Prenatal Hyperglycemia Exposure and Cellular Stress, a Sugar-Coated View of Early Programming of Metabolic Diseases



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
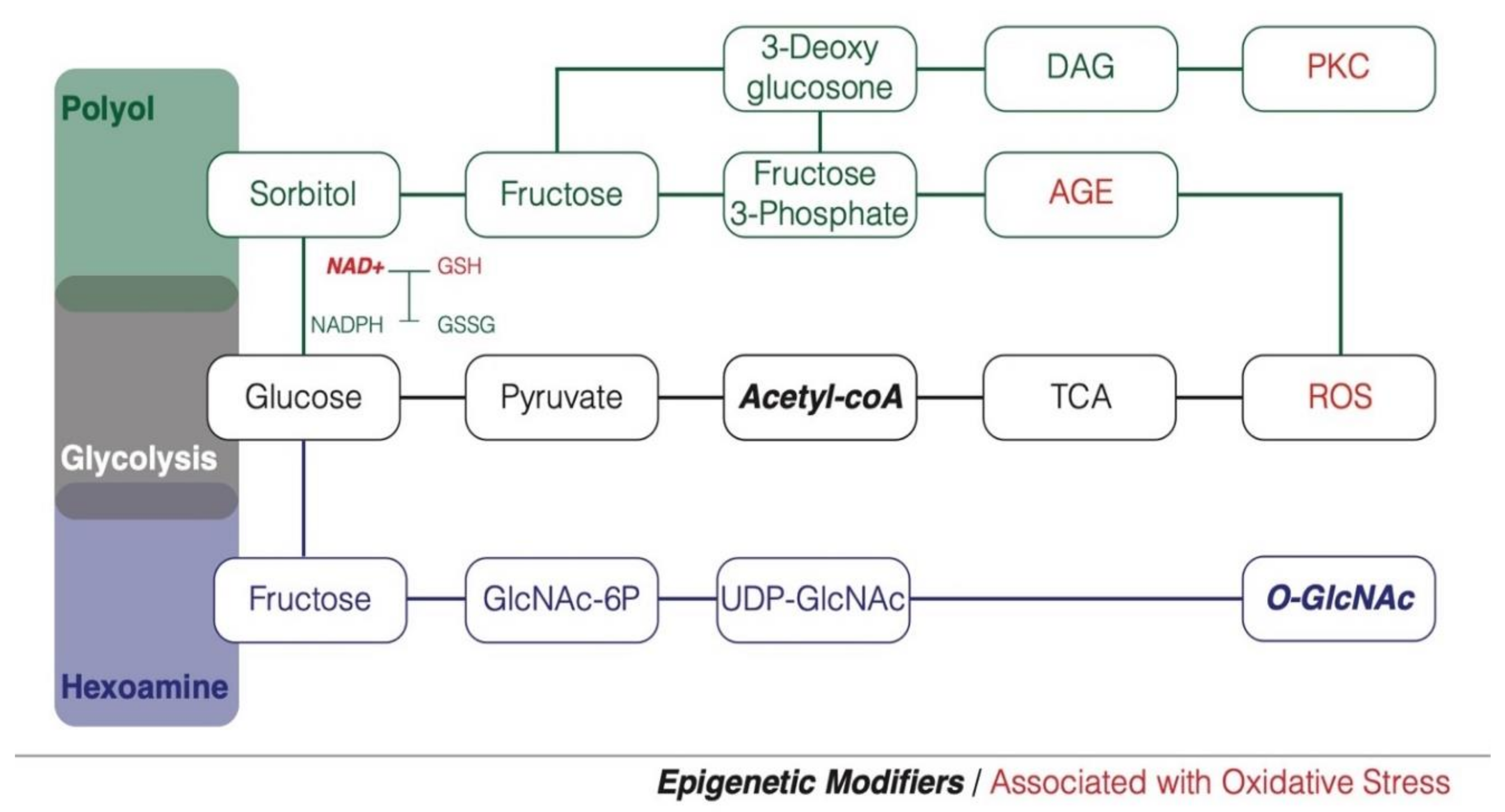
2. Hyperglycemia and Glucose Metabolism
3. Hyperglycemia, Oxidative Stress and Diabetes Susceptibility
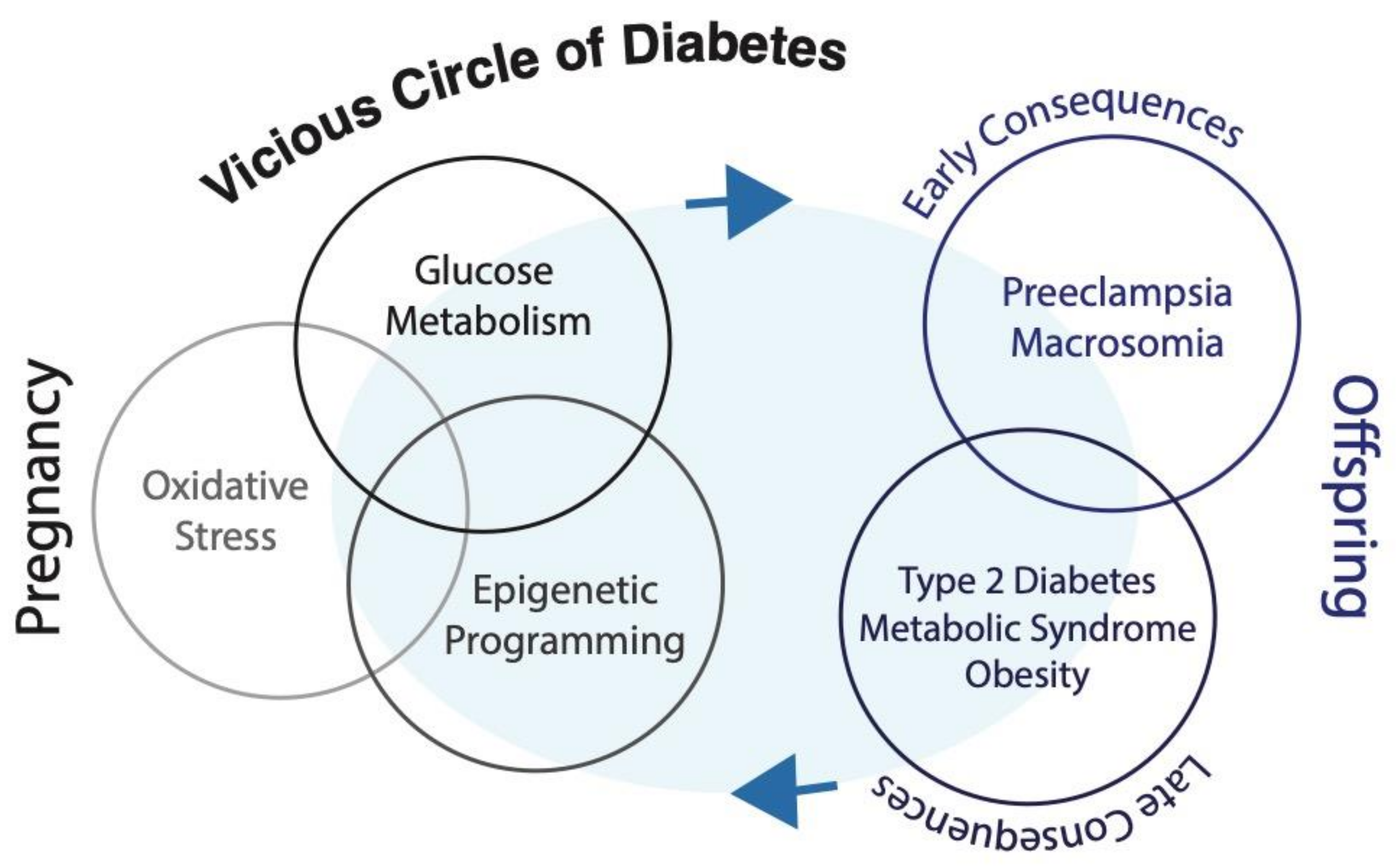
4. Hyperglycemia and Early Programming of Metabolic Diseases
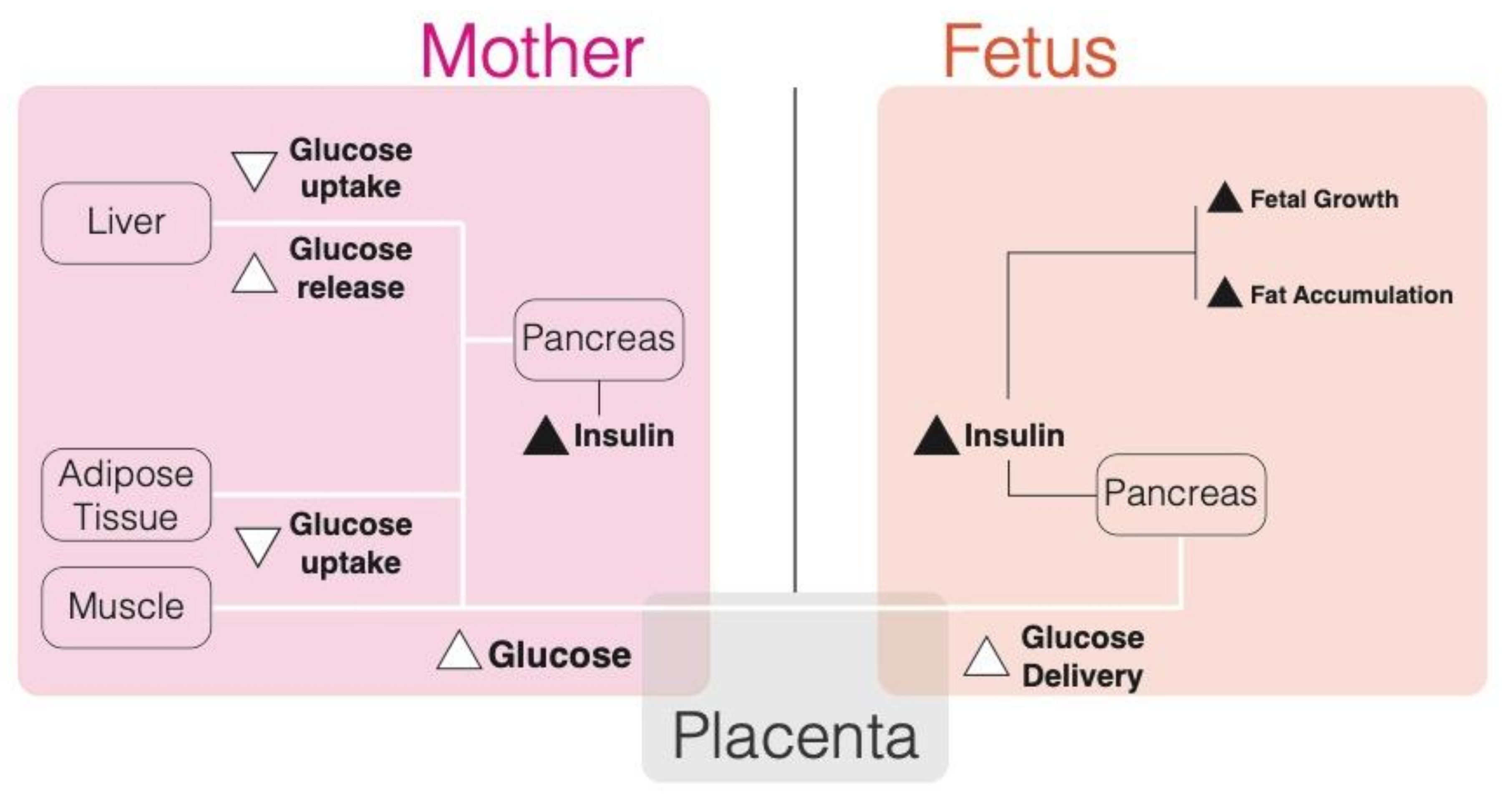
4.1. Maternal Glucose Metabolism during Pregnancy
4.2. Hyperglycemia and Pregnancy
5. Glycemic Memory and Long-term Consequences
6. Sexual Dimorphism in Glycemic Memory
7. Epigenetic Glycemic Memory
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Barker, D.; Osmond, C. Infant Mortality, Childhood Nutrition, and Ischaemic Heart Disease in England And Wales. Lancet 1986, 327, 1077–1081. [Google Scholar] [CrossRef]
- Pettitt, D.J.; Jovanovic, L. Birth weight as a predictor of type 2 diabetes mellitus: The U-shaped curve. Curr. Diabetes Rep. 2001, 1, 78–81. [Google Scholar] [CrossRef]
- Curhan, G.C.; Chertow, G.M.; Willett, W.C.; Spiegelman, D.; Colditz, G.A.; Manson, J.E.; Speizer, F.E.; Stampfer, M.J. Birth weight and adult hypertension and obesity in women. Circulation 1996, 94, 1310–1315. [Google Scholar] [CrossRef] [PubMed]
- Curhan, G.C.; Willett, W.C.; Rimm, E.B.; Spiegelman, D.; Ascherio, A.L.; Stampfer, M.J. Birth Weight and Adult Hypertension, Diabetes Mellitus, and Obesity in US Men. Circulation 1996, 94, 3246–3250. [Google Scholar] [CrossRef] [PubMed]
- Urakawa, H.; Katsuki, A.; Sumida, Y.; Gabazza, E.C.; Murashima, S.; Morioka, K.; Maruyama, N.; Kitagawa, N.; Tanaka, T.; Hori, Y.; et al. Oxidative Stress Is Associated with Adiposity and Insulin Resistance in Men. J. Clin. Endocrinol. Metab. 2003, 88, 4673–4676. [Google Scholar] [CrossRef] [PubMed]
- Kaneto, H.; Xu, G.; Song, K.-H.; Suzuma, K.; Bonner-Weir, S.; Sharma, A.; Weir, G.C. Activation of the Hexosamine Pathway Leads to Deterioration of Pancreatic β-Cell Function through the Induction of Oxidative Stress. J. Boil. Chem. 2001, 276, 31099–31104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tozour, J.N.; Delahaye, F.; Suzuki, M.; Praiss, A.; Zhao, Y.; Cai, L.; Heo, H.J.; Greally, J.M.; Hughes, F.; Cui, L. Intrauterine Hyperglycemia Is Associated with an Impaired Postnatal Response to Oxidative Damage. Stem Cells Dev. 2018, 27, 683–691. [Google Scholar] [CrossRef]
- Chung, S.S.; Ho, E.C.; Lam, K.S. Contribution of Polyol Pathway to Diabetes-Induced Oxidative Stress. J. Am. Soc. Nephrol. 2003, 14, 233–236. [Google Scholar] [CrossRef] [Green Version]
- Brownlee, M. The Pathobiology of Diabetic Complications: A Unifying Mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [Green Version]
- Patti, M.E.; Virkamaki, A.; Landaker, E.J.; Kahn, C.R.; Yki-Jarvinen, H. Activation of the hexosamine pathway by glucosamine in vivo induces insulin resistance of early postreceptor insulin signaling events in skeletal msucle. Diabetes 1999, 48, 1562–1571. [Google Scholar] [CrossRef]
- Yanagida, K.; Maejima, Y.; Santoso, P.; Otgon-Uul, Z.; Yang, Y.; Sakuma, K.; Shimomura, K.; Yada, T. Hexosamine pathway but not interstitial changes mediates glucotoxicity in pancreatic Beta-cells as assessed by cytosolic Ca2+ respone to glucose. Aging 2014, 6, 207–215. [Google Scholar] [CrossRef]
- Fontayne, A.; Dang, P.M.-C.; Gougerot-Pocidalo, M.-A.; El-Benna, J. Phosphorylation of p47phox sites by PKC alpha, beta II, delta, and zeta: Effect on binding to p22phox and on NADPH oxidase activation. Biochemistry 2002, 41, 7743–7750. [Google Scholar] [CrossRef] [PubMed]
- Robertson, R.P. Chronic Oxidative Stress as a Central Mechanism for Glucose Toxicity in Pancreatic Islet Beta Cells in Diabetes. J. Boil. Chem. 2004, 279, 42351–42354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, S.P.; Dean, R.T. Glucose autoxidation and protein modification. Biochem. J. 1987, 245, 243–250. [Google Scholar] [CrossRef]
