, the prefrontal cortex and hippocampus are connected neuronal systems and dysregulated prefrontal and hippocampal activity may underlie some of the symptoms of SZ [18].
2.2. Disturbances in Neurotransmission in SZ
Dopaminergic hypothesis of SZ supplemented by the glutamatergic hypothesis are still the most important regarding the neurobiology of this disease (see Figure 1) [19].
The original dopaminergic hypothesis claims that overactivity of the dopamine system causes schizophrenic symptoms, in particular hallucinations and delusions. On the other hand, anhedonia and social withdrawal result from reduced activation of the D1 receptor [20]. The most commonly accepted hypothesis today is aberrant silent hypothesis which underlies the symptoms of SZ with abnormalities in the dopaminergic neurotransmission. This hypothesis is on the incentive salience hypothesis [21] which proposes that the mesolimbic dopaminergic neurotransmission is critical in the attribution of salience governing attention and affects decision making and functioning [19,22]. The aberrant salience hypothesis suggests that the attribution of salience is hindered by excessive dopamine firing in psychosis, while in healthy individuals, dopamine is responsible for mediating contextually appropriate saliences [19,23].
Another possible mechanism that can be involved in SZ is dysfunction of the glutamatergic system that affects synaptic plasticity and—among others—N-methyl-D-aspartate (NMDA) receptor signaling [24]. These receptors play an important role in neurotransmission and plasticity, and their underactivity leads to morphological changes that can cause structural changes in the brain and chronic psychosis [25]. A significant importance for glutamatergic system in SZ was first proposed about 30 years ago. It was based on the observation that the psychotomimetics, phencyclidine (PCP), and ketamine induce psychosis and cognitive deterioration similar to SZ by antagonizing neurotransmission at NMDA-type glutamate receptors [26]. The combination of NMDA hypofunction and presynaptic dopamine dysfunction can provide a very good explanation for most clinical aspects of SZ [27]. Further support for the glutamatergic hypothesis of SZ is supplied by the investigated possibility to use ligands of metabotropic glutamate receptors (mGluRs) [28,29], in particular their positive allosteric modulators [30] to alleviate SZ symptoms.
Other neurotransmitter systems are also involved in SZ. In particular, changes in serotoninergic neurotransmission may lead to negative and cognitive symptoms of the disease [19,31]. The serotonin hypothesis of SZ is based on the reports about the hallucinogenic drug lysergic acid diethylamide (LSD) and its mechanism of action linked to serotonin [32]. It is well known that in particular second-generation antipsychotics, such as olanzapine or risperidone, display better affinity to the serotonin 5-HT2A receptors than to the dopamine D2 receptors, which is also a determinant of their atypicality. Atypical antipsychotics exert fewer extrapyramidal symptoms and are more efficient in the treatment of negative and cognitive symptoms than the classical first-generation drugs. Antagonism of 5-HT2A receptor and agonism or partial agonism of 5-HT1A receptor are favorable for antipsychotic activity of novel compounds [33].
Other aminergic neurotransmitter systems, such as adrenergic, muscarinic and histaminergic also have some role in SZ. They are particularly useful for the treatment of cognitive disturbances [34,35,36,37].
It has been also reported that GABA-ergic (γ-aminobutyric acid) neurotransmission is altered in SZ [38]. The GABA system is one of the main inhibitory systems in the brain and is capable of diminishing the dopaminergic system neurotransmission. It has been found in clinical studies that GABA receptor agonists alleviate some SZ symptoms [39]. Further studies are needed to determine if ligands acting on the inhibitory GABA system may supply a sensory filter for the brain in flames as it was suggested.
2.3. Genetic Origins of SZ
Despite many years of research, the knowledge of the etiology and pathogenetic development of SZ is still limited. First reports about heritability of SZ are derived from family studies. It is estimated that if both parents of the patient suffer from SZ, the probability that the patient will develop SZ is about 50%. Studies involving identical twins indicate 60%–90% heritability [48,49,50,51]. These studies tried to indirectly assessed the heritability of SZ, however newer investigations enable direct genomic approaches (comparative genomic hybridization; single nucleotide polymorphism (SNP) chips; next-generation sequencing (NGS); genome-wide association studies (GWAS), and the clustered regularly interspaced short palindromic repeats-associated nuclease 9 (CRISPR/Cas9) genomic editing system) [52]. These modern approaches also have their shortcomings, which are mostly consequences of the complexity of the pathomechanism of SZ [52].
The first specific gene which was associated with SZ is disrupted-in-schizophrenia 1 (DISC1) [53]. It was demonstrated that a balanced translocation disrupting this gene co-segregated with SZ; however, the mechanism of it and linkage to SZ symptoms remains unclear. Since 2014 when GWAS produced a number of hits, some genetic changes have been associated with SZ comprising complement component 4 (C4), neurexin 1 (NRXN1), RNA-binding motif 12 (RBM12), and SETD1A [54]. It should be also stressed that copy number variants (CNVs) have been strongly implicated in SZ risk, as previously reviewed [54,55].
It is assumed that genetics is one of the keys factor leading to the emergence of SZ; however, the occurrence of the disease depends also on environmental stress and neurodevelopmental factors. Genetics may predispose a patient to the disease; however, genetic factors solely do not lead to the development of SZ. Whereas proofs from twin studies suggests a strong heritable component, few individual loci have been found in genome-wide screens, which suggests a role for epigenetic effects [56]. It is supposed that large numbers of weakly acting loci may cumulatively increase disease risk, including several mapping to epigenetic pathways and contributing to the complexity of the disease [56].
2.4. Neurodevelopmental Hypothesis of SZ
According to the neurodevelopmental hypothesis, the etiology of SZ includes pathological processes of the central nervous system that take place during fetal life and then in the perinatal period and early childhood [57,58]. A very important phenomenon in the neurodevelopmental hypothesis is excessive pruning of synapses and loss of brain plasticity. Much evidence indicating the neurodevelopmental origin of SZ has been found in in vitro, in vivo, and post mortem studies which reveal the quantitative loss of neurons, changes in the size of some brain structures, and cytoarchitectonic changes in cells. The first model which took into account neurodevelopmental abnormalities was termed the “single hit model” which was followed by the “second hit model” and was finally developed into the “multiple hit model” [59]. The multiple hit model best represents complex genetic, social, and environmental interactions that have been explored as contributing to the development of SZ [59].
3. In Vitro Models of SZ
Advances in the field of stem cell genetics and biology make in vitro research important for understanding central nervous system diseases. The use of in vitro techniques with a selected cell model makes it possible to carry out molecular, developmental, and pathophysiological studies with high credibility. In this type of research, the selection of an appropriate in vitro model is very important, but because of the large access to many types of cell lines this task is difficult [60].
One of the important models for studying SZ are nerve cell cultures. In 1977, Banker and Cowan obtained for the first time the primary culture of nerve cells from the rat hippocampus. This line turned out to be an ideal model of neural cultures used to date [61]. Some populations of nerve cells can be obtained directly from living people and cultivated as primary cultures. However, ethical views limit the use of primary human brain tissue for research. Due to the fact that the primary nerve cells are also difficult to obtain (especially human neurons), cell lines which can be differentiated into neural cells have been used in in vitro research. What is more, stem cells have also found widespread use in research. The presented cell lines are very good models for the study of intracellular mechanisms as a result of drug administration, for molecular research and genetic susceptibility as well [62,63].
Continuous cell lines established from human tumors are widely used as in vitro models. Some of the tumor cell lines have the characteristics of nerve cells. Currently, the most frequently used cell line with neuronal features is the SH-SY5Y line (Figure 2)—cells of the human neuroblastoma tumors. The SH-SY5Y cell line was first obtained in the seventies of the last century from the bone marrow of a patient with a neuroma. Cells show biochemical and functional characteristics of neurons, which can include: neurite growth, synthesis of neurotransmitters, and activity of neuronal enzyme markers. An additional advantage of this cell line is the expression of specific proteins and isoforms of proteins that are not naturally present in primary rodent cultures [63]. Thanks to these properties, this line is the most frequently used in vitro model for Parkinson’s disease, SZ, Alzheimer’s disease and Huntington’s disease research as well as for the study of the mechanisms of antidepressants and antipsychotics. Furthermore, SH-SY5Y cells can be differentiated using substances such as retinoic acid, resulting in different phenotypes of nerve cells. Non-differentiated cells proliferate continuously and are characterized by the presence of immature neurons. Often undifferentiated SH-SY5Y cells are considered to be immature catecholaminergic neurons which cannot be counted among their positive attributes. After application of differentiation-inducing substances, the cells exhibit characteristics similar to primary neurons. There is a decrease in the rate of proliferation and an increase in c neuron-specific enolase (NSE) activity, which is one of the tumor markers. An important advantage of SH-SY5Y cells can be differentiation into neuronal phenotypes such as dopaminergic, adrenergic and cholinergic phenotypes. One of the most commonly used methods of inducing cell differentiation is the addition of retinoic acid (RA) to the culture medium [64,65].
In addition to the numerous advantages of the SH-SY5Y cell line used in the study of central nervous system diseases, neurotoxicity, and neuroprotection, several disadvantages should be mentioned. The described cell line was derived from tumor cells, which underwent numerous neoplastic transformations as a result of multiple passages. Therefore, this line has a diploid number of chromosomes and is characterized by unlimited growth time. To sum up, due to numerous genetic aberrations, SH-SY5Y cells may change their current function. In addition, cells show little sensitivity to neurotoxins and are not synchronized, therefore not all have markers that are characteristic of mature neurons.
Another important model to study the mechanisms of central nervous system diseases are stem cells. Stem cells must have two main features. The first feature is the ability to proliferate in the unlimited manner. This characteristic also applies to the cancer cells described above, which are divided in an uncontrolled manner, while stem cell division is strictly regulated. The second very important feature is the ability to share and differentiate into mature cells. There are two main classes of stem cells used as a model for in vitro studies of neurodegenerative diseases: pluripotent, which can become any cell in the body, and multipotent, which can turn into any type of cell but a more limited population.
Multipotent stem cells are a very good option for clinical use. Thanks to their properties, they can become any cell type found in the human body. Multipotent cells together with the respective compounds differentiate into particular cell lines [66]. Neuronal stem cells (NSCs) are an excellent in vitro model for studying the mechanisms of SZ. Cells occur in the central nervous system in the subgranular zone (SGZ) of the hippocampal dentate gyrus and in the periventricular zone. Neuronal stem cells are characterized by an unlimited ability to self-renew, the ability to differentiate into specialized cells and also have a normal number of chromosomes compared to cancer-derived cells. NSCs provide a very good model for in vitro research, in particular to explore the mechanisms of drugs treatment and to identify susceptibility genes for mental disorders. Additional features of neuronal stem cells are strong plasticity, easy isolation, and in vitro culture, as well as low immunogenicity and high immigration capacity. Neuronal stem cells were used to study pathogenic changes in the expression of the DISC1 gene, which is important in the development of SZ. Application of the NSC model in in vitro studies allows for investigation of the molecular functions of susceptibility genes for SZ [62,66,67].
In addition, induced pluripotent stem cells (iPSCs) are reported as a very useful cell technology that has been applied to cells taken from psychiatric patients. IPSCs can be transformed into all cells in the human body, including central nervous system cells [66]. In particular, skin fibroblasts were taken from SZ patients and healthy controls, “reprogrammed”, and then neurons grew from pluripotent stem cells. Interestingly, neurons from SZ patients presented altered expression of genes involved in signaling of glutamate, cyclic adenosine monophosphate (cAMP) and the WNT protein family, as well as reduced synaptic connectivity and neurite number [68]. IPSCs might be also used to study in vitro neurodevelopmental features of psychiatric disorders. It is also possible to provide a source of models of neurons from individual patients by reprogramming human fibroblasts directly into neuronal cells (i.e., “induced neuronal cells”) [69]. This will help to investigate mechanisms of drug response as well as to study mature cellular pathophysiology of SZ.
Despite the many advantages of application of in vitro models of stem cells, there are several limitations that have to be mentioned. At the moment, current research involving stem cells is based on several samples; it is still necessary to optimize the conditions of isolation and stem cell differentiation. Protocols for induction of stem cells are still in the development phase, so when choosing them as a model for research, we need to take into account not only their advantages but also the disadvantages and the fact that they have not been fully characterized [62].
