Large Scale Conversion of Trilobolide into the Payload of Mipsagargin: 8-O-(12-Aminododecanoyl)-8-O-Debutanoylthapsigargin

 ,
,  , , , and
, , , and
Abstract
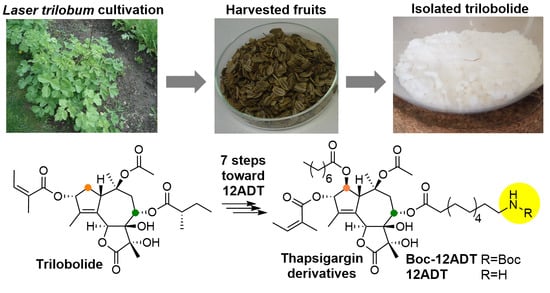
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
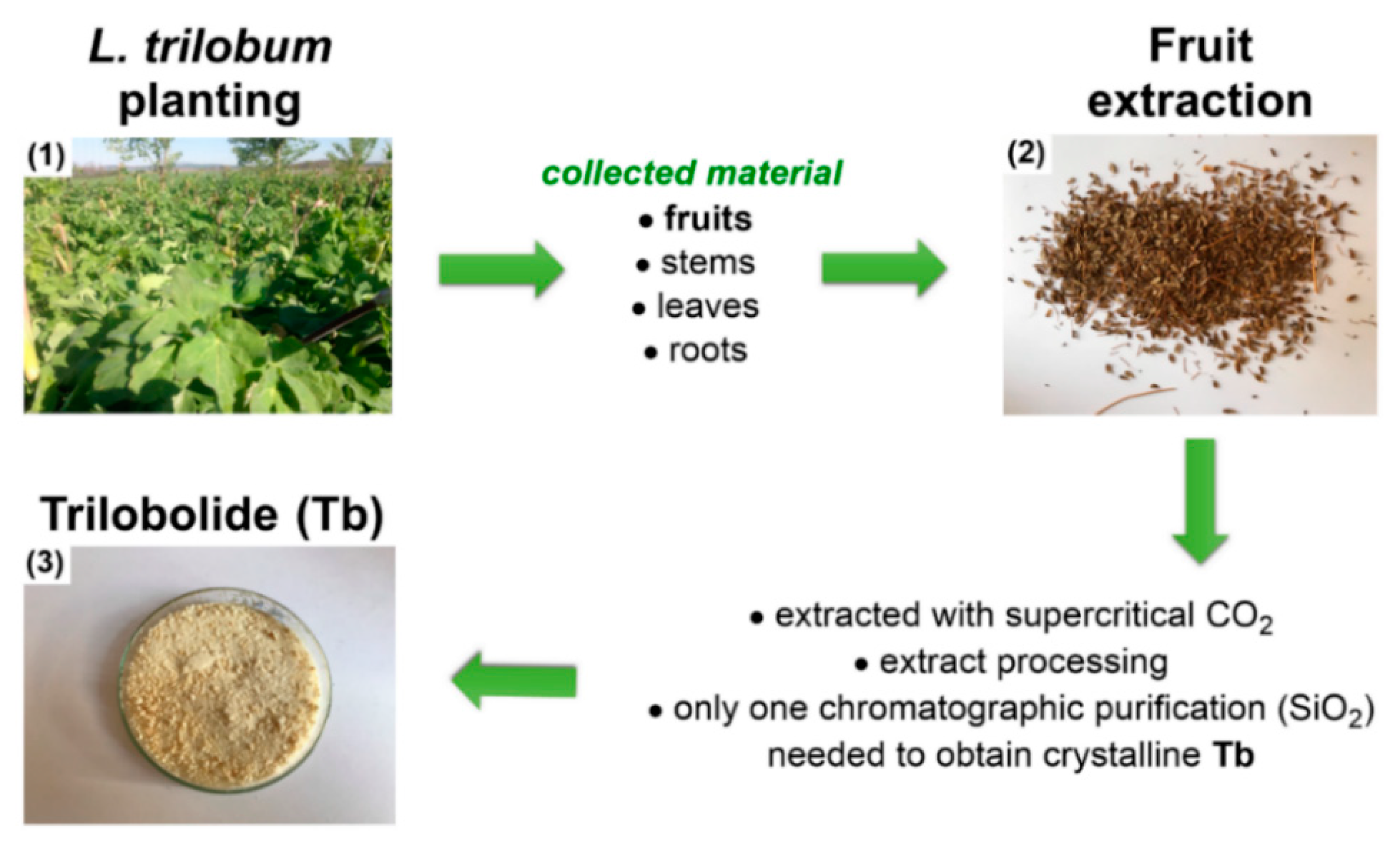
2.1. Supercritical Carbon Dioxide Extraction of Trilobolide from L. Trilobum Fruits
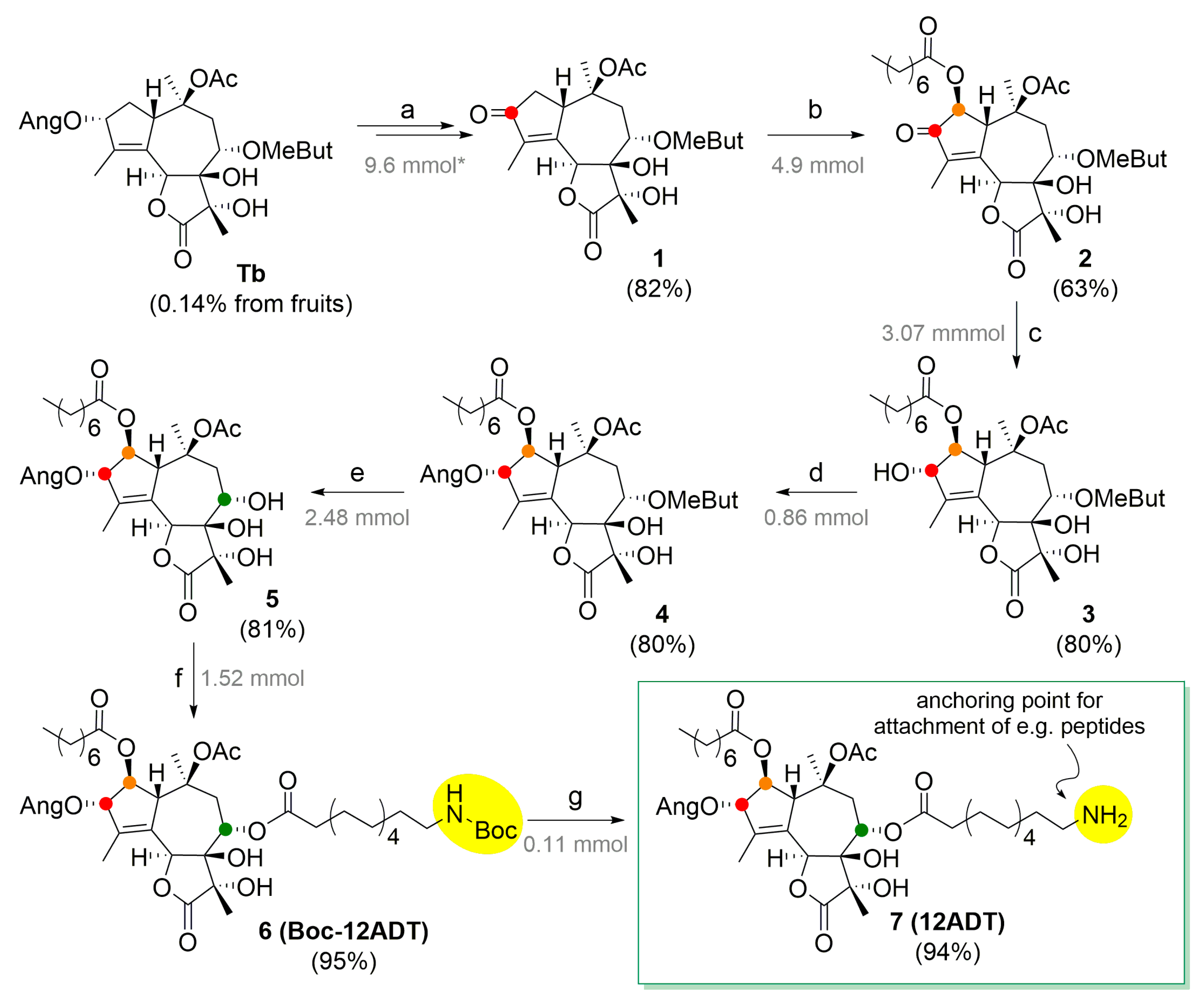
2.2. Scale-Up Synthesis of 8-O-(12-Aminododecanoyl)-8-O-Debutanoyl Thapsigargin (12ADT) from Tb
2.2.1. Synthesis of 3-oxo-3-Desangeloyl Trilobolide (1)
2.2.2. Synthesis of 3-Oxo-2-Octanoyl-3-Desangeloyl Trilobolide (2)
2.2.3. Synthesis of (3S)-Hydroxy-2-Octanoyl-3-Desangeloyl Trilobolide (3)
2.2.4. Preparation of 2,4,6-Trichlorobenzoic (Z)-2-Methylbut-2-Enoic Anhydride
2.2.5. Synthesis of 8-O-(2-(S)-Methylbutanoyl)-8-O-Debutanoyl Thapsigargin (4)
2.2.6. Synthesis of 8-O-Debutanoyl Thapsigargin (5)
2.2.7. Preparation of Boc-N-12-Aminododecanoic Acid
2.2.8. Synthesis of 8-O-(Boc-N-12-Aminododecanoyl)-8-O-Debutanoyl Thapsigargin (6)
2.2.9. Deprotection of Boc group to afford 8-O-(12-aminododecanoyl)-8-O-debutanoyl thapsigargin (7)
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Doan, N.T.Q.; Paulsen, E.S.; Sehgal, P.; Moeller, J.V.; Nissen, P.; Denmeade, S.R.; Isaacs, J.T.; Dionne, C.A.; Christensen, S.B. Targeting thapsigargin towards tumors. Steroids 2015, 97, 2–7. [Google Scholar] [CrossRef]
- Ma, Z.; Fan, C.; Yang, Y.; Di, S.; Hu, W.; Li, T.; Zhu, Y.; Han, J.; Xin, Z.; Wu, G.; et al. Thapsigargin sensitizes human esophageal cancer to TRAIL induced apoptosis via AMPK activation. Sci. Rep. 2016, 6, 35196. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.; Wang, P.; Wang, X. Thapsigargin induces apoptosis of prostate cancer through cofilin-1 and paxillin. Oncol. Lett. 2018, 16, 1975–1980. [Google Scholar] [CrossRef]
- Wu, L.; Huang, X.; Kuang, Y.; Xing, Z.; Deng, X.; Luo, Z. Thapsigargin induces apoptosis in adrenocortical carcinoma by activating endoplasmic reticulum stress and the JNK signaling pathway: An in vitro and in vivo study. Drug Des. Devel. Ther. 2019, 13, 2787–2798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denmeade, S.R.; Mhaka, A.M.; Rosen, D.M.; Brennen, W.N.; Dalrymple, S.; Dach, I.; Olesen, C.; Gurel, B.; DeMarzo, A.M.; Wilding, G.; et