- Zhang, M.; Kho, A.L.; Anilkumar, N.; Chibber, R.; Pagano, P.J.; Shah, A.M.; Cave, A.; Leger, A.J.; Jacques, S.L.; Badar, J.; et al. Glycated Proteins Stimulate Reactive Oxygen Species Production in Cardiac Myocytes. Circulation 2006, 113, 1235–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, L.-J. Pathogenesis of Chronic Hyperglycemia: From Reductive Stress to Oxidative Stress. J. Diabetes Res. 2014, 2014, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Matsumura, T.; Edelstein, D.; Rossetti, L.; Zsengeller, Z.; Szabo, C.; Brownlee, M. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J. Clin. Investig. Oct. 2003, 112, 1049–1057. [Google Scholar] [CrossRef] [Green Version]
- Tiedge, M.; Lortz, S.; Drinkgern, J.; Lenzen, S. Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes 1997, 46, 1733–1742. [Google Scholar] [CrossRef]
- Kawamori, D.; Kajimoto, Y.; Kaneto, H.; Umayahara, Y.; Fujitani, Y.; Miyatsuka, T.; Watada, H.; Leibiger, I.B.; Yamasaki, Y.; Hori, M. Oxidative stress induces nucleo-cytoplasmic translocation of pancreatic transcription factor PDX-1 through activation of c-Jun NH(2)-terminal kinase. Diabetes 2003, 52, 2896–2904. [Google Scholar] [CrossRef] [Green Version]
- Kaneto, H.; Katakami, N.; Matsuhisa, M.; Matsuoka, T.-A. Role of Reactive Oxygen Species in the Progression of Type 2 Diabetes and Atherosclerosis. Mediat. Inflamm. 2010, 2010, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Del Guerra, S.; Lupi, R.; Marselli, L.; Masini, M.; Bugliani, M.; Sbrana, S.; Torri, S.; Pollera, M.; Boggi, U.; Mosca, F.; et al. Functional and Molecular Defects of Pancreatic Islets in Human Type 2 Diabetes. Diabetes 2005, 54, 727–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudich, A.; Tirosh, A.; Potashnik, R.; Hemi, R.; Kanety, H.; Bashan, N. Prolonged oxidative stress impairs insulin-induced GLUT4 translocation in 3T3-L1 adipocytes. Diabetes 1998, 47, 1562–1569. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, J.W. Metabolic stress in insulin’s target cells leads to ROS accumulation—A hypothetical common pathway causing insulin resistance. FEBS Lett. 2007, 581, 3734–3742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawahito, S.; Kitahata, H.; Oshita, S. Problems associated with glucose toxicity: Role of hyperglycemia-induced oxidative stress. World J. Gastroenterol. 2009, 15, 4137–4142. [Google Scholar] [CrossRef]
- Barbour, L.A.; McCurdy, C.E.; Hernandez, T.L.; Kirwan, J.P.; Catalano, P.M.; Friedman, J.E. Cellular Mechanisms for Insulin Resistance in Normal Pregnancy and Gestational Diabetes. Diabetes Care 2007, 30, S112–S119. [Google Scholar] [CrossRef] [Green Version]
- Catalano, P.M.; Huston, L.; Amini, S.B.; Kalhan, S.C. Longitudinal changes in glucose metabolism during pregnancy in obese women with normal glucose tolerance and gestational diabetes mellitus. Am. J. Obstet. Gynecol. 1999, 180, 903–916. [Google Scholar] [CrossRef]
- Catalano, P.M.; Tyzbir, E.; Roman, N. Longitudinal changes in insulin release and insulin resistance in non-obese pregnant women. Am. J. Obstet. Gynecol. 1991, 165, 1667–1672. [Google Scholar] [CrossRef]
- Catalano, P.M.; Tyzbir, E.D.; Wolfe, R.R.; Calles, J.; Roman, N.M.; Amini, S.B.; Sims, E.A. Carbohydrate metabolism during pregnancy in control subjects and women with gestational diabetes. Am. J. Physiol. Metab. 1993, 264, E60–E67. [Google Scholar] [CrossRef]
- Catalano, P.M.; Tyzbir, E.D.; Wolfe, R.R.; Roman, N.M.; Amini, S.B.; Sims, E.A. Longitudinal changes in basal hepatic glucose production and suppression during insulin infusion in normal pregnant women. Am. J. Obstet. Gynecol. 1992, 167, 913–919. [Google Scholar] [CrossRef]
- Girard, J. The Inhibitory Effects of Insulin on Hepatic Glucose Production Are Both Direct and Indirect. Diabetes 2006, 55. [Google Scholar] [CrossRef] [Green Version]