In recent years, a line of mouse hippocampal neurons (HT22, Figure 3) has been used as the main and very good model for in vitro studies of mental diseases such as SZ. More specifically, HT-22 is an immortalized mouse hippocampal cell line subcloned from the HT-4 cell line. The HT-4 cells were originally immortalized from a primary mouse hippocampal neuronal culture [70].
This line is a valuable tool for understanding the molecular and cellular processes important in SZ and other neuropsychological diseases [70]. As it is commonly known, the hippocampus is essential for the proper functioning of the brain mainly to encode and transmit information in the sensory system. Neuropsychiatric disorders are associated with structural and functional abnormalities of hippocampal neurons. HT22 cells can provide a simple way to identify molecules and complex cellular mechanisms in SZ. HT22 cells provide an in vitro model for cytotoxicity studies, mainly cytotoxicity of glutamate, which is an indicator of many neurodegenerative diseases [71]. More specifically, glutamate is a neurotransmitter that is used as a signaling molecule between nerve cells and is mainly responsible for the proper functioning of the brain, including learning and memory. Small levels of glutamate are necessary for the proper functioning of the brain, whereas elevated levels of glutamate can lead to excessive stimulation of nerve cells which will damage cells or activate the intrinsic pathway of apoptosis. Cytotoxicity, which is induced by glutamate, plays a pivotal role in the development of neurodegenerative disorders such as Parkinson’s disease, Alzheimer’s disease, SZ, and depressive disorders. In this regard, inhibition or reduction of glutamate level is known as effective treatment strategy of the above mentioned disorders. However, this is not the case of SZ, which is characterized by the reduced level of glutamate. Moreover, hippocampal neuronal cells are useful for testing drugs for many diseases of the central nervous system [72,73,74]. An additional advantage of culturing HT22 cells is the feasibility of conducting and performing experimental procedures. The hippocampal cells, as a result of numerous passages, retain their cytoarchitectonic properties and are useful to study many aspects of cellular and molecular mechanisms [74]. Available literature data does not show disadvantages of implementation of the mouse hippocampal neurons as an in vitro test model.
Application of the right in vitro cell model is very important aspect in the drug discovery process. One of the greatest achievements in cell culture techniques was obtaining three-dimensional (3D) culture systems, which are one of the best models for in vitro studies. Until now, in most tests traditional two-dimensional, single-layer cell cultures (2D) on rigid and flat surfaces have been used [63]. Although 2D cultures are routinely used in laboratories around the world, they do not reflect the natural environment of cells. Culture cells on flat media have changed metabolism, gene expression and response to stimulus. Recent studies have shown that 3D cultures more accurately reflect the conditions that prevail in the body, which increases productivity of the cell cultured. Furthermore, these cells have an increased physiological response to bioactive substances compared to two-dimensional cultures. However, the most important difference between 2D and 3D cell culture is the difference in their morphology. In two-dimensional cultures, cells adhere to the substrate only on one side of the cell. However, in the case of three-dimensional cultures, the cells adhere to the substrate with their whole surface. More specifically, the substrate surface mimics the extracellular matrix by formation a tissue scaffold that provides structure for cells to attach and grow [75]. In 3D cultures, a commercial culture medium is used, which includes: collagen, lamins, proteoglycan, entactin, and growth factors. The 3D in vitro model is the best model to study Alzheimer’s and Parkinson’s diseases as well as SZ. Next, three-dimensional cultures are a cell model that provides better interactions between cells and cells and extracellular matrix; 3D systems do not have complex vascular systems that support tissues in vivo (oxidize, provide nutrients, remove waste). Cells in three-dimensional cultures are suitable for this function by diffusion. In the case of larger spheroids, the cells at various depths (from the surface of the spheroid) are differently nourished and thus found in other phases of the cell cycle [75,76]. In contrast to 3D cultures in a two-dimensional monolayer culture, all cells are evenly nourished and oxygenated. In order to get to know all the mechanisms that underlie neurodegenerative diseases one should create and choose a cellular model that best reflects the physiological conditions that prevail in the human brain and three-dimensional cell cultures are an example of it [63,75].
4. Animal Models of SZ
4.1. Pharmacological Models of SZ
Two main pharmacological models of SZ include the amphetamine model and phencyclidine model. An amphetamine model of SZ, developed in 1950s, is based on the hyperfunction of the dopaminergic neurotransmission in the mesolimbic neural system. However, D2 receptor antagonists block amphetamine-induced psychosis, including persecutory delusions and auditory hallucinations that are considered as positive symptoms of SZ. It has been presented that single amphetamine administration enhances spontaneous locomotor activity and stereotyped movements as well. In addition, repeated amphetamine administration induces locomotor activity sensitization and then exaggerating hyperlocomotion (i.e., index that has translational relevance to positive symptoms) related to amphetamine re-challenge after withdrawal [77].
It has also been shown that chronic amphetamine treatment did not induce deficits in social interaction in rats [78]. In addition, amphetamine sensitization may be accompanied by deficits in prefrontal cortex (PFC)-dependent cognitive tasks, such as impairments in reversal learning and the extra-dimensional shift in the attentional set-shifting task [77,79], and reduced accuracy with shorter stimulus duration, as well as an increase in omissions in the five-choice serial reaction time task [80]. However, pretreatment with a low dose of either clozapine or haloperidol prevents the induction of sensitization [81], and both drugs attenuate amphetamine-induced impairment in attention performance [82]. Moreover, repeated amphetamine has no effect on acquisition or retention of spatial visual learning and memory (i.e., hippocampal dependent cognition) determined in the Morris water maze [77]. In this regard, cognitive impairment induced by chronic amphetamine administration is restricted to some PFC-dependent tasks; however, hippocampal function is unaltered.
Increasing evidence indicates that hypofunction of glutamatergic neuronal system has been observed in schizophrenic patients [83]. In particular, administration of an NMDA-type ionotropic glutamate receptor–antagonist, for example ketamine or PCP, induced symptoms (i.e., delusions and hallucinations) observed in patients with SZ [84]. PCP induces positive symptoms of SZ in both acute and stabilized chronic patients. In addition, psychotic symptoms are observed in normal volunteers even after administration of low doses of PCP accompanied by negative symptoms of SZ, such as progressive withdrawal and poverty of speech [85]. Additionally, PCP induces cognitive impairments after acute (low dose) and chronic treatment [85]. In the acute PCP animal model of SZ hyperlocomotion, social withdrawal, and cognition and prepulse inhibition (PPI) impairments are observed [78,86,87,88]. However, chronic PCP treatment is used to more accurately mimic the SZ symptoms in rodents. There are some variations in the subchronic PCP treatment that affect the PCP peak concentration in the brain and may induce some differences in the cognitive paradigm. In particular, differences in subchronic PCP treatment, including PCP dose, gender, and strain as well as period of administration [89].
Typical and atypical antipsychotics (e.g., haloperidol and clozapine) attenuate PCP-induced sensitization that confirms the possible modulation of the positive symptoms of SZ [90]. However, negative symptoms are also observed in rodents, as seen in patients with SZ. For instance, social impairments are induced by chronic PCP treatment in rats [91] and mice [92]. They are reversed by haloperidol and clozapine administration in rats [88], and by clozapine but not haloperidol in mice [92]. It is also known that a subchronic PCP dosage regimen impairs reversal learning in rodents, as observed in SZ patients. A reversal learning task is composed of an initial phase (it requires memory of a previously learned reward), followed by second reversal phase. Animals have to acquire the new strategy when a previously rewarded strategy is blocked. In this regard, animals are required to present flexibility and motivation as well as attention so they can implement a new response and inhibit a response that was previously learned [93]. Novel antipsychotic drugs can attenuate the executive dysfunction that is observed in patients with SZ. For example, acute treatment with clozapine, lamotrigine, and phenytoin [94] but not with haloperidol [95] reversed the acute PCP-induced selective deficit in reversal learning. Treatment with asenapine [96] and sertindole [97] as well as clozapine but not haloperidol and chlorpromazine [98] ameliorated deficits in reversal learning induced by subchronic PCP administration. However, it was also shown that acute treatment with ketamine and dizocilpine (MK-801) i.e., NMDA antagonists, produces a pharmacological deficit in reversal learning [99] as observed in SZ.
It has been shown that cortical function impairment is one of the hallmarks of SZ. Deficits are moderate to severe across several domains, including attention, working memory, verbal learning and memory, and executive functions [100]. Below some tests to study cognitive abilities and impairments are presented.
The prefrontal cortex is responsible for many of our cognitive abilities and executive functions such as the ability to modify behavior in response to the altering relevance of stimuli (i.e., cognitive flexibility). SZ patients poorly performed the intradimensional shift/extradimensional shift (ID/ED) task [101] as well as the Wisconsin Card Sorting Test (WCST) [102]. The rodent version of the ID/ED task and WCST is known as the attentional set-shifting task (ASST) [103]. In the ASST rodents must select a bowl containing a food reward based on the ability to discriminate the media covering the bait or the odor. This test requires animals to initially learn a rule and form an attentional ‘set’ within the same stimulus dimensions. In one ED phase of the ASST test, regarded as the index of cognitive flexibility, the rodent must switch their attention to a new, previously irrelevant stimulus dimension and discriminate between the odors and no longer between the media covering the bait [104]. Rats must perform a series of seven discriminations, including simple discrimination (SD), compound discrimination (CD), reversal 1 (R1), intra-dimensional shift (IDS), reversal 2 (R2), extra-dimensional shift (EDS), and reversal 3 (R3). Administration of NMDA receptors antagonists (e.g., ketamine) induces a broad range of SZ-like impairments, including psychotic-like behaviors as well as negative symptoms and cognitive deficits. More specifically, administration of ketamine for 10 consecutive days produces distinct deficits in the ASST that may be attenuated by antipsychotic drugs [105] and nicotinic receptors ligands as well [104]. It was also demonstrated that selective impairments are found in the EDS phase after subchronic treatment with PCP (for seven days followed by seven days washout period). This deficit was ameliorated by 7-day administration of clozapine and risperidone but not by haloperidol [106].
Novel object recognition (NOR) in rodents is used to study declarative (episodic) memory, one of the several cognitive domains that are abnormal in SZ. NOR evaluates the spontaneous exploratory behavior of rodents. In this test, NMDA receptor antagonists, including PCP, MK-801, or ketamine induce deficits in NOR [107]. In the NOR test two identical objects are exposed to animal for short time (3–10 min) in a restricted chamber without any additional cues that may assist learning and memory. First, the time spent to explore each of two identical objects is recorded (i.e., acquisition). The inter-trial interval should be included, mostly between 3 min and 1–3 h, but not greater than 24 h. During second exposure (i.e., retention trial) the rodent is placed back in the chamber and one of familiar objects is replaced by novel object, in the same place as familiar one. The Discrimination Index (DI), enables discrimination between the new and known objects, i.e., it applies the difference in exploration time for familiar objects, and then divides this value by the total amount of time of exploration of the new and known objects. It is expressed by the formula: (DI) = (Novel Object Exploration Time/Total Exploration Time) – (Familiar Object Exploration Time/Total Exploration Time) × 100.
It is known that schizophrenic patients show deficits in spatial working memory recognition tasks that are relevant to the NOR paradigm [108]. All atypical antipsychotic drugs (i.e., clozapine, asenapine, olanzapine, quetiapine, risperidone, and sertindole) reverse NOR impairments induced by subchronic NMDA receptor antagonist (e.g., PCP or MK-801) treatment [109].
The Morris water maze (MWM) is known as a visual learning and memory task that depends on coordinated action of neurotransmitter systems and several brain regions [110]. The MWM depends on many cognitive substrates such as learning, working and long-term memory, attention, and retention as well. They all are relevant for the deficits found in schizophrenic patients. In the MWM, a round pool filled with water is defined as testing area. There is also a hidden platform that is submerged below the water surface. The animal learns how to escape by finding the platform, in most cases with the help of visual cues. Alternatively, the platform can be placed in another quadrant, or removed during another phase of the experiment. This way, memory retention and extinction can be investigated. Subchronic treatment with PCP induced a significant learning and memory impairment (i.e., increase in swimming distance to the platform). However, PCP-induced deficits are reversed by second-generation antipsychotics such as sertindole, clozapine, and risperidone [111].