al. Engineering a prostate specific membrane antigen activated tumor endothelial cell prodrug for cancer therapy. Sci. Transl. Med. 2012, 4, 140–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Škubník, J.; Jurášek, M.; Ruml, T.; Rimpelová, S. Mitotic poisons in research and medicine. Molecules 2020, 25, 4632. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.S.; Koh, C.G.; Li, H.Y. Mitosis targeted anti-cancer therapies: Where they stand. Cell Death Discov. 2012, 3. [Google Scholar] [CrossRef] [Green Version]
- Denmeade, S.R.; Jakobsen, C.M.; Janssen, S.; Khan, S.R.; Garrett, E.S.; Lilja, H.; Christensen, S.B.; Isaacs, J.T. Prostate specific antigen activated thapsigargin prodrug as targeted therapy for prostate cancer. J. Natl. Cancer Inst. 2003, 95, 990–1000. [Google Scholar] [CrossRef] [Green Version]
- Peterková, L.; Kmoníčková, E.; Ruml, T.; Rimpelová, S. Sarco/endoplasmic reticulum calcium ATPase inhibitors: Beyond anticancer perspective. J. Med. Chem. 2020, 63, 1937–1963. [Google Scholar] [CrossRef]
- Sohoel, H.; Lund, J.A.-M.; Moller, J.V.; Nissen, P.; Denmeade, S.R.; Isaacs, J.T.; Olsen, C.E.; Christensen, S.B. Natural products as starting materials for development of second-generation SERCA inhibitors targeted towards prostate cancer cells. Bioorg. Med. Chem. 2006, 14, 2810–2815. [Google Scholar] [CrossRef]
- Sehgal, P.; Szalai, P.; Olesen, C.; Praetorius, H.A.; Nissen, P.; Christensen, S.B.; Engedal, N.; Møller, J.V. Inhibition of the sarco/endoplasmic reticulum (ER) Ca(2+) ATPase by thapsigargin analogs induces cell death via ER Ca(2+) depletion and the unfolded protein response. J. Biol. Chem. 2017, 292, 19656–19673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, T.B.; Lopez, C.Q.; Manczak, T.; Martinez, K.; Simonsen, H.T. Thapsigargin from Thapsia, L. to mipsagargin. Molecules 2015, 20, 6113–6127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kmoníčková, E.; Harmatha, J.; Vokáč, K.; Kostecká, P.; Farghali, H.; Zídek, Z. Sesquiterpene lactone trilobolide activates production of interferon-γ and nitric oxide. Fitoterapia 2010, 81, 1213–1219. [Google Scholar] [CrossRef]