- Kalhan, S.C.; D’Angelo, L.J.; Savin, S.M.; Adam, P.A.J. Glucose Production in Pregnant Women at Term Gestation. J. Clin. Investig. 1979, 63, 388–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coate, K.C.; Smith, M.S.; Shiota, M.; Irimia, J.M.; Roach, P.J.; Farmer, B.; Williams, P.E.; Moore, M.C. Hepatic Glucose Metabolism in Late Pregnancy. Diabetes 2013, 62, 753–761. [Google Scholar] [CrossRef] [Green Version]
- Butte, N.F. Carbohydrate and lipid metabolism in pregnancy: Normal compared with gestational diabetes mellitus. Am. J. Clin. Nutr. 2000, 71, 1256S–1261S. [Google Scholar] [CrossRef] [PubMed]
- Lesser, K.B.; Carpenter, M.W. Metabolic changes associated with normal pregnancy and pregnancy complicated by diabetes mellitus. Semin. Perinatol. 1994, 18, 399–406. [Google Scholar] [PubMed]
- Sonagra, A.D.; Biradar, S.M.; Dattatreya, K.; DS, J.M. Normal Pregnancy- A State of Insulin Resistance. J. Clin. Diagn. Res. 2014, 8, CC01–CC03. [Google Scholar] [CrossRef] [PubMed]
- Barbour, L.A.; Shao, J.; Qiao, L.; Pulawa, L.K.; Jensen, D.R.; Bartke, A.; Garrity, M.; Draznin, B.; Friedman, J.E. Human placental growth hormone causes severe insulin resistance in transgenic mice. Am. J. Obstet. Gynecol. 2002, 186, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Brelje, T.C.; Scharp, D.W.; Lacy, P.E.; Ogren, L.; Talamantes, F.; Robertson, M.; Friesen, H.G.; Sorenson, R.L. Effect of homologous placental lactogens, prolactins, and growth hormones on islet B-cells division and insulin secretion in rat, mouse, and human islets: Implication for placental lactogen regulation of islet function during pregnancy. Endocrinology 1993, 132, 879–887. [Google Scholar] [CrossRef]
- Hotamisligil, G.; Peraldi, P.; Budavari, A.; Ellis, R.; White, M.; Spiegelman, B. IRS-1-mediated inhibition of insulin receptor tyrosine kindase activity in TNF-alpha and obesity-induced insulin resistance. Science 1996, 271, 665–668. [Google Scholar] [CrossRef]
- Peraldi, P.; Hotamisligil, G.; Buurman, W.; White, M.; Spiegelman, B. Tumor necrosis factor (TNF)-alpha inhibits insulin signaling thorugh stimulation of the p55 TNF receptor and activation of sphingomyelinase. J. Biol. Chem. 1996, 271, 13018–13022. [Google Scholar] [CrossRef] [Green Version]
- Hara, K.; Yamauchi, T.; Kadowaki, T. Adiponectin: An adipokine linking adipocytes and type 2 diabetes in humans. Curr. Diabetes Rep. 2005, 5, 136–140. [Google Scholar] [CrossRef]
- Catalano, P.M.; Hoegh, M.; Minium, J.; Huston-Presley, L.; Bernard, S.; Kalhan, S.C.; Mouzon, S.H.-D. Adiponectin in human pregnancy: Implications for regulation of glucose and lipid metabolism. Diabetol. 2006, 49, 1677–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, J.; Catalano, P.M.; Yamashita, H.; Ishizuka, T.; Friedman, J.E. Vanadate enhances but does not normalize glucose transport and insulin receptor phosphorylation in skeletal muscle from obese women with gestational diabetes mellitus. Am. J. Obstet. Gynecol. 2000, 183, 1263–1270. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.E.; Ishizuka, T.; Shao, J.; Huston, L.; Highman, T.; Catalano, P. Impaired glucose transport and insulin receptor tyrosine phosphorylation in skeletal muscle from obese women with gestational diabetes. Diabetes 1999, 48, 1807–1814. [Google Scholar] [CrossRef] [PubMed]
- Okuno, S.; Akazawo, S.; Yasuhi, I.; Kawasaki, E.; Matsumoto, K.; Yamasaki, H.; Matsuo, H.; Yamaguchi, Y.; Nagataki, S. Decreased Expression of the GLUT4 Glucose Transporter Protein in Adipose Tissue During Pregnancy. Horm. Metab. Res. 1995, 27, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Joosen, A.M.C.P.; Bakker, A.H.F.; Zorenc, A.H.G.; Kersten, S.; Schrauwen, P.; Westerterp, K.R. PPARγ activity in subcutaneous abdominal fat tissue and fat mass gain during short-term overfeeding. Int. J. Obes. 2005, 30, 302–307. [Google Scholar] [CrossRef] [Green Version]
- Catalano, P.M.; Nizielski, S.E.; Shao, J.; Preston, L.; Qiao, L.; Friedman, J.E. Downregulated IRS-1 and PPARγ in obese women with gestational diabetes: Relationship to FFA during pregnancy. Am. J. Physiol. Metab. 2002, 282, E522–E533. [Google Scholar] [CrossRef] [Green Version]