Attention has been also identified as being typically impaired in SZ patients. Attention allows an individual to detect, select, and process relevant stimuli and in the same time is responsible for filtering out unnecessary stimuli from all possible environmental stimuli. The 5-Choice Serial Reaction Time Task (5-CSR) has been developed to evaluate attention (sustained and divided) impairments in rodents [112]. In this test animal has to detect the light stimulus (illumination) presented in one of five apertures of a dark operant chamber. After successful identification (via a nose poke) of the correct illuminated aperture, a reward (food pellet) is delivered and the next trial is initiated. If the animal detects incorrect aperture (without illumination) or does not respond within limited time period, then rodent does not receive food reward and house light is illuminated (during 5 s). If the animal responds during inter-trial interval (i.e., before light stimulus is presented), a time out period is initiated as it is an incorrect response. An acute administration of PCP produces a decrease in percent of correct responding and choice accuracy as well as an increase in the correct latency [113]. However, repeated treatment with PCP induces cognitive impairment that leads to a significant reduction in correct responses and accuracy, and increased premature responding. In addition, it causes a significant increase in correct latency, without any influence on completed number of total trials or latency to collect the food reward. Moreover, chronic administration of clozapine partially ameliorated the performance disruptions that were induced by repeated treatment with PCP, significantly reducing the accuracy impairment as well as the increase in premature responding [113].
It has been reported that negative symptom of asociality (i.e., withdrawal from social contact or lack of desire to have social contact) is a core behavioral feature in SZ [114]. Some literature data reveals that NMDA receptor antagonists, such as PCP, MK-801, and ketamine induce social interaction deficits [112]. In particular, subchronic PCP administration impairs social interaction in adult female rats [115]. Social behavior impairments have been reported in mice during PCP withdrawal for up to 28 days after 2 weeks of treatment [92]. However, there are some discrepancies in social interaction evaluation, for example a significantly increased time in which rats actively engaged in social interaction [116]. This discrepancy seems to be due to the differences in the dose, time between the PCP final injection and the social interaction test that was applied. In particular, increased social activity was observed from 24 h until 6 weeks after the final PCP injection [116].
Social interaction is typically evaluated by placing a pair of unfamiliar rodents into an arena under bright conditions, and the amount of time when animals spend engaged with one another is calculated [117]. To minimize anxiety-related components of social interactions several simple changes were described, such as low lighting conditions, habituation to the arena, close weight-matching of the animals, and ad-libitum food availability [114]. A significant decrease in social interaction by test subjects is mostly interpreted as social withdrawal [78,88]. In a different version of social test an arena is divided into several chambers and the time the rodent spends in the chamber with a caged unfamiliar conspecific and the empty chambers is scored [118]. It reflects social avoidance based on calculated preference for the empty chamber. It has been reported that several brain regions, including the hippocampus, amygdala, prefrontal cortex, and cerebellum have been implicated in both social interaction and negative symptoms of SZ [114].
4.2. Genetic Animal Models of SZ
Animal models of disruption can be obtained for almost all human genes [119]. The mouse genome is almost as well characterized as the human and mouse models are cost effective, straightforward to produce, and allow investigation at the molecular, cellular, circuit, and behavioral levels [119]. The disadvantage of rat models is the cost of maintenance and it is not balanced by obvious advantages of rat models. The high-throughput and low-cost models involve zebrafish (Danio rerio) and fruit fly (Drosophila melanogaster), but obviously these systems are not applicable for reproducing more complex human behaviors [119]. It should be stressed that there are a number of shortcomings of usage of genetic animal models to study neuropsychiatric disorders. Interpreting SZ-like phenotypes in mice, including complex symptoms such as paranoia and delusional beliefs, is challenging: they can only be inferred indirectly from disordered mice behavior, a major limitation of modelling SZ in animals [119].
Two most popular genetic mice models of SZ involve DISC1 mouse models [120,121] and neuroregulin-1 models [121].
DISC1 is an intracellular scaffolding protein which possesses a number of interacting proteins, facilitating the formation of protein complexes [121]. The DISC1 gene was found as a risk factor for mental diseases in a Scottish pedigree that displayed a translocation between chromosomes 1 and 11 (q42;q14.3). There are three main types of DISC1 mice models of SZ: haploinsufficiency models, point-mutation models, and transgenic (dominant negative) models.
The chromosomal translocation identified in the Scottish family disrupts the DISC1 gene at intron 8 on one chromosome, whereas this gene on the other chromosome is kept intact [120]. The disruption of the gene can cause loss-of-function, most probably because of nonsense-mediated mRNA decay [120]. Thus, the total result caused by this genetic mutation can be haploinsufficiency [120]. Three variants of this model have been obtained, termed Δ25bp [122], Δex2/3 [123], and Disc1 locus-impairment (Disc1-LI) [124]. In spite of the fact that Δ25bp mice display working memory impairment and Δex2/3 mice are characterized by a higher impulsivity phenotype, these models exhibit no additional behavioral endophenotypes which typically occur in most severe mental illnesses. However, recent detailed studies found abnormalities at the cellular level in these mice which were reviewed elsewhere [120]. It remains an open question as to what extent the haploinsufficiency models can contribute to the understanding of the molecular biology of the disease, in particular in the light of the complex nature of the DISC1 gene.
Regarding point-mutation models based on DISC1 gene, it is worth mentioning that two missense mutation models were obtained, Q31L and L100P, which lead to depressive-like and schizophrenic-like behavioral phenotypes, respectively [125]. Further studies on the L100P model revealed a number of abnormalities, such as increased dopamine function, changed neurexin function, deregulated glycogen synthase kinase-3α (GSK3α) activities in synapses, and abnormalities in interneuron development [120]. It should be stressed that these models are sensitive to the animal housing conditions and subtle factors such as diet might change behavioral phenotypes typical for these models which should be taken into consideration while interpreting data from these models.
An alternative option to point-mutation based models is to express putative dominant-negative (DN) isoforms of DISC1 in mouse brains to obtain transgenic models [120,126,127,128,129]. In such models a number of cellular and anatomical functions is disturbed with consequences for mental disease like animal behavior, including e.g. social withdrawal. There are a few issues which should be considered while using transgenic models to model SZ. Importantly, cell functioning and communication (including astrocytes and oligodendrocytes) are impaired, so these models may be applied to study disease-related impairment at the cellular level. Next, transient disruption of DISC1 functioning during a neonatal period disrupted long-term potentiation/long term depression (LTP/LTD) later in adulthood, which supplies a basis for cognitive disfunction and its delayed onset in mental disorders [120]. This model can also help to link oxidative stress with human mental diseases and can be used to study SZ-related impairments in different neurotransmitter systems.
The second important animal genetic model of SZ is neuregulin-1 based model. Neuregulins are epidermal growth factors which activate ErbB receptor tyrosine kinases, and have significant roles in normal developmental processes, plasticity, and oncogenesis [121]. In the central nervous system the best characterized neuregulin-1 (NRG1) regulates neuronal migration, glial development and differentiation, and neurotransmission and plasticity [121]. It was suggested in 2002 that NRG1 is a candidate gene linked with SZ [130] and down-regulation of neuregulin1/ErbB4 signaling in the hippocampus is critical for learning and memory [131]. As a consequence a number of mice models were constructed to investigate the functional and behavioral consequences of targeting NRG1 and its receptor, ErbB [121]. In a new study Papaleo et al. [132] developed a transgenic mouse model (NRG1-IV/NSE-tTA) in which human NRG1-IV is selectively overexpressed in a neuronal specific manner. They showed that NRG1-IV/NSE-tTA mice display disturbed behaviors relevant to SZ, such as impaired sensorimotor gating, discrimination memory, and social behaviors. Olaya et al. [133] generated transgenic mice with forebrain-driven Nrg1 III overexpression (Nrg1 III tg) and stated that these mice exhibited several SZ-relevant behaviors including social and cognitive deficits as well as impaired sensorimotor gating. Finally, there are also reports about the role of neuregulin-3 in SZ [134] which may also be a base of animal genetic models.
4.3. Neurodevelopmental Models of SZ
According to human epidemiologic studies infection, malnutrition, or hypoxia in the fetus are the factors which can contribute to the development of SZ. In this context, neurodevelopmental animal models of this disease have been generated where the exposure to harmful prenatal factors was highly controlled [142]. Among neurodevelopmental and lesion models, the prenatal polyI:C injection model, the prenatal methylazoxymethanol acetate (MAM) injection model, the prenatal lipopolysaccharide (LPS) model, and the neonatal ventral hippocampal lesion model will be shortly characterized.
PolyI:C is a synthetic analogue of double-stranded RNA. Prenatal injection of PolyI:C to the pregnant rodent dams results in SZ-like behavior of their adolescent offspring. The observed changes include enhanced locomotor activity as a reaction to psychostimulant (but not increased spontaneous locomotor activity), impaired PPI in auditory startle responses, selective impairment in non-spatial memory processing, social withdrawal, and anhedonia [142]. It is worth mentioning that pretreatment with antipsychotic or antidepressive drugs reversed these behavioral changes in this model by decreasing positive and cognitive symptoms. Less evidence is available regarding the drug efficacy for negative symptoms in this model.
MAM is a neurotoxin which administered at a gestational point of time to rat dams interferes with behavioral phenotypes in their offspring. The behavioral changes include increased spontaneous locomotor activity and locomotor sensitivity to the psychostimulant and cognitive and negative symptoms, such as the reduction of social interaction before puberty and deficits in PPI, spatial learning, and cognitive function after puberty [142]. The literature concerning the efficacy of antipsychotics in this model is rather limited. Gill et al. [143] investigated the effect of haloperidol and subsequent response to a novel α5 gamma-aminobutyric acid (GABA(A)) receptor-positive allosteric modulator (α5PAM), on the activity of the dopaminergic system in the MAM neurodevelopmental model of SZ. They found that MAM rats withdrawn from haloperidol exhibited reduced spontaneous dopaminergic system activity and increased locomotor sensitivity to amphetamine compared with control; however they was unresponsive to α5PAM treatment. Neves and Grace [144] reported that modulators of α7 nicotinic receptor reverse the hyperdopaminergic tone in the MAM model of SZ. Thus, MAM model of SZ causes a disturbance in the dopaminergic neurotransmission in accordance with the dopaminergic hypothesis of SZ. It is also important to mention that the MAM model of SZ involves to some extent negative symptoms of the disease and may be used to search for novel drugs efficient against the negative symptoms.
Bacterial infections during pregnancy can contribute to the development of SZ in the offspring. This is the basis of prenatal LPS model of SZ which involves prenatal exposure to a bacterial endotoxin, LPS, in rodents. LPS binds to TLR4 (toll-like receptor 4) in immune cells, and induces a systemic innate immune response similar to PolyI:C [145]. LPS mice exhibit numerous anatomical and cellular abnormalities, in particular in the dopaminergic system, which manifest as behavioral endpoints (positive, negative, and cognitive symptoms). Wischhof et al. [146] reported that some of the prenatal immune activation effects are sex-dependent, which indicates the problem of gender differences in animal model of SZ. Moreover, it has been also postulated that maternal bacterial infections have additive effect with malnutrition on the development of SZ as studied in the LPS model in the conditions of iron deficiency [147].
As has been demonstrated in post-mortem and neuroimaging studies, the hippocampus displays significant structural and functional alterations in patients with SZ [142]. In this context, the ventral hippocampal lesion animal model was proposed. It is based on pathological presence of ventricular enlargement and hippocampal atrophy in individuals with SZ [148,149]. A neonatal lesion of the ventral hippocampus in rodents by microinjection of ibotenic acid (excitatory toxin) induces abnormal behavioral phenotypes after puberty [142], including positive, cognitive, and negative symptoms. Interestingly, the rodents display a higher consumption of sucrose in respective tests, which is a manifestation of changes in the reward system and it is in contrast to the anhedonia in SZ. In addition to the MAM model, the ventral hippocampal lesion animal model can be used to search for drugs against SZ that are efficient to treat negative symptoms.