- Winther, A.-M.L.; Liu, H.; Sonntag, Y.; Olesen, C.; Le, M.M.; Soehoel, H.; Olsen, C.-E.; Christensen, S.B.; Nissen, P.; Moller, J.V. Critical roles of hydrophobicity and orientation of side chains for inactivation of sarcoplasmic reticulum Ca2+ ATPase with thapsigargin and thapsigargin analogs. J. Biol. Chem. 2010, 285, 28883–28892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harmatha, J.; Buděšínský, M.; Jurášek, M.; Zimmermann, T.; Drašar, P.; Zídek, Z.; Kmoníčková, E.; Vejvodová, L. Structural modification of trilobolide for upgrading its immunobiological properties and reducing its cytotoxic action. Fitoterapia 2019, 134, 88–95. [Google Scholar] [CrossRef]
- Christensen, S.B.; Andersen, A.; Kromann, H.; Treiman, M.; Tombal, B.; Denmeade, S.; Isaacs, J.T. Thapsigargin analogs for targeting programmed death of androgen independent prostate cancer cells. Bioorg. Med. Chem. 1999, 7, 1273–1280. [Google Scholar] [CrossRef]
- Janssen, S.; Rosen, D.M.; Ricklis, R.M.; Dionne, C.A.; Lilja, H.; Christensen, S.B.; Isaacs, J.T.; Denmeade, S.R. Pharmacokinetics, biodistribution, and antitumor efficacy of a human glandular kallikrein 2 (hK2)-activated thapsigargin prodrug. Prostate 2006, 66, 358–368. [Google Scholar] [CrossRef]
- Zimmermann, T.; Christensen, S.; Franzyk, H. Preparation of enzyme-activated thapsigargin prodrugs by solid-phase synthesis. Molecules 2018, 23, 1463. [Google Scholar] [CrossRef] [Green Version]
- Mahalingam, D.; Peguero, J.; Cen, P.; Arora, S.P.; Sarantopoulos, J.; Rowe, J.; Allgood, V.; Tubb, B.; Campos, L. A phase II, multicenter, single-arm study of mipsagargin (G-202) as a second-line therapy following sorafenib for adult patients with progressive advanced hepatocellular carcinoma. Cancers 2019, 11, 833. [Google Scholar] [CrossRef] [Green Version]
- Mahalingam, D.; Wilding, G.; Denmeade, S.; Sarantopoulas, J.; Cosgrove, D.; Cetnar, J.; Azad, N.; Bruce, J.; Kurman, M.; Allgood, V.E.; et al. Mipsagargin, a novel thapsigargin-based PSMA-activated prodrug: Results of a first-in-man phase I clinical trial in patients with refractory, advanced or metastatic solid tumours. Br. J. Cancer 2016, 114, 986–994. [Google Scholar] [CrossRef]
- Tarvainen, I.; Zimmermann, T.; Heinonen, P.; Jäntti, M.H.; Yli-Kauhaluoma, J.; Talman, V.; Franzyk, H.; Tuominen, R.K.; Christensen, S.B. Missing selectivity of targeted 4β-phorbol prodrugs expected to be potential chemotherapeutics. ACS Med. Chem. Lett. 2019. [Google Scholar] [CrossRef] [PubMed]