- Vargas, R.; Repke, J.T.; Ural, S.H. Type 1 Diabetes Mellitus and Pregnancy. Rev. Obstet. Gynecol. 2010, 3, 92–100. [Google Scholar]
- Dunne, F. Type 2 diabetes and pregnancy. Semin. Fetal Neonatal Med. 2005, 10, 333–339. [Google Scholar] [CrossRef]
- Plasencia, W.; Garcia, R.; Pereira, S.; Akolekar, R.; Nicolaides, K.H. Criteria for Screening and Diagnosis of Gestational Diabetes Mellitus in the First Trimester of Pregnancy. Fetal Diagn. Ther. 2011, 30, 108–115. [Google Scholar] [CrossRef]
- Coustan, D.R.; Lowe, L.P.; Metzger, B.E.; Dyer, A.R. International Association of Diabetes and Pregnancy Study Groups the Hyperglycemia and Adverse Pregnancy Outcome (HAPO) study: Paving the way for new diagnostic criteria for gestational diabetes mellitus. Am. J. Obstet. Gynecol. 2010, 202, 654.e1–654.e6. [Google Scholar] [CrossRef] [Green Version]
- Kwak, S.H.; Jang, H.C.; Park, K.S. Finding Genetic Risk Factors of Gestational Diabetes. Genom. Informatics 2012, 10, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Garvey, W.T.; Maianu, L.; Zhu, J.H.; Brechtel-Hook, G.; Wallace, P.; Baron, A.D. Evidence for defects in the trafficking and translocation of GLUT4 glucose transporters in skeletal muscle as a cause of human insulin resistance. J. Clin. Investig. 1998, 101, 2377–2386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ACOG. Pregestational Diabetes Mellitus; ACOG Practice Bulletin No 60; ACOG: Washington, DC, USA, 2005; Volume 105, pp. 675–685. [Google Scholar]
- Sheffield, J.; Butler-Koster, E.; Casey, B.; McIntire, D.D.; Leveno, K.J. Maternal diabetes mellitus and infant malformations. Obstet. Gynecol. 2002, 100, 925–930. [Google Scholar] [PubMed]
- Nold, J.L.; Georgieff, M.K. Infants of diabetic mothers. Pediatr. Clin. North Am. 2004, 51, 619–637. [Google Scholar] [CrossRef] [PubMed]
- Boskovic, R.; Feig, D.S.; Derewlany, L.; Knie, B.; Portnoi, G.; Koren, G. Transfer of insulin lisro across the human placenta. Diabetes Care 2003, 26, 1390–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, W.W. Placental-Fetal Glucose Exchange and Fetal Glucose Metabolism. Trans. Am. Clin. Clim. Assoc. 2006, 117, 321–340. [Google Scholar]
- Gittes, G.K. Developmental biology of the pancreas: A comprehensive review. Dev. Biol. 2009, 326, 4–35. [Google Scholar] [CrossRef] [Green Version]
- Kc, K.; Shakya, S.; Zhang, H. Gestational Diabetes Mellitus and Macrosomia: A Literature Review. Ann. Nutr. Metab. 2015, 66, 14–20. [Google Scholar] [CrossRef]
- Metzger, E.B.; Lowe, L.P.; Dyer, A.R.; Trimble, E.R.; Chaovarindr, U.; Coustan, D.R.; Hadden, D.R.; McCance, D.R.; Hod, M.; McIntyre, H.D.; et al. Hyperglycemia and Adverse Pregnancy Outcomes. N. Engl. J. Med. 2008, 358, 1991–2002. [Google Scholar] [CrossRef] [Green Version]
- Yogev, Y.; Xenakis, E.M.; Langer, O. The association between preeclampsia and the severity of gestational diabetes: The impact of glycemic control. Am. J. Obstet. Gynecol. 2004, 191, 1655–1660. [Google Scholar] [CrossRef]
- Kim, C.; Newton, K.M.; Knopp, R.H. Gestational diabetes and the incidence of type 2 diabetes: A systematic review. Diabetes Care 2002, 25, 1862–1868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yessoufou, A.; Moutairou, K. Maternal Diabetes in Pregnancy: Early and Long-Term Outcomes on the Offspring and the Concept of “Metabolic Memory”. Exp. Diabetes Res. 2011, 2011, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Effect of Intensive Treatment of Diabetes on the Development and Progression of Long-Term Complications in Insulin-Dependent Diabetes Mellitus. N. Engl. J. Med. 1993, 329, 977–986. [CrossRef]
- Holman, R.R.; Paul, S.; Bethel, M.A.; Matthews, D.R.; Neil, H.A.W. 10-Year Follow-up of Intensive Glucose Control in Type 2 Diabetes. N. Engl. J. Med. 2008, 359, 1577–1589. [Google Scholar] [CrossRef] [Green Version]