5. Summary of Discussed Models
6. Conclusions
In spite of many years of studies, the pathomechanism of SZ is still elusive and the treatment of this disease is far from perfect. Novel antipsychotics, efficient against not only positive but also cognitive and negative symptoms are urgently needed. In order to design and test novel chemical compounds for potential treatment of SZ, in vitro and in vivo models of the disease are required. The difficulty of animal models in case of mental diseases lies in the fact that the disorder manifests fully only in humans and in animals the conclusions are drawn based on their behavior, which can be challenging especially in the case of paranoic thinking or delusion. Nevertheless, the in vitro and in vivo models of SZ are better than currently available in silico approaches and methods of choice before human studies. In order to obtain better effects, the models may be used in combination, which is particularly important to model-treatment-resistant SZ.
Funding
The research was performed under OPUS grant from National Science Center (NCN, Poland), grant number 2017/27/B/NZ7/01767 (to A.A.K).
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| 5-CSR | 5-Choice Serial Reaction Time Task |
| ASST | Attentional set-shifting task |
| C4 | Schizophrenia comprising complement component 4 |
| cAMP | Cyclic adenosine monophosphate |
| CRISP/Cas9 | Clustered regularly interspaced short palindromic repeats-associated nuclease 9 |
| CT | Computer tomography |
| DI | Discrimination Index |
| DISC1 | Disrupted-in-schizophrenia 1 |
| DN | Dominant negative |
| GABA | γ-Aminobutyric acid |
| GSK3α | Glycogen synthase kinase-3α |
| GWAS | Genome-wide association studies |
| iPSCs | Induced pluripotent stem cells |
| ID/ED | Intradimensional shift/extradimensional shift |
| LPS | Lipopolysaccharide |
| LSD | Lysergic acid diethylamide |
| LTD | Long-term depression |
| LTP | Long-term potentiation |
| MAM | Methylazoxymethanol acetate |
| mGluR | Metabotropic glutamate receptor |
| MK-801 | Dizocilpine |
| MWM | Morris Water Maze |
| NGS | Next generation sequencing |
| NMDA | N-methyl-D-aspartate |
| NOR | Novel object recognition |
| NRG1 | Neuregulin-1 |
| NRXN1 | Neurexin 1 |
| NSCs | Neuronal stem cells |
| NSE | Neuron-specific enolase |
| PAM | Positive allosteric modulator |
| PCP | Phencyclidine |
| PET | Positron emission tomography |
| PFC | Prefrontal cortex |
| PPI | Prepulse inhibition |
| R1, R2, and R3 | Reversal 1, 2, and 3 |
| RA | Retinoic acid |
| RBM12 | RNA-binding motif 12 |
| SD | Simple discrimination |
| SGZ | Subgranular zone |
| SNP | Single nucleotide polymorphism |
| SPECT | Single photon emission computed tomography |
| SZ | Schizophrenia |
| TLR4 | Toll-like receptor 4 |
| WCST | Wisconsin Card Sorting Test |
References
- McCutcheon, R.A.; Abi-Dargham, A.; Howes, O.D. Schizophrenia, Dopamine and the Striatum: From Biology to Symptoms. Trends Neurosci. 2019, 42, 205–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Early Development of Kraepelin’s Ideas on Classification: A Conceptual History. PubMed—NCBI. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/pubmed/3078049 (accessed on 8 November 2018).
- Jackson, J.H. Remarks on Evolution and Dissolution of the Nervous System. J. Ment. Sci. 1887, 33, 25–48. [Google Scholar] [CrossRef]
- Jablensky, A. The diagnostic concept of schizophrenia: Its history, evolution, and future prospects. Dialogues Clin. Neurosci. 2010, 12, 271–287. [Google Scholar] [PubMed]
- Ashok, A.H.; Baugh, J.; Yeragani, V.K. Paul Eugen Bleuler and the origin of the term schizophrenia (SCHIZOPRENIEGRUPPE). Indian J. Psychiatry 2012, 54, 95–96. [Google Scholar] [PubMed]
- Maatz, A.; Hoff, P.; Angst, J. Eugen Bleuler’s schizophrenia—A modern perspective. Dialogues Clin. Neurosci. 2015, 17, 43–49. [Google Scholar] [PubMed]
- Seidman, L.J.; Mirsky, A.F. Evolving Notions of Schizophrenia as a Developmental Neurocognitive Disorder. J. Int. Neuropsychol. Soc. 2017, 23, 881–892. [Google Scholar] [CrossRef] [Green Version]
- Davison, J.; O’Gorman, A.; Brennan, L.; Cotter, D.R. A systematic review of metabolite biomarkers of schizophrenia. Schizophr. Res. 2018, 195, 32–50. [Google Scholar] [CrossRef] [Green Version]
- Elkis, H.; Buckley, P.F. Treatment-Resistant Schizophrenia. Psychiatr. Clin. N. Am. 2016, 39, 239–265. [Google Scholar] [CrossRef]
- Harrison, G.; Hopper, K.; Craig, T.; Laska, E.; Siegel, C.; Wanderling, J.; Dube, K.C.; Ganev, K.; Giel, R.; der Heiden, W.; et al. Recovery from psychotic illness: A 15- and 25-year international follow-up study. Br. J. Psychiatry 2001, 178, 506–517. [Google Scholar] [CrossRef] [Green Version]
- McClure, R.J.; Keshavan, M.S.; Pettegrew, J.W. Chemical and physiologic brain imaging in schizophrenia. Psychiatr. Clin. N. Am. 1998, 21, 93–122. [Google Scholar] [CrossRef]
- Weinstein, J.J.; Chohan, M.O.; Slifstein, M.; Kegeles, L.S.; Moore, H.; Abi-Dargham, A. Pathway-Specific Dopamine Abnormalities in Schizophrenia. Biol. Psychiatry 2017, 81, 31–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchsbaum, M.S.; Hazlett, E.A. Positron emission tomography studies of abnormal glucose metabolism in schizophrenia. Schizophr. Bull. 1998, 24, 343–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makara-Studzińska, M.; Łos, R. Neuroanatomical structural changes seen in patients with schizophrenia and their healthy siblings. Pol. Merkur. Lek. 2012, 33, 51–53. [Google Scholar]
- Gong, Q.; Dazzan, P.; Scarpazza, C.; Kasai, K.; Hu, X.; Marques, T.R.; Iwashiro, N.; Huang, X.; Murray, R.M.; Koike, S.; et al. A Neuroanatomical Signature for Schizophrenia Across Different Ethnic Groups. Schizophr. Bull. 2015, 41, 1266–1275. [Google Scholar] [CrossRef] [Green Version]
- Chakos, M.H.; Schobel, S.A.; Gu, H.; Gerig, G.; Bradford, D.; Charles, C.; Lieberman, J.A. Duration of illness and treatment effects on hippocampal volume in male patients with schizophrenia. Br. J. Psychiatry 2005, 186, 26–31. [Google Scholar] [CrossRef]
- Kolomeets, N.S.; Orlovskaya, D.D.; Rachmanova, V.I.; Uranova, N.A. Ultrastructural alterations in hippocampal mossy fiber synapses in schizophrenia: A postmortem morphometric study. Synapse 2005, 57, 47–55. [Google Scholar] [CrossRef]
- Donegan, J.J.; Lodge, D.J. Cell-based therapies for the treatment of schizophrenia. Brain Res. 2017, 1655, 262–269. [Google Scholar] [CrossRef] [Green Version]
- Stępnicki, P.; Kondej, M.; Kaczor, A.A. Current Concepts and Treatments of Schizophrenia. Molecules 2018, 23, 2087. [Google Scholar] [CrossRef] [Green Version]
- Brisch, R.; Saniotis, A.; Wolf, R.; Bielau, H.; Bernstein, H.G.; Steiner, J.; Bogerts, B.; Braun, K.; Jankowski, Z.; Kumaratilake, J.; et al. The Role of Dopamine in Schizophrenia from a Neurobiological and Evolutionary Perspective: Old Fashioned, but Still in Vogue. Front. Psychiatry 2014, 5, 47. [Google Scholar]
- Berridge, K.C.; Robinson, T.E. What is the role of dopamine in reward: Hedonic impact, reward learning, or incentive salience? Brain Res. Brain Res. Rev. 1998, 28, 309–369. [Google Scholar] [CrossRef]
- Lau, C.I.; Wang, H.C.; Hsu, J.L.; Liu, M.E. Does the dopamine hypothesis explain schizophrenia? Rev. Neurosci. 2013, 24, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Kapur, S. Psychosis as a state of aberrant salience: A framework linking biology, phenomenology, and pharmacology in schizophrenia. Am. J. Psychiatry 2003, 160, 13–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uno, Y.; Coyle, J.T. Glutamate hypothesis in schizophrenia. Psychiatry Clin. Neurosci. 2019, 73, 204–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsman, A.; van den Heuvel, M.P.; Klomp, D.W.J.; Kahn, R.S.; Luijten, P.R.; Hulshoff Pol, H.E. Glutamate in Schizophrenia: A Focused Review and Meta-Analysis of 1H-MRS Studies. Schizophr. Bull. 2013, 39, 120–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javitt, D.C. Glutamate and schizophrenia: Phencyclidine, N-methyl-D-aspartate receptors, and dopamine-glutamate interactions. Int. Rev. Neurobiol. 2007, 78, 69–108. [Google Scholar]
- Howes, O.; McCutcheon, R.; Stone, J. Glutamate and dopamine in schizophrenia: An update for the 21st century. J. Psychopharmacol. 2015, 29, 97–115. [Google Scholar] [CrossRef] [Green Version]
- Maksymetz, J.; Moran, S.P.; Conn, P.J. Targeting metabotropic glutamate receptors for novel treatments of schizophrenia. Mol. Brain 2017, 10, 15. [Google Scholar] [CrossRef] [Green Version]
- Maj, C.; Minelli, A.; Giacopuzzi, E.; Sacchetti, E.; Gennarelli, M. The Role of Metabotropic Glutamate Receptor Genes in Schizophrenia. Curr. Neuropharmacol. 2016, 14, 540–550. [Google Scholar] [CrossRef] [Green Version]
- Trabanco, A.A.; Bartolomé, J.M.; Cid, J.M. mGluR2 positive allosteric modulators: An updated patent review (2013–2018). Expert Opin. Ther. Pat. 2019, 29, 497–507. [Google Scholar] [CrossRef]
- Kondej, M.; Stępnicki, P.; Kaczor, A.A. Multi-Target Approach for Drug Discovery against Schizophrenia. Int. J. Mol. Sci. 2018, 19, 3105. [Google Scholar] [CrossRef] [Green Version]
- Aghajanian, G.K.; Marek, G.J. Serotonin model of schizophrenia: Emerging role of glutamate mechanisms. Brain Res. Brain Res. Rev. 2000, 31, 302–312. [Google Scholar] [CrossRef]
- Kaczor, A.A.; Targowska-Duda, K.M.; Budzyńska, B.; Biała, G.; Silva, A.G.; Castro, M. In vitro, molecular modeling and behavioral studies of 3-{[4-(5-methoxy-1H-indol-3-yl)-1,2,3,6-tetrahydropyridin-1-yl]methyl}-1,2-dihydroquinolin-2-one (D2AAK1) as a potential antipsychotic. Neurochem. Int. 2016, 96, 84–99. [Google Scholar] [CrossRef] [PubMed]