- Akinboye, E.S.; Rogers, O.C.; Isaacs, J.T. 2-Fluoro-5-maleimidobenzoic acid-linked albumin drug (MAD) delivery for selective systemic targeting of metastatic prostate cancer. Prostate 2018, 78, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Akinboye, E.S.; Brennen, W.N.; Denmeade, S.R.; Isaacs, J.T. Albumin-linked prostate-specific antigen-activated thapsigargin and niclosamide based molecular grenades targeting the microenvironment in metastatic castration-resistant prostate cancer. Asian J. Urol. 2019, 6, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Smith, J.M.; Felding, J.; Baran, P.S. Scalable Synthesis of (−)-Thapsigargin. ACS Cent. Sci. 2017, 3, 47–51. [Google Scholar] [CrossRef]
- Ley, S.V.; Antonello, A.; Balskus, E.P.; Booth, D.T.; Christensen, S.B.; Cleator, E.; Gold, H.; Högenauer, K.; Hünger, U.; Myers, R.M.; et al. Synthesis of the thapsigargins. Proc. Natl. Acad. Sci. USA 2004, 101, 12073–12078. [Google Scholar] [CrossRef] [Green Version]
- Crestey, F.; Toma, M.; Christensen, S.B. Concise synthesis of thapsigargin from nortrilobolide. Tetrahedron Lett. 2015, 56, 5896–5898. [Google Scholar] [CrossRef]
- Doan, N.T.Q.; Crestey, F.; Olsen, C.E.; Christensen, S.B. Chemo-and regioselective functionalization of nortrilobolide: Application for semisynthesis of the natural product 2-acetoxytrilobolide. J. Nat. Prod. 2015, 78, 1406–1414. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S.P.; Ball, M.; Wierschem, F.; Cleator, E.; Oliver, S.; Hogenauer, K.; Simic, O.; Antonello, A.; Hunger, U.; Smith, M.D.; et al. Total synthesis of five thapsigargins: Guaianolide natural products exhibiting sub-nanomolar SERCA inhibition. Chemistry 2007, 13, 5688–5712. [Google Scholar] [CrossRef]
- Jakobsen, C.M.; Denmeade, S.R.; Isaacs, J.T.; Gady, A.; Olsen, C.E.; Christensen, S.B. Design, synthesis, and pharmacological evaluation of thapsigargin analogues for targeting apoptosis to prostatic cancer cells. J. Med. Chem. 2001, 44, 4696–4703. [Google Scholar] [CrossRef]
- Wang, D.S.; Wagner, M.; Butt, H.J.; Wu, S. Supramolecular hydrogels constructed by red-light-responsive host-guest interactions for photo-controlled protein release in deep tissue. Soft Matter 2015, 11, 7656–7662. [Google Scholar] [CrossRef] [Green Version]