- Nathan, D.M.; Cleary, P.A.; Backlund, J.-Y.C.; Genuth, S.M.; Lachin, J.M.; Orchard, T.J.; Raskin, P.; Zinman, B. Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Study Research Group Intensive Diabetes Treatment and Cardiovascular Disease in Patients with Type 1 Diabetes. N. Engl. J. Med. 2005, 353, 2643–2653. [Google Scholar] [CrossRef]
- Olsen, A.S.; Sarras, M.P.; Leontovich, A.; Intine, R.V. Heritable Transmission of Diabetic Metabolic Memory in Zebrafish Correlates with DNA Hypomethylation and Aberrant Gene Expression. Diabetes 2012, 61, 485–491. [Google Scholar] [CrossRef] [Green Version]
- Caramori, M.L.; Kim, Y.; Natarajan, R.; Moore, J.H.; Rich, S.S.; Mychaleckyj, J.C.; Kuriyama, R.; Kirkpatrick, D.; Mauer, M. Differential Response to High Glucose in Skin Fibroblasts of Monozygotic Twins Discordant for Type 1 Diabetes. J. Clin. Endocrinol. Metab. 2015, 100, E883–E889. [Google Scholar] [CrossRef] [Green Version]
- Dabelea, D.; Hanson, R.L.; Lindsay, R.S.; Pettitt, D.J.; Imperatore, G.; Gabir, M.M.; Roumain, J.; Bennett, P.H.; Knowler, W.C. Intrauterine exposure to diabetes conveys risks for type 2 diabetes and obesity: A study of discordant sibships. Diabetes 2000, 49, 2208–2211. [Google Scholar] [CrossRef] [Green Version]
- Lawlor, D.A.; Lichtenstein, P.; Langström, N. The association of maternal diabetes in pregnancy with offspring adiposity into early adulthood: Sibling study in a prospective cohort of 280,866 men from 248,293 families. Circulation 2011, 123, 258–265. [Google Scholar] [CrossRef]
- Kaufman, F.R. Type 2 diabetes mellitus in children and youth: A new epidemic. J. Pediatr. Endocrinol. Metab. 2002, 15, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Boney, C.M. Metabolic Syndrome in Childhood: Association with Birth Weight, Maternal Obesity, and Gestational Diabetes Mellitus. Pediatrics 2005, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillier, T.A.; Pedula, K.L.; Schmidt, M.M.; Mullen, J.A.; Charles, M.-A.; Pettitt, D.J. Childhood Obesity and Metabolic Imprinting: The ongoing effects of maternal hyperglycemia. Diabetes Care 2007, 30, 2287–2292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dabelea, D.; Mayer-Davis, E.J.; Lamichhane, A.P.; D’Agostino, R.B.; Liese, A.D.; Vehik, K.S.; Narayan, K.V.; Zeitler, P.; Hamman, R.F. Association of Intrauterine Exposure to Maternal Diabetes and Obesity with Type 2 Diabetes in Youth. Diabetes Care 2008, 31, 1422–1426. [Google Scholar] [CrossRef] [Green Version]
- Cho, N.H.; Silverman, B.L.; Rizzo, T.A.; Metzger, B.E. Correlations between the intrauterine metabolic environment and blood pressure in adolescent offspring of diabetic mothers. J. Pediatr. 2000, 136, 587–592. [Google Scholar] [CrossRef]
- Boloker, J.; Gertz, S.J.; Simmons, R.A. Gestational diabetes leads to the development of diabetes in adulthood in the rat. Diabetes 2002, 51, 1499–1506. [Google Scholar] [CrossRef] [Green Version]
- Merzouk, H.; Madani, S.; Hichami, A.; Prost, J.; Belleville, J.; Khan, N. Age-related changes in fatty acids in obese offspring of streptozocin-induced diabetic rats. Obes. Res. 2002, 10, 703–714. [Google Scholar] [CrossRef]
- Geary, M.P.P.; Pringle, P.J.; Rodeck, C.H.; Kingdom, J.C.P.; Hindmarsh, P.C. Sexual Dimorphism in the Growth Hormone and Insulin-Like Growth Factor Axis at Birth. J. Clin. Endocrinol. Metab. 2003, 88, 3708–3714. [Google Scholar] [CrossRef]
- Xiao, L.; Zhao, J.P.; Nuyt, A.M.; Fraser, W.D.; Luo, Z.-C. Female fetus is associated with greater maternal insulin resistance in pregnancy. Diabet. Med. 2014, 31, 1696–1701. [Google Scholar] [CrossRef]
- Lingwood, B.; Henry, A.M.; D’Emden, M.C.; Fullerton, A.-M.; Mortimer, R.H.; Colditz, P.B.; Cao, K.-A.L.; Callaway, L.K. Determinants of Body Fat in Infants of Women with Gestational Diabetes Mellitus Differ With Fetal Sex. Diabetes Care 2011, 34, 2581–2585. [Google Scholar] [CrossRef] [Green Version]