- Ellenbroek, B.A.; Ghiabi, B. Do Histamine receptor 3 antagonists have a place in the therapy for schizophrenia? Curr. Pharm. Des. 2015, 21, 3760–3770. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Chen, Z. The roles of histamine and its receptor ligands in central nervous system disorders: An update. Pharmacol. Ther. 2017, 175, 116–132. [Google Scholar] [CrossRef] [PubMed]
- Arnsten, A.F.T. Adrenergic targets for the treatment of cognitive deficits in schizophrenia. Psychopharmacology 2004, 174, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Dean, B.; Scarr, E. Possible involvement of muscarinic receptors in psychiatric disorders: A focus on schizophrenia and mood disorders. Curr. Mol. Med. 2015, 15, 253–264. [Google Scholar] [CrossRef]
- Schmidt, M.J.; Mirnics, K. Neurodevelopment, GABA system dysfunction, and schizophrenia. Neuropsychopharmacology 2015, 40, 190–206. [Google Scholar] [CrossRef] [Green Version]
- Wassef, A.; Baker, J.; Kochan, L.D. GABA and schizophrenia: A review of basic science and clinical studies. J. Clin. Psychopharmacol. 2003, 23, 601–640. [Google Scholar] [CrossRef]
- Girgis, R.R.; Zoghbi, A.W.; Javitt, D.C.; Lieberman, J.A. The past and future of novel, non-dopamine-2 receptor therapeutics for schizophrenia: A critical and comprehensive review. J. Psychiatr. Res. 2019, 108, 57–83. [Google Scholar] [CrossRef]
- Lucatch, A.M.; Lowe, D.J.E.; Clark, R.C.; Kozak, K.; George, T.P. Neurobiological Determinants of Tobacco Smoking in Schizophrenia. Front. Psychiatry 2018, 9, 672. [Google Scholar] [CrossRef] [Green Version]
- Stevens, K.E.; Zheng, L.; Floyd, K.L.; Stitzel, J.A. Maximizing the effect of an α7 nicotinic receptor PAM in a mouse model of schizophrenia-like sensory inhibition deficits. Brain Res. 2015, 1611, 8–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potasiewicz, A.; Hołuj, M.; Kos, T.; Popik, P.; Arias, H.R.; Nikiforuk, A. 3-Furan-2-yl-N-p-tolyl-acrylamide, a positive allosteric modulator of the α7 nicotinic receptor, reverses schizophrenia-like cognitive and social deficits in rats. Neuropharmacology 2017, 113, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Espejo, E.; Viveros, M.-P.; Núñez, L.; Ellenbroek, B.A.; Rodriguez de Fonseca, F. Role of cannabis and endocannabinoids in the genesis of schizophrenia. Psychopharmacology 2009, 206, 531–549. [Google Scholar] [CrossRef]
- Müller, N. Inflammation in Schizophrenia: Pathogenetic Aspects and Therapeutic Considerations. Schizophr. Bull. 2018, 44, 973–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.; Matsuda, T.; Hayes, L.N.; Yang, S.; Rodriguez, K.; Severance, E.G.; Yolken, R.H.; Sawa, A.; Eaton, W.W. Infection and inflammation in schizophrenia and bipolar disorder. Neurosci. Res. 2017, 115, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Maas, D.A.; Vallès, A.; Martens, G.J.M. Oxidative stress, prefrontal cortex hypomyelination and cognitive symptoms in schizophrenia. Transl. Psychiatry 2017, 7, e1171. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, P.F.; Kendler, K.S.; Neale, M.C. Schizophrenia as a complex trait: Evidence from a meta-analysis of twin studies. Arch. Gen. Psychiatry 2003, 60, 1187–1192. [Google Scholar] [CrossRef] [Green Version]
- Kallmann, F.J. The genetic theory of schizophrenia; an analysis of 691 schizophrenic twin index families. Am. J. Psychiatry 1946, 103, 309–322. [Google Scholar] [CrossRef]
- Lichtenstein, P.; Yip, B.H.; Björk, C.; Pawitan, Y.; Cannon, T.D.; Sullivan, P.F.; Hultman, C.M. Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: A population-based study. Lancet 2009, 373, 234–239. [Google Scholar] [CrossRef] [Green Version]
- Wray, N.R.; Gottesman, I.I. Using summary data from the danish national registers to estimate heritabilities for schizophrenia, bipolar disorder, and major depressive disorder. Front. Genet. 2012, 3, 118. [Google Scholar] [CrossRef] [Green Version]
- Zhuo, C.; Hou, W.; Li, G.; Mao, F.; Li, S.; Lin, X.; Jiang, D.; Xu, Y.; Tian, H.; Wang, W.; et al. The genomics of schizophrenia: Shortcomings and solutions. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 93, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Millar, J.K.; Wilson-Annan, J.C.; Anderson, S.; Christie, S.; Taylor, M.S.; Semple, C.A.; Devon, R.S.; St Clair, D.M.; Muir, W.J.; Blackwood, D.H.; et al. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum. Mol. Genet. 2000, 9, 1415–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coelewij, L.; Curtis, D. Mini-review: Update on the genetics of schizophrenia. Ann. Hum. Genet. 2018, 82, 239–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, P.J. Recent genetic findings in schizophrenia and their therapeutic relevance. J. Psychopharmacol. 2015, 29, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Föcking, M.; Doyle, B.; Munawar, N.; Dillon, E.T.; Cotter, D.; Cagney, G. Epigenetic Factors in Schizophrenia: Mechanisms and Experimental Approaches. Mol. Neuropsychiatry 2019, 5, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, S.H.; Folsom, T.D. The Neurodevelopmental Hypothesis of Schizophrenia, Revisited. Schizophr. Bull. 2009, 35, 528–548. [Google Scholar] [CrossRef]
- Limosin, F. Neurodevelopmental and environmental hypotheses of negative symptoms of schizophrenia. BMC Psychiatry 2014, 14, 88. [Google Scholar] [CrossRef] [Green Version]
- Davis, J.; Eyre, H.; Jacka, F.N.; Dodd, S.; Dean, O.; McEwen, S.; Debnath, M.; McGrath, J.; Maes, M.; Amminger, P.; et al. A review of vulnerability and risks for schizophrenia: Beyond the two hit hypothesis. Neurosci. Biobehav. Rev. 2016, 65, 185–194. [Google Scholar] [CrossRef] [Green Version]
- Schlachetzki, J.C.M.; Saliba, S.W.; Oliveira, A.C.P. de Studying neurodegenerative diseases in culture models. Braz. J. Psiquiatr. 2013, 35, S92–S100. [Google Scholar] [CrossRef] [Green Version]
- Banker, G.A.; Cowan, W.M. Rat hippocampal neurons in dispersed cell culture. Brain Res. 1977, 126, 397–425. [Google Scholar] [CrossRef]
- Bray, N.J.; Kapur, S.; Price, J. Investigating schizophrenia in a “dish”: Possibilities, potential and limitations. World Psychiatry 2012, 11, 153–155. [Google Scholar] [CrossRef] [PubMed]
- Słońska, A.; Cymerys, J. Application of three-dimensional neuronal cell cultures in the studies of mechanisms of neurodegenerative diseases. Postepy Hig. Med. Dosw. 2017, 71, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Kovalevich, J.; Langford, D. Considerations for the Use of SH-SY5Y Neuroblastoma Cells in Neurobiology. Methods Mol. Biol. 2013, 1078, 9–21. [Google Scholar] [PubMed] [Green Version]
- Shipley, M.M.; Mangold, C.A.; Szpara, M.L. Differentiation of the SH-SY5Y Human Neuroblastoma Cell Line. J. Vis. Exp. 2016, e53193. [Google Scholar] [CrossRef]
- Biehl, J.K.; Russell, B. Introduction to Stem Cell Therapy. J. Cardiovasc. Nurs. 2009, 24, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Oh, J.H.; Jung, C.R.; Lee, M.O.; Kim, J.; Son, M.Y. Comparative analysis of human embryonic stem cell-derived neural stem cells as an in vitro human model. Int. J. Mol. Med. 2018, 41, 783–790. [Google Scholar] [CrossRef]
- Brennand, K.J.; Simone, A.; Jou, J.; Gelboin-Burkhart, C.; Tran, N.; Sangar, S.; Li, Y.; Mu, Y.; Chen, G.; Yu, D.; et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature 2011, 473, 221–225. [Google Scholar] [CrossRef]
- Pang, Z.P.; Yang, N.; Vierbuchen, T.; Ostermeier, A.; Fuentes, D.R.; Yang, T.Q.; Citri, A.; Sebastiano, V.; Marro, S.; Südhof, T.C.; et al. Induction of human neuronal cells by defined transcription factors. Nature 2011, 476, 220–223. [Google Scholar] [CrossRef]
- Liu, J.; Li, L.; Suo, W.Z. HT22 hippocampal neuronal cell line possesses functional cholinergic properties. Life Sci. 2009, 84, 267–271. [Google Scholar] [CrossRef]
- Rolando, C.; Taylor, V. Neural stem cell of the hippocampus: Development, physiology regulation, and dysfunction in disease. Curr. Top. Dev. Biol. 2014, 107, 183–206. [Google Scholar]
- Fukui, M.; Song, J.H.; Choi, J.; Choi, H.J.; Zhu, B.T. Mechanism of glutamate-induced neurotoxicity in HT22 mouse hippocampal cells. Eur. J. Pharmacol. 2009, 617, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bambrick, L.L.; Yarowsky, P.J.; Krueger, B.K. Glutamate as a hippocampal neuron survival factor: An inherited defect in the trisomy 16 mouse. Proc. Natl. Acad. Sci. USA 1995, 92, 9692–9696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heckers, S.; Konradi, C. Hippocampal neurons in schizophrenia. J. Neural. Transm. 2002, 109, 891–905. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, R.; Broglie, J.J.; Adcock, A.F.; Yang, L. Three-Dimensional Cell Culture Systems and Their Applications in Drug Discovery and Cell-Based Biosensors. Assay Drug Dev. Technol. 2014, 12, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Eglen, R.M. Three-Dimensional Cell Cultures in Drug Discovery and Development. SLAS Discov. 2017, 22, 456–472. [Google Scholar]
- Featherstone, R.E.; Rizos, Z.; Kapur, S.; Fletcher, P.J. A sensitizing regimen of amphetamine that disrupts attentional set-shifting does not disrupt working or long-term memory. Behav. Brain Res. 2008, 189, 170–179. [Google Scholar] [CrossRef]
- Sams-Dodd, F. Distinct effects of d-amphetamine and phencyclidine on the social behaviour of rats. Behav. Pharmacol. 1995, 6, 55–65. [Google Scholar] [CrossRef]
- Fletcher, P.J.; Tenn, C.C.; Rizos, Z.; Lovic, V.; Kapur, S. Sensitization to amphetamine, but not PCP, impairs attentional set shifting: Reversal by a D1 receptor agonist injected into the medial prefrontal cortex. Psychopharmacology 2005, 183, 190–200. [Google Scholar] [CrossRef]
- Fletcher, P.J.; Tenn, C.C.; Sinyard, J.; Rizos, Z.; Kapur, S. A sensitizing regimen of amphetamine impairs visual attention in the 5-choice serial reaction time test: Reversal by a D1 receptor agonist injected into the medial prefrontal cortex. Neuropsychopharmacology 2007, 32, 1122–1132. [Google Scholar] [CrossRef]