- Amorim, M.H.R.; Gil da Costa, R.M.; Lopes, C.; Bastos, M.M.S.M. Sesquiterpene lactones: Adverse health effects and toxicity mechanisms. Crit. Rev. Toxicol. 2013, 43, 559–579. [Google Scholar] [CrossRef] [PubMed]
- Kishkentayeva, A.S.; Adekenov, S.M.; Drašar, P.B. Production technologies of pharmacologically active sesquiterpene lactones. Eurasian Chem. Technol. J. 2018, 20, 325–333. [Google Scholar] [CrossRef]
- de Melo, M.M.R.; Silvestre, A.J.D.; Silva, C.M. Supercritical fluid extraction of vegetable matrices: Applications, trends and future perspectives of a convincing green technology. J. Supercrit. Fluids 2014, 92, 115–176. [Google Scholar] [CrossRef]
- Liang, X.; Grue-Soerensen, G.; Petersen, A.K.; Hogberg, T. Semisynthesis of ingenol 3-angelate (PEP005): Efficient stereoconservative angeloylation of alcohols. Synlett 2012, 23, 2647–2652. [Google Scholar] [CrossRef] [Green Version]
- Jurášek, M.; Rimpelová, S.; Kmoníčková, E.; Drašar, P.; Ruml, T. Tailor-made fluorescent trilobolide to study its biological relevance. J. Med. Chem. 2014, 57, 7947–7954. [Google Scholar] [CrossRef]
- Huml, L.; Jurášek, M.; Mikšátková, P.; Zimmermann, T.; Tomanová, P.; Buděšínský, M.; Rottnerová, Z.; Šimková, M.; Harmatha, J.; Kmoníčková, E.; et al. Immunoassay for determination of trilobolide. Steroids 2017, 117, 105–111. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zimmermann, T.; Drašar, P.; Rimpelová, S.; Christensen, S.B.; Khripach, V.A.; Jurášek, M. Large Scale Conversion of Trilobolide into the Payload of Mipsagargin: 8-O-(12-Aminododecanoyl)-8-O-Debutanoylthapsigargin. Biomolecules 2020, 10, 1640. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10121640
Zimmermann T, Drašar P, Rimpelová S, Christensen SB, Khripach VA, Jurášek M. Large Scale Conversion of Trilobolide into the Payload of Mipsagargin: 8-O-(12-Aminododecanoyl)-8-O-Debutanoylthapsigargin. Biomolecules. 2020; 10(12):1640. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10121640
Chicago/Turabian StyleZimmermann, Tomáš, Pavel Drašar, Silvie Rimpelová, Søren Brøgger Christensen, Vladimir A. Khripach, and Michal Jurášek. 2020. "Large Scale Conversion of Trilobolide into the Payload of Mipsagargin: 8-O-(12-Aminododecanoyl)-