- Ricart, W.; López, J.; Mozas, J.; Pericot, A.; Sancho, M.; González, N.; Balsells, M.; Luna, R.; Cortázar, A.; Navarro, P.; et al. Maternal glucose tolerance status influences the risk of macrosomia in male but not in female fetuses. J. Epidemiol. Commun. Health 2009, 63, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Tam, W.H.; Ma, R.C.W.; Ozaki, R.; Li, A.M.; Chan, M.H.M.; Yuen, L.Y.; Lao, T.T.H.; Yang, X.; Ho, C.S.; Tutino, G.E.; et al. In Utero Exposure to Maternal Hyperglycemia Increases Childhood Cardiometabolic Risk in Offspring. Diabetes Care 2017, 40, 679–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahado-Singh, R.O.; Mele, L.; Landon, M.B.; Ramin, S.M.; Carpenter, M.W.; Casey, B.; Wapner, R.J.; Varner, M.W.; Rouse, D.J.; Thorp, J.M.; et al. Fetal male gender and the benefits of treatment of mild gestational diabetes mellitus. Am. J. Obstet. Gynecol. 2012, 206, 422.e1–422.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dearden, L.; Balthasar, N. Sexual dimorphism in offspring glucose-sensitive hypothalamic gene expression and physiological responses to maternal high-fat diet feeding. Endocrinology 2014, 155, 2144–2154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuelsson, A.-M.; Matthews, P.A.; Jansen, E.; Taylor, P.D.; Poston, L. Sucrose feeding in mouse pregnancy leads to hypertension, and sex-linked obesity and insulin resistance in female offspring. Front. Physiol. 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- Vickers, M.H.; Clayton, Z.; Yap, C.; Sloboda, D.M. Maternal Fructose Intake during Pregnancy and Lactation Alters Placental Growth and Leads to Sex-Specific Changes in Fetal and Neonatal Endocrine Function. Endocrinology 2011, 152, 1378–1387. [Google Scholar] [CrossRef] [Green Version]
- Katkhuda, R.; Peterson, E.S.; Roghair, R.D.; Norris, A.W.; Scholz, T.D.; Segar, J.L. Sex-specific programming of hypertension in offspring of late-gestation diabetic rats. Pediatr. Res. 2012, 72, 352–361. [Google Scholar] [CrossRef]
- De Sá, F.G.; De Queiroz, D.B.; Ramos-Alves, F.E.; Santos-Rocha, J.; Da Silva, O.A.; Moreira, H.S.; Leal, G.A.; Da Rocha, M.A.; Duarte, G.P.; Xavier, F.E. Hyperglycaemia in pregnant rats causes sex-related vascular dysfunction in adult offspring: Role of cyclooxygenase-2. Exp. Physiol. 2017, 102, 1019–1036. [Google Scholar] [CrossRef] [Green Version]
- Grasemann, C.; Devlin, M.J.; Rzeczkowska, P.A.; Herrmann, R.; Horsthemke, B.; Hauffa, B.P.; Grynpas, M.; Alm, C.; Bouxsein, M.L.; Palmert, M.R. Parental Diabetes: The Akita Mouse as a Model of the Effects of Maternal and Paternal Hyperglycemia in Wildtype Offspring. PLoS ONE 2012, 7, e50210. [Google Scholar] [CrossRef] [Green Version]
- Ide, T.; Tsutsui, H.; Ohashi, N.; Hayashidani, S.; Suematsu, N.; Tsuchihashi, M.; Tamai, H.; Takeshita, A. Greater oxidative stress in healthy young men compared with premenopausal women. Arter. Thromb. Vasc. Biol. 2002, 22, 438–442. [Google Scholar] [CrossRef] [Green Version]
- Barp, J.; Araujo, A.S.D.R.; Fernandes, T.; Rigatto, K.; Llesuy, S.; Belló-Klein, A.; Singal, P. Myocardial antioxidant and oxidative stress changes due to sex hormones. Braz. J. Med Biol. Res. 2002, 35, 1075–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matarrese, P.; Colasanti, T.; Ascione, B.; Margutti, P.; Franconi, F.; Alessandri, C.; Conti, F.; Riccieri, V.; Rosano, G.; Ortona, E.; et al. Gender Disparity in Susceptibility to Oxidative Stress and Apoptosis Induced by Autoantibodies Specific to RLIP76 in Vascular Cells. Antioxid. Redox Signal. 2011, 15, 2825–2836. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, K.; Elmarakby, A.A.; El-Remessey, A.; Sullivan, J.C. Oxidative stress contributes to sex differences in angiotensin II-mediated hypertension in spontaneously hypertensive rats. Am. J. Physiol. Integr. Comp. Physiol. 2012, 302, R274–R282. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, H. Gender difference in glutathione metabolism during aging in mice. Exp. Gerontol. 2003, 38, 507–517. [Google Scholar] [CrossRef]