- Meng, Z.H.; Feldpaush, D.L.; Merchant, K.M. Clozapine and haloperidol block the induction of behavioral sensitization to amphetamine and associated genomic responses in rats. Brain Res. Mol. Brain Res. 1998, 61, 39–50. [Google Scholar] [CrossRef]
- Martinez, V.; Sarter, M. Detection of the moderately beneficial cognitive effects of low-dose treatment with haloperidol or clozapine in an animal model of the attentional impairments of schizophrenia. Neuropsychopharmacology 2008, 33, 2635–2647. [Google Scholar] [CrossRef] [PubMed]
- Konradi, C.; Heckers, S. Molecular aspects of glutamate dysregulation: Implications for schizophrenia and its treatment. Pharmacol. Ther. 2003, 97, 153–179. [Google Scholar] [CrossRef] [Green Version]
- Cohen, B.D.; Rosenbaum, G.; Luby, E.D.; Gottlieb, J.S. Comparison of phencyclidine hydrochloride (Sernyl) with other drugs. Simulation of schizophrenic performance with phencyclidine hydrochloride (Sernyl), lysergic acid diethylamide (LSD-25), and amobarbital (Amytal) sodium; II. Symbolic and sequential thinking. Arch. Gen. Psychiatry 1962, 6, 395–401. [Google Scholar] [PubMed]
- Javitt, D.C.; Zukin, S.R. Recent advances in the phencyclidine model of schizophrenia. Am. J. Psychiatry 1991, 148, 1301–1308. [Google Scholar]
- Kalinichev, M.; Robbins, M.J.; Hartfield, E.M.; Maycox, P.R.; Moore, S.H.; Savage, K.M.; Austin, N.E.; Jones, D.N.C. Comparison between intraperitoneal and subcutaneous phencyclidine administration in Sprague-Dawley rats: A locomotor activity and gene induction study. Prog. Neuropsychopharmacol. Biol. Psychiatry 2008, 32, 414–422. [Google Scholar] [CrossRef]
- Mansbach, R.S.; Geyer, M.A. Effects of phencyclidine and phencyclidine biologs on sensorimotor gating in the rat. Neuropsychopharmacology 1989, 2, 299–308. [Google Scholar] [CrossRef]
- Sams-Dodd, F. A test of the predictive validity of animal models of schizophrenia based on phencyclidine and D-amphetamine. Neuropsychopharmacology 1998, 18, 293–304. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.A.; Watson, D.J.G.; Fone, K.C.F. Animal models of schizophrenia. Br. J. Pharmacol. 2011, 164, 1162–1194. [Google Scholar] [CrossRef]
- Phillips, M.; Wang, C.; Johnson, K.M. Pharmacological characterization of locomotor sensitization induced by chronic phencyclidine administration. J. Pharmacol. Exp. Ther. 2001, 296, 905–913. [Google Scholar]
- Lee, P.R.; Brady, D.L.; Shapiro, R.A.; Dorsa, D.M.; Koenig, J.I. Social interaction deficits caused by chronic phencyclidine administration are reversed by oxytocin. Neuropsychopharmacology 2005, 30, 1883–1894. [Google Scholar] [CrossRef] [Green Version]
- Qiao, H.; Noda, Y.; Kamei, H.; Nagai, T.; Furukawa, H.; Miura, H.; Kayukawa, Y.; Ohta, T.; Nabeshima, T. Clozapine, but not haloperidol, reverses social behavior deficit in mice during withdrawal from chronic phencyclidine treatment. Neuroreport 2001, 12, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.H.; Marsden, C.A.; Robbins, T.W. Behavioural rigidity and rule-learning deficits following isolation-rearing in the rat: Neurochemical correlates. Behav. Brain Res. 1991, 43, 35–50. [Google Scholar] [CrossRef]
- Idris, N.F.; Neill, J.C.; Large, C.H. Comparison of the efficacy of two anticonvulsants, phenytoin and valproate to improve PCP and d-amphetamine induced deficits in a reversal learning task in the rat. Front. Behav. Neurosci. 2009, 3, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Idris, N.F.; Repeto, P.; Neill, J.C.; Large, C.H. Investigation of the effects of lamotrigine and clozapine in improving reversal-learning impairments induced by acute phencyclidine and D-amphetamine in the rat. Psychopharmacology 2005, 179, 336–348. [Google Scholar] [CrossRef]
- McLean, S.L.; Neill, J.C.; Idris, N.F.; Marston, H.M.; Wong, E.H.F.; Shahid, M. Effects of asenapine, olanzapine, and risperidone on psychotomimetic-induced reversal-learning deficits in the rat. Behav. Brain Res. 2010, 214, 240–247. [Google Scholar] [CrossRef] [Green Version]
- Idris, N.; Neill, J.; Grayson, B.; Bang-Andersen, B.; Witten, L.M.; Brennum, L.T.; Arnt, J. Sertindole improves sub-chronic PCP-induced reversal learning and episodic memory deficits in rodents: Involvement of 5-HT(6) and 5-HT (2A) receptor mechanisms. Psychopharmacology 2010, 208, 23–36. [Google Scholar] [CrossRef]
- Abdul-Monim, Z.; Reynolds, G.P.; Neill, J.C. The effect of atypical and classical antipsychotics on sub-chronic PCP-induced cognitive deficits in a reversal-learning paradigm. Behav. Brain Res. 2006, 169, 263–273. [Google Scholar] [CrossRef]
- Idris, N.F.; Grayson, B.; Neill, J.C. The emerging role of NMDA receptor hypofunction in mediation of reversal learning deficits in the rat. J. Psychopharmacol. 2006, 20, A68. [Google Scholar]
- Bowie, C.R.; Harvey, P.D. Cognitive deficits and functional outcome in schizophrenia. Neuropsychiatr. Dis. Treat. 2006, 2, 531–536. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.C.; Robbins, T.W.; Everitt, B.J. The effects of intradimensional and extradimensional shifts on visual discrimination learning in humans and non-human primates. Q. J. Exp. Psychol. B 1988, 40, 321–341. [Google Scholar]
- Berg, E.A. A simple objective technique for measuring flexibility in thinking. J. Gen. Psychol. 1948, 39, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Birrell, J.M.; Brown, V.J. Medial frontal cortex mediates perceptual attentional set shifting in the rat. J. Neurosci. 2000, 20, 4320–4324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potasiewicz, A.; Golebiowska, J.; Popik, P.; Nikiforuk, A. Procognitive effects of varenicline in the animal model of schizophrenia depend on α4β2- and α 7-nicotinic acetylcholine receptors. J. Psychopharmacol. 2018, 33, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Nikiforuk, A.; Popik, P. Effects of quetiapine and sertindole on subchronic ketamine-induced deficits in attentional set-shifting in rats. Psychopharmacology 2012, 220, 65–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLean, S.L.; Beck, J.P.; Woolley, M.L.; Neill, J.C. A preliminary investigation into the effects of antipsychotics on sub-chronic phencyclidine-induced deficits in attentional set-shifting in female rats. Behav. Brain Res. 2008, 189, 152–158. [Google Scholar] [CrossRef] [Green Version]
- Meltzer, H.Y.; Rajagopal, L.; Huang, M.; Oyamada, Y.; Kwon, S.; Horiguchi, M. Translating the N-methyl-D-aspartate receptor antagonist model of schizophrenia to treatments for cognitive impairment in schizophrenia. Int. J. Neuropsychopharmacol. 2013, 16, 2181–2194. [Google Scholar] [CrossRef] [Green Version]
- Tek, C.; Gold, J.; Blaxton, T.; Wilk, C.; McMahon, R.P.; Buchanan, R.W. Visual perceptual and working memory impairments in schizophrenia. Arch. Gen. Psychiatry 2002, 59, 146–153. [Google Scholar] [CrossRef] [Green Version]
- Rajagopal, L.; Massey, B.W.; Huang, M.; Oyamada, Y.; Meltzer, H.Y. The novel object recognition test in rodents in relation to cognitive impairment in schizophrenia. Curr. Pharm. Des. 2014, 20, 5104–5114. [Google Scholar] [CrossRef]
- D’Hooge, R.; De Deyn, P.P. Applications of the Morris water maze in the study of learning and memory. Brain Res. Brain Res. Rev. 2001, 36, 60–90. [Google Scholar] [CrossRef]
- Didriksen, M.; Skarsfeldt, T.; Arnt, J. Reversal of PCP-induced learning and memory deficits in the Morris’ water maze by sertindole and other antipsychotics. Psychopharmacology 2007, 193, 225–233. [Google Scholar] [CrossRef]
- Neill, J.C.; Barnes, S.; Cook, S.; Grayson, B.; Idris, N.F.; McLean, S.L.; Snigdha, S.; Rajagopal, L.; Harte, M.K. Animal models of cognitive dysfunction and negative symptoms of schizophrenia: Focus on NMDA receptor antagonism. Pharmacol. Ther. 2010, 128, 419–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amitai, N.; Semenova, S.; Markou, A. Cognitive-disruptive effects of the psychotomimetic phencyclidine and attenuation by atypical antipsychotic medications in rats. Psychopharmacology 2007, 193, 521–537. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.A.; Koenig, J.I. Social interaction and social withdrawal in rodents as readouts for investigating the negative symptoms of schizophrenia. Eur. Neuropsychopharmacol. 2014, 24, 759–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snigdha, S.; Neill, J.C. Efficacy of antipsychotics to reverse phencyclidine-induced social interaction deficits in female rats—A preliminary investigation. Behav. Brain Res. 2008, 187, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, T.A.; Harte, M.K.; McKibben, C.E.; Elliott, J.J.; Reynolds, G.P. Disturbances in social interaction occur along with pathophysiological deficits following sub-chronic phencyclidine administration in the rat. Behav. Brain Res. 2008, 194, 230–235. [Google Scholar] [CrossRef]
- File, S.E. The use of social interaction as a method for detecting anxiolytic activity of chlordiazepoxide-like drugs. J. Neurosci. Methods 1980, 2, 219–238. [Google Scholar] [CrossRef]
- Moy, S.S.; Nadler, J.J.; Perez, A.; Barbaro, R.P.; Johns, J.M.; Magnuson, T.R.; Piven, J.; Crawley, J.N. Sociability and preference for social novelty in five inbred strains: An approach to assess autistic-like behavior in mice. Genes Brain Behav. 2004, 3, 287–302. [Google Scholar] [CrossRef]
- St. Clair, D.; Johnstone, M. Using mouse transgenic and human stem cell technologies to model genetic mutations associated with schizophrenia and autism. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20170037. [Google Scholar] [CrossRef] [Green Version]
- Tomoda, T.; Sumitomo, A.; Jaaro-Peled, H.; Sawa, A. Utility and validity of DISC1 mouse models in biological psychiatry. Neuroscience 2016, 321, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Leung, C.; Jia, Z. Mouse Genetic Models of Human Brain Disorders. Front. Genet. 2016, 7, 40. [Google Scholar] [CrossRef] [Green Version]
- Koike, H.; Arguello, P.A.; Kvajo, M.; Karayiorgou, M.; Gogos, J.A. Disc1 is mutated in the 129S6/SvEv strain and modulates working memory in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 3693–3697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuroda, K.; Yamada, S.; Tanaka, M.; Iizuka, M.; Yano, H.; Mori, D.; Tsuboi, D.; Nishioka, T.; Namba, T.; Iizuka, Y.; et al. Behavioral alterations associated with targeted disruption of exons 2 and 3 of the Disc1 gene in the mouse. Hum. Mol. Genet. 2011, 20, 4666–4683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahani, N.; Seshadri, S.; Jaaro-Peled, H.; Ishizuka, K.; Hirota-Tsuyada, Y.; Wang, Q.; Koga, M.; Sedlak, T.W.; Korth, C.; Brandon, N.J.; et al. DISC1 regulates trafficking and processing of APP and Aβ generation. Mol. Psychiatry 2015, 20, 874–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clapcote, S.J.; Lipina, T.V.; Millar, J.K.; Mackie, S.; Christie, S.; Ogawa, F.; Lerch, J.P.; Trimble, K.; Uchiyama, M.; Sakuraba, Y.; et al. Behavioral phenotypes of Disc1 missense mutations in mice. Neuron 2007, 54, 387–402. [Google Scholar] [CrossRef] [Green Version]
- Hikida, T.; Jaaro-Peled, H.; Seshadri, S.; Oishi, K.; Hookway, C.; Kong, S.; Wu, D.; Xue, R.; Andradé, M.; Tankou, S.; et al. Dominant-negative DISC1 transgenic mice display schizophrenia-associated phenotypes detected by measures translatable to humans. Proc. Natl. Acad. Sci. USA 2007, 104, 14501–14506. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Zhou, Y.; Jentsch, J.D.; Brown, R.A.M.; Tian, X.; Ehninger, D.; Hennah, W.; Peltonen, L.; Lönnqvist, J.; Huttunen, M.O.; et al. Specific developmental disruption of disrupted-in-schizophrenia-1 function results in schizophrenia-related phenotypes in mice. Proc. Natl. Acad. Sci. USA 2007, 104, 18280–18285. [Google Scholar] [CrossRef] [Green Version]
- Pletnikov, M.V.; Ayhan, Y.; Nikolskaia, O.; Xu, Y.; Ovanesov, M.V.; Huang, H.; Mori, S.; Moran, T.H.; Ross, C.A. Inducible expression of mutant human DISC1 in mice is associated with brain and behavioral abnormalities reminiscent of schizophrenia. Mol. Psychiatry 2008, 13, 173–186. [Google Scholar] [CrossRef]
- Shen, S.; Lang, B.; Nakamoto, C.; Zhang, F.; Pu, J.; Kuan, S.L.; Chatzi, C.; He, S.; Mackie, I.; Brandon, N.J.; et al. Schizophrenia-related neural and behavioral phenotypes in transgenic mice expressing truncated Disc1. J. Neurosci. 2008, 28, 10893–10904. [Google Scholar] [CrossRef] [Green Version]
- Harrison, P.J.; Law, A.J. Neuregulin 1 and schizophrenia: Genetics, gene expression, and neurobiology. Biol. Psychiatry 2006, 60, 132–140. [Google Scholar] [CrossRef]
- Tian, J.; Geng, F.; Gao, F.; Chen, Y.H.; Liu, J.H.; Wu, J.L.; Lan, Y.J.; Zeng, Y.N.; Li, X.W.; Yang, J.M.; et al. Down-Regulation of Neuregulin1/ErbB4 Signaling in the Hippocampus Is Critical for Learning and Memory. Mol. Neurobiol. 2017, 54, 3976–3987. [Google Scholar] [CrossRef]
- Papaleo, F.; Yang, F.; Paterson, C.; Palumbo, S.; Carr, G.V.; Wang, Y.; Floyd, K.; Huang, W.; Thomas, C.J.; Chen, J.; et al. Behavioral, Neurophysiological, and Synaptic Impairment in a Transgenic Neuregulin1 (NRG1-IV) Murine Schizophrenia Model. J. Neurosci. 2016, 36, 4859–4875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olaya, J.C.; Heusner, C.L.; Matsumoto, M.; Shannon Weickert, C.; Karl, T. Schizophrenia-relevant behaviours of female mice overexpressing neuregulin 1 type III. Behav. Brain Res. 2018, 353, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Avramopoulos, D. Neuregulin 3 and its roles in schizophrenia risk and presentation. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2018, 177, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Fradley, R.L.; O’Meara, G.F.; Newman, R.J.; Andrieux, A.; Job, D.; Reynolds, D.S. STOP knockout and NMDA NR1 hypomorphic mice exhibit deficits in sensorimotor gating. Behav. Brain Res. 2005, 163, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Mohn, A.R.; Gainetdinov, R.R.; Caron, M.G.; Koller, B.H. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell 1999, 98, 427–436. [Google Scholar] [CrossRef] [Green Version]
- Belforte, J.E.; Zsiros, V.; Sklar, E.R.; Jiang, Z.; Yu, G.; Li, Y.; Quinlan, E.M.; Nakazawa, K. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat. Neurosci. 2010, 13, 76–83. [Google Scholar] [CrossRef] [Green Version]
- Papaleo, F.; Yang, F.; Garcia, S.; Chen, J.; Lu, B.; Crawley, J.N.; Weinberger, D.R. Dysbindin-1 modulates prefrontal cortical activity and schizophrenia-like behaviors via dopamine/D2 pathways. Mol. Psychiatry 2012, 17, 85–98. [Google Scholar] [CrossRef] [Green Version]
- Karlsgodt, K.H.; Robleto, K.; Trantham-Davidson, H.; Jairl, C.; Cannon, T.D.; Lavin, A.; Jentsch, J.D. Reduced dysbindin expression mediates N-methyl-D-aspartate receptor hypofunction and impaired working memory performance. Biol. Psychiatry 2011, 69, 28–34. [Google Scholar] [CrossRef] [Green Version]
- Tueting, P.; Doueiri, M.-S.; Guidotti, A.; Davis, J.M.; Costa, E. Reelin down-regulation in mice and psychosis endophenotypes. Neurosci. Biobehav. Rev. 2006, 30, 1065–1077. [Google Scholar] [CrossRef]
- Krueger, D.D.; Howell, J.L.; Hebert, B.F.; Olausson, P.; Taylor, J.R.; Nairn, A.C. Assessment of cognitive function in the heterozygous reeler mouse. Psychopharmacology 2006, 189, 95–104. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, Y.; Nitta, A. Behavioral phenotypes for negative symptoms in animal models of schizophrenia. J. Pharmacol. Sci. 2014, 126, 310–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill, K.M.; Cook, J.M.; Poe, M.M.; Grace, A.A. Prior antipsychotic drug treatment prevents response to novel antipsychotic agent in the methylazoxymethanol acetate model of schizophrenia. Schizophr. Bull. 2014, 40, 341–350. [Google Scholar] [CrossRef] [Green Version]
- Neves, G.A.; Grace, A.A. α7 Nicotinic receptor-modulating agents reverse the hyperdopaminergic tone in the MAM model of schizophrenia. Neuropsychopharmacology 2018, 43, 1712–1720. [Google Scholar] [CrossRef] [PubMed]
- Mattei, D.; Schweibold, R.; Wolf, S.A. Brain in flames—Animal models of psychosis: Utility and limitations. Neuropsychiatr. Dis. Treat. 2015, 11, 1313–1329. [Google Scholar] [PubMed] [Green Version]
- Wischhof, L.; Irrsack, E.; Osorio, C.; Koch, M. Prenatal LPS-exposure—A neurodevelopmental rat model of schizophrenia--differentially affects cognitive functions, myelination and parvalbumin expression in male and female offspring. Prog. Neuropsychopharmacol. Biol. Psychiatry 2015, 57, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Harvey, L.; Boksa, P. Additive effects of maternal iron deficiency and prenatal immune activation on adult behaviors in rat offspring. Brain Behav. Immun. 2014, 40, 27–37. [Google Scholar] [CrossRef]
- Lipska, B.K.; Jaskiw, G.E.; Weinberger, D.R. Postpubertal emergence of hyperresponsiveness to stress and to amphetamine after neonatal excitotoxic hippocampal damage: A potential animal model of schizophrenia. Neuropsychopharmacology 1993, 9, 67–75. [Google Scholar] [CrossRef] [Green Version]
- Lipska, B.K. Using animal models to test a neurodevelopmental hypothesis of schizophrenia. J. Psychiatry Neurosci. 2004, 29, 282–286. [Google Scholar] [PubMed]
- Zhu, D.; Zhang, J.; Wu, J.; Li, G.; Yao, W.; Hao, J.; Sun, J. Paliperidone Protects SH-SY5Y Cells Against MK-801-Induced Neuronal Damage Through Inhibition of Ca2+ Influx and Regulation of SIRT1/miR-134 Signal Pathway. Mol. Neurobiol. 2016, 53, 2498–2509. [Google Scholar] [CrossRef]
- Mackay-Sim, A. Patient-derived stem cells: Pathways to drug discovery for brain diseases. Front. Cell Neurosci. 2013, 7, 29. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, R.; Sportelli, V.; Ziller, M.; Spengler, D.; Hoffmann, A. Tracing Early Neurodevelopment in Schizophrenia with Induced Pluripotent Stem Cells. Cells 2018, 7, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, J.; Ma, L.; Wu, M.; Zhang, L.; Zhang, X.; Zhai, Q.; Jiang, T.; Wang, Q.; Xiong, L. Anandamide Protects HT22 Cells Exposed to Hydrogen Peroxide by Inhibiting CB1 Receptor-Mediated Type 2 NADPH Oxidase. Oxid. Med. Cell Longev. 2014, 2014, 893516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langhans, S.A. Three-Dimensional in Vitro Cell Culture Models in Drug Discovery and Drug Repositioning. Front. Pharmacol. 2018, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- Featherstone, R.E.; Kapur, S.; Fletcher, P.J. The amphetamine-induced sensitized state as a model of schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2007, 31, 1556–1571. [Google Scholar] [CrossRef] [PubMed]
- Moore, H.; Jentsch, J.D.; Ghajarnia, M.; Geyer, M.A.; Grace, A.A. A neurobehavioral systems analysis of adult rats exposed to methylazoxymethanol acetate on E17: Implications for the neuropathology of schizophrenia. Biol. Psychiatry 2006, 60, 253–264. [Google Scholar] [CrossRef] [Green Version]
- Tseng, K.Y.; Chambers, R.A.; Lipska, B.K. The neonatal ventral hippocampal lesion as a heuristic neurodevelopmental model of schizophrenia. Behav. Brain Res. 2009, 204, 295–305. [Google Scholar] [CrossRef] [Green Version]
- Jaaro-Peled, H. Gene models of schizophrenia: DISC1 mouse models. Prog. Brain Res. 2009, 179, 75–86. [Google Scholar]
- Mei, L.; Xiong, W.C. Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat. Rev. Neurosci. 2008, 9, 437–452. [Google Scholar] [CrossRef]
Figure 1.
Dopaminergic and glutamatergic system abnormalities in schizophrenia (SZ). NMDA: N-methyl-D-aspartate.
Figure 1.
Dopaminergic and glutamatergic system abnormalities in schizophrenia (SZ). NMDA: N-methyl-D-aspartate.

Figure 2.
SH-SY5Y cells.

Figure 3.
Mouse hippocampal neurons (HT22) cells.