- Tondreau, M.Y.; Boucher, E.; Simard, M.; Tremblay, Y.; Bilodeau, J.-F. Sex-specific perinatal expression of glutathione peroxidases during mouse lung development. Mol. Cell. Endocrinol. 2012, 355, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Hunaiti, A.A.; Al-Shareef, M. Interplay between Glutathione-S-Transferase and Glucose-6-Phosphate Dehydrogenase in Neonatal Cord Blood. Neonatology 1997, 72, 273–278. [Google Scholar] [CrossRef]
- Lavoie, J.-C.; Chessex, P. Gender and Maturation Affect Glutathione Status in Human Neonatal Tissues. Free. Radic. Biol. Med. 1997, 23, 648–657. [Google Scholar] [CrossRef]
- Franzago, M.; Fraticelli, F.; Stuppia, L.; Vitacolonna, E. Nutrigenetics, epigenetics and gestational diabetes: Consequences in mother and child. Epigenetics 2019, 14, 215–235. [Google Scholar] [CrossRef] [Green Version]
- El Hajj, N.; Pliushch, G.; Schneider, E.; Dittrich, M.; Müller, T.; Korenkov, M.; Aretz, M.; Zechner, U.; Lehnen, H.; Haaf, T. Metabolic Programming of MEST DNA Methylation by Intrauterine Exposure to Gestational Diabetes Mellitus. Diabetes 2013, 62, 1320–1328. [Google Scholar] [CrossRef] [Green Version]
- Reichetzeder, C.; Putra, S.E.D.; Pfab, T.; Slowinski, T.; Neuber, C.; Kleuser, B.; Hocher, B. Increased global placental DNA methylation levels are associated with gestational diabetes. Clin. Epigenetics 2016, 8, 82. [Google Scholar] [CrossRef] [Green Version]
- Bouchard, L.; Thibault, S.; Guay, S.-P.; Santure, M.; Monpetit, A.; St-Pierre, J.; Perron, P.; Brisson, D. Leptin Gene Epigenetic Adaptation to Impaired Glucose Metabolism During Pregnancy. Diabetes Care 2010, 33, 2436–2441. [Google Scholar] [CrossRef] [Green Version]
- Bouchard, L.; Hivert, M.-F.; Guay, S.-P.; St-Pierre, J.; Perron, P.; Brisson, D. Placental Adiponectin Gene DNA Methylation Levels Are Associated with Mothers’ Blood Glucose Concentration. Diabetes 2012, 61, 1272–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houde, A.-A.; Guay, S.-P.; Desgagné, V.; Hivert, M.-F.; Baillargeon, J.-P.; St-Pierre, J.; Perron, P.; Gaudet, D.; Brisson, D.; Bouchard, L. Adaptations of placental and cord bloodABCA1 DNA methylation profile to maternal metabolic status. Epigenetics 2013, 8, 1289–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houde, A.A.; St-Pierre, J.; Hivert, M.F.; Baillargeon, J.P.; Perron, P.; Gaudet, D.; Brisson, D.; Bouchard, L. Placental lipoprotein lipase DNA methylation levels are associated with gestational diabetes mellitus and maternal and cord blood lipid profiles. J. Dev. Orig. Health Dis. 2014, 5, 132–141. [Google Scholar] [CrossRef] [PubMed]
- García-Cardona, M.C.; Huang, F.; García-Vivas, J.M.; Lopez-Camarillo, C.; Navarro, B.E.D.R.; Olivos, E.N.; Hong-Chong, E.; Bolaños-Jiménez, F.; Marchat, L.A. DNA methylation of leptin and adiponectin promoters in children is reduced by the combined presence of obesity and insulin resistance. Int. J. Obes. 2014, 38, 1457–1465. [Google Scholar] [CrossRef] [PubMed]
- Miao, F.; Chen, Z.; Genuth, S.; Paterson, A.D.; Zhang, L.; Wu, X.; Li, S.M.; Cleary, P.; Riggs, A.; Harlan, D.M.; et al. Evaluating the Role of Epigenetic Histone Modifications in the Metabolic Memory of Type 1 Diabetes. Diabetes 2014, 63, 1748–1762. [Google Scholar] [CrossRef] [Green Version]
- El-Osta, A.; Brasacchio, D.; Yao, D.; Pocai, A.; Jones, P.L.; Roeder, R.G.; Cooper, M.E.; Brownlee, M. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J. Exp. Med. 2008, 205, 2409–2417. [Google Scholar] [CrossRef]
- Yun, J.-M.; Jialal, I.; Devaraj, S. Epigenetic regulation of high glucose-induced proinflammatory cytokine production in monocytes by curcumin. J. Nutr. Biochem. 2011, 22, 450–458. [Google Scholar] [CrossRef] [Green Version]
- Raza, H.; John, A. Glutathione metabolism and oxidative stress in neonatal rat tissues from streptozotocin-induced diabetic mothers. Diabetes Metab. Res. Rev. 2004, 20, 72–78. [Google Scholar] [CrossRef]