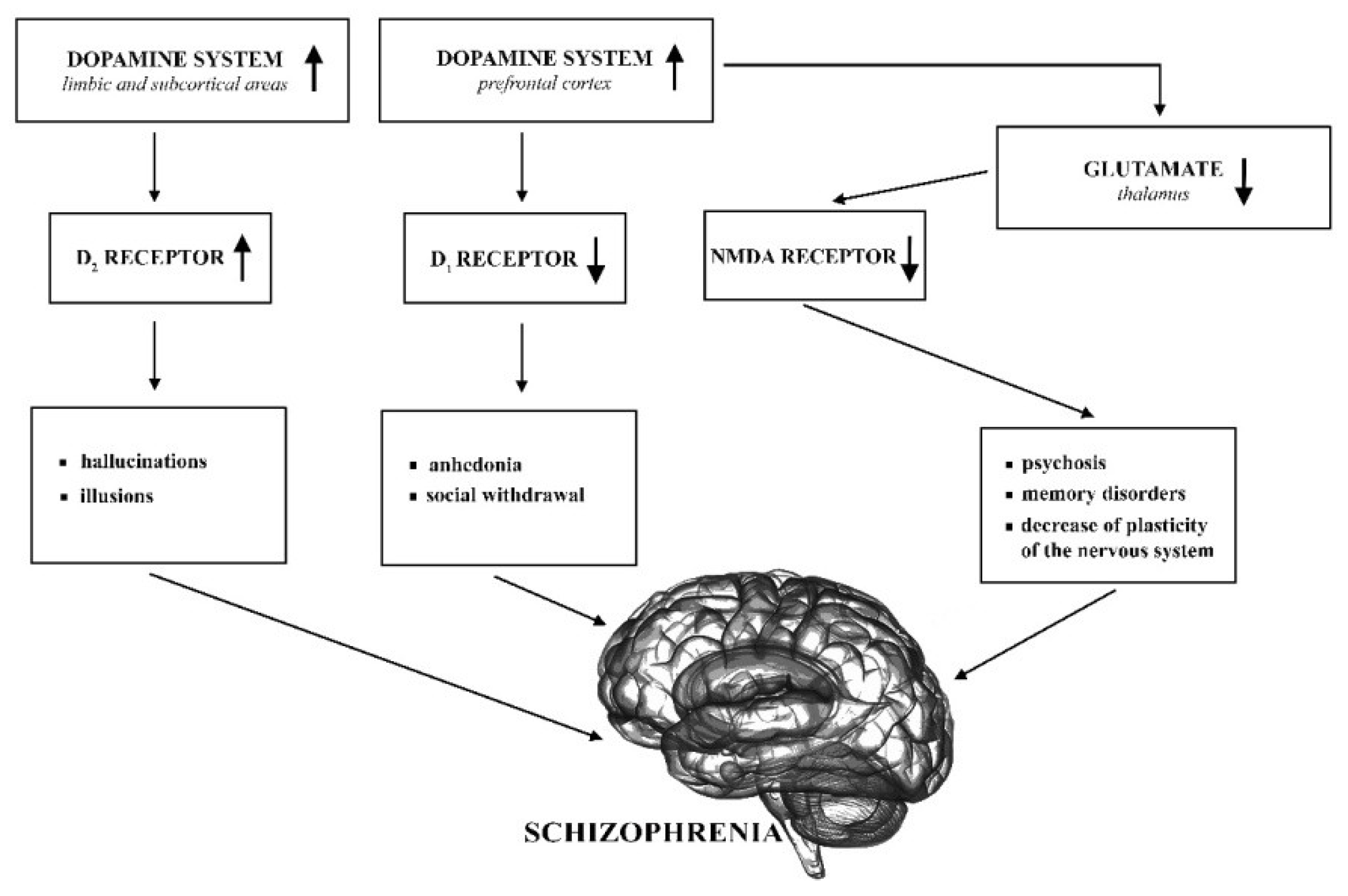{kind=link}
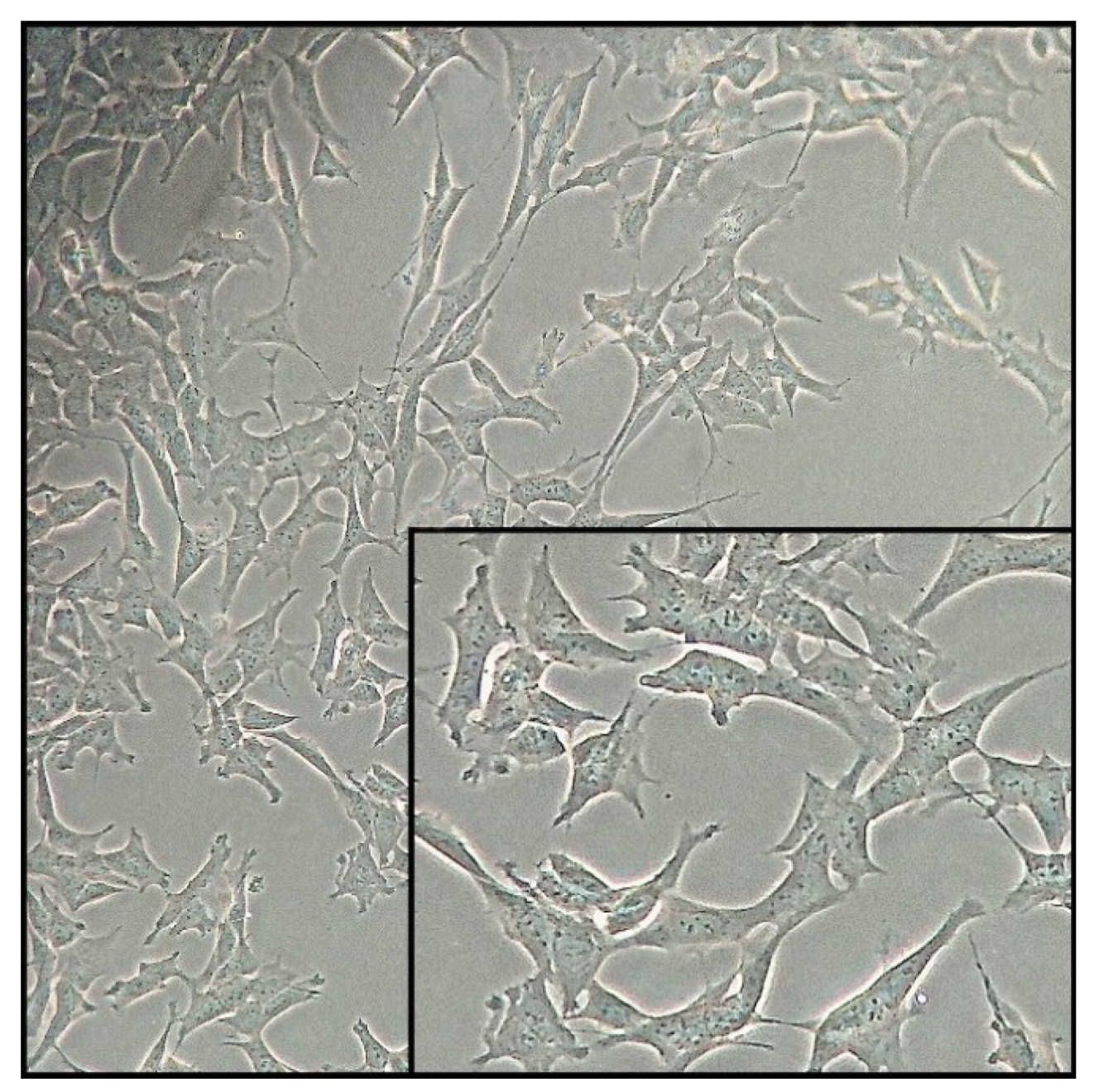{kind=link}
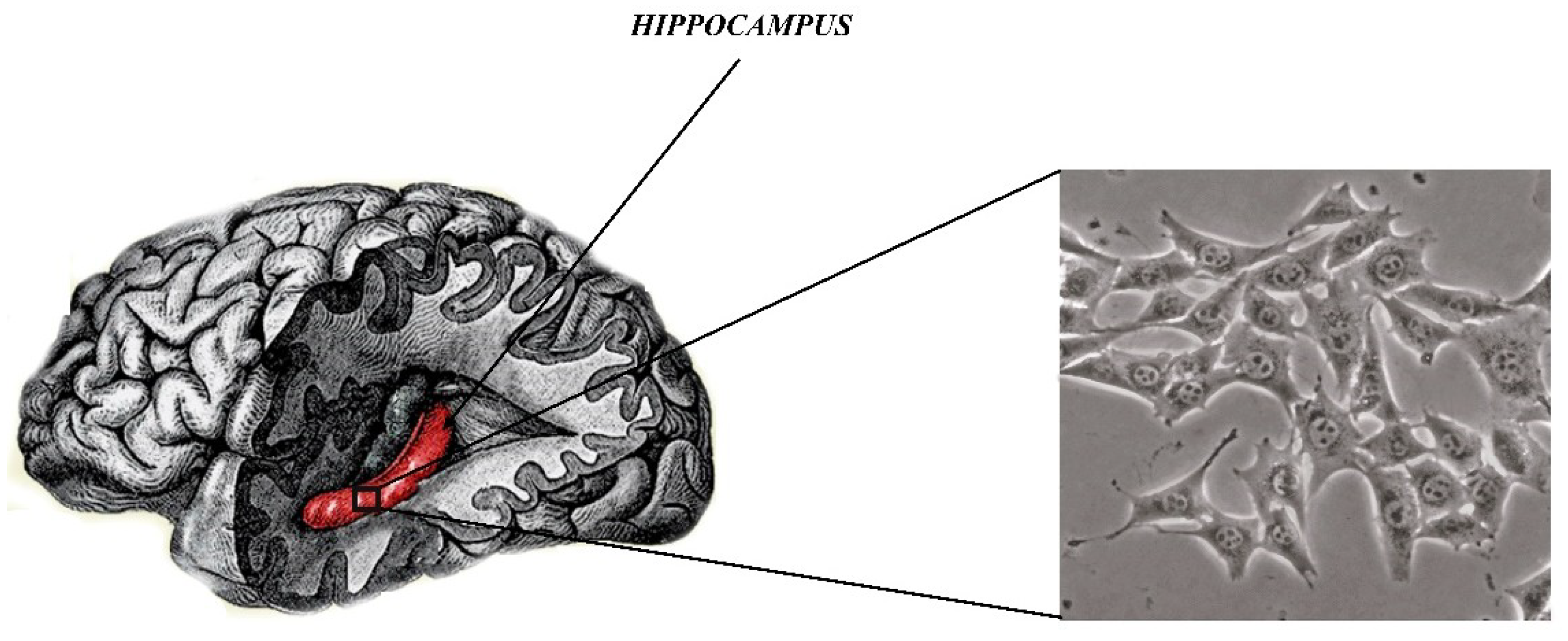{kind=link}
Table 1.
Examples of studies using in vitro and in vivo models of SZ (partially elaborated based on reference [89]).
Table 1.
Examples of studies using in vitro and in vivo models of SZ (partially elaborated based on reference [89]).
| In Vitro Model | Study |
| SH-SY5Y cell line | Study of molecular mechanisms and study of transmission in signaling pathways [150] |
| Multipotent stem cells | Gene expression studies and pathway dysfunctions associated with mitochondrial metabolism and oxidative stress [151] Analysis of gene and receptor expression, study of neurodevelopmental pathways [152] |
| Pluripotent stem cells | |
| HT-22 cell line | Study of biochemical basis of cellular function and disease processes and neurodevelopmental pathways [153] |
| Three-dimensional culture systems (3D) | Toxicity studies and determination of the biological or biochemical activity of the compounds [154] |
| In Vivo Model | Study/Effect |
| Amphetamine model Phencyclidine model | Locomotor sensitization; increased mesolimbic dopamine response; persistent deficit in prepulse inhibition (PPI); cognitive impairments [77,155] Enhanced mesolimbic dopamine response; no sustained deficit in PPI; reduced social interaction [90,112] |
| MAM model Neonatal ventral hippocampal lesion model | Spontaneous hyperactivity; amphetamine- and NMDA antagonist-induced hyperactivity; deficits in PPI; cognitive impairment; reduced social interaction [143,156] Amphetamine- and NMDA antagonist-induced locomotor hyperactivity; cognitive impairments; deficits in PPI and social interaction [149,157] |
| DISC-1 knock-out Neuregulin1 and ErbB4 knock-out | Increased sensitivity to psychostimulants; cognitive deficits; reduced social interaction; depressive-like behavior; deficits in PPI in some mutants [120,125,158] Spontaneous locomotor hyperactivity; social interaction impairment; PPI deficits in Neuregulin1 but not ErbB4 mutants [130,159] |
Table 2.
Advantages and disadvantages of in vitro and in vivo models of SZ (partially elaborated based on reference [89]).
Table 2.
Advantages and disadvantages of in vitro and in vivo models of SZ (partially elaborated based on reference [89]).
| In Vitro model | Disadvantages | Advantages |
| SH-SY5Y cell line | Genetic aberrations No synchronization | Biochemical and functional characteristics of neurons Expression of specific proteins and isoforms of proteins Ability to differentiate |
| Multipotent stem cells | Protocols for differentiation and isoallocation conditions are under development | Differentiate into individual cell lines Unlimited self-renewal capacity Correct number of chromosomes Easy isolation and in vitro culture Low immunogenicity High immigration capacity |
| Pluripotent stem cells | Differentiation into all types of cells | |
| HT-22 cell line | - | Model for glutamate cytotoxicity studies Ease in conducting experimental procedures Preserving cytoarchitectonic properties |
| Three-dimensional culture systems (3D) | Lack of even nutrition and oxygenation | Increased physiological response to bioactive substances Better interaction between cells and cells and the extracellular matrix |
| In Vivo model | Disadvantages | Advantages |
| Pharmacological (phencyclidine and amphetamine models) | There is no current “gold standard” medication to treat all the symptoms that can be used as a positive control (e.g., haloperidol and clozapine should reverse only positive symptoms of this disease) Models should have an appropriate symptoms homology, construct (replicate pathology and theoretical neurobiological response), and predictive validity to the clinical SZ disorder. | Animal models are very valuable preclinical tools used to investigate the neurobiological basis of SZ Resemble “positive-like” symptoms of SZ Resemble negative and cognitive symptoms of SZ by showing altered social deficits and cognitive impairments Resemble altered mesolimbic dopamine function |
| Neurodevelopmental models | ||
| Genetic models |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Koszła, O.; Targowska-Duda, K.M.; Kędzierska, E.; Kaczor, A.A. In Vitro and In Vivo Models for the Investigation of Potential Drugs Against Schizophrenia. Biomolecules 2020, 10, 160. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10010160
AMA Style
Koszła O, Targowska-Duda KM, Kędzierska E, Kaczor AA. In Vitro and In Vivo Models for the Investigation of Potential Drugs Against Schizophrenia. Biomolecules. 2020; 10(1):160. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10010160
Chicago/Turabian StyleKoszła, Oliwia, Katarzyna M. Targowska-Duda, Ewa Kędzierska, and Agnieszka A. Kaczor. 2020. "In Vitro and In Vivo Models for the Investigation of Potential Drugs Against Schizophrenia" Biomolecules 10, no. 1: 160. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10010160
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.