- Cerda, S.R.; Weitzman, S.A. Influence of oxygen radical injury on DNA methylation. Mutat. Res. Mutat. Res. 1997, 386, 141–152. [Google Scholar] [CrossRef]
- Madugundu, G.S.; Cadet, J.; Wagner, J. Hydroxyl-radical-induced oxidation of 5-methylcytosine in isolated and cellular DNA. Nucleic Acids Res. 2014, 42, 7450–7460. [Google Scholar] [CrossRef] [PubMed]
- Kreuz, S.; Fischle, W. Oxidative stress signaling to chromatin in health and disease. Epigenomics 2016, 8, 843–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.A.; Alam, K.; Dixit, K.; Rizvi, M.M.A. Role of peroxynitrite induced structural changes on H2B histone by physicochemical method. Int. J. Biol. Macromol. 2016, 82, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Durning, S.P.; Flanagan-Steet, H.; Prasad, N.; Wells, L. O-Linked β-N-acetylglucosamine (O-GlcNAc) Acts as a Glucose Sensor to Epigenetically Regulate the Insulin Gene in Pancreatic Beta Cells. J. Biol. Chem. 2015, 291, 2107–2118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, B.A.; Hanover, J.A. O-GlcNAc and the Epigenetic Regulation of Gene Expression. J. Biol. Chem. 2014, 289, 34440–34448. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Yin, X.; Yang, H.; Xu, Y. Histone Demethylase LSD2 Acts as an E3 Ubiquitin Ligase and Inhibits Cancer Cell Growth through Promoting Proteasomal Degradation of OGT. Mol. Cell 2015, 58, 47–59. [Google Scholar] [CrossRef] [Green Version]
- Vella, P.; Scelfo, A.; Jammula, S.; Chiacchiera, F.; Williams, K.; Cuomo, A.; Roberto, A.; Christensen, J.; Bonaldi, T.; Helin, K.; et al. Tet Proteins Connect the O-Linked N-acetylglucosamine Transferase Ogt to Chromatin in Embryonic Stem Cells. Mol. Cell 2013, 49, 645–656. [Google Scholar] [CrossRef] [Green Version]
- Deplus, R.; Delatte, B.; Schwinn, M.K.; Defrance, M.; Méndez, J.; Murphy, N.; Dawson, M.A.; Volkmar, M.; Putmans, P.; Calonne, E.; et al. TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J. 2013, 32, 645–655. [Google Scholar] [CrossRef]
- Cai, L.; Sutter, B.M.; Li, B.; Tu, B.P. Acetyl-CoA Induces Cell Growth and Proliferation by Promoting the Acetylation of Histones at Growth Genes. Mol. Cell 2011, 42, 426–437. [Google Scholar] [CrossRef] [Green Version]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-Citrate Lyase Links Cellular Metabolism to Histone Acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef] [Green Version]
- Mulligan, P.; Yang, F.; Di Stefano, L.; Ji, J.-Y.; Ouyang, J.; Nishikawa, J.L.; Toiber, D.; Kulkarni, M.; Wang, Q.; Najafi-Shoushtari, H.; et al. A SIRT1-LSD1 Corepressor Complex Regulates Notch Target Gene Expression and Development. Mol. Cell 2011, 42, 689–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brawerman, G.M.; Kereliuk, S.M.; Brar, N.; Cole, L.K.; Seshadri, N.; Pereira, T.J.; Xiang, B.; Hunt, K.L.; Fonseca, M.A.; Hatch, G.M.; et al. Maternal resveratrol administration protects against gestational diabetes-induced glucose intolerance and islet dysfunction in the rat offspring. J. Physiol. 2019, 597, 4175–4192. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tozour, J.; Hughes, F.; Carrier, A.; Vieau, D.; Delahaye, F. Prenatal Hyperglycemia Exposure and Cellular Stress, a Sugar-Coated View of Early Programming of Metabolic Diseases. Biomolecules 2020, 10, 1359. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10101359
Tozour J, Hughes F, Carrier A, Vieau D, Delahaye F. Prenatal Hyperglycemia Exposure and Cellular Stress, a Sugar-Coated View of Early Programming of Metabolic Diseases. Biomolecules. 2020; 10(10):1359. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10101359
Chicago/Turabian StyleTozour, Jessica, Francine Hughes, Arnaud Carrier, Didier Vieau, and Fabien Delahaye. 2020. "Prenatal Hyperglycemia Exposure and Cellular Stress, a Sugar-Coated View of Early Programming of Metabolic Diseases" Biomolecules 10, no. 10: 1359. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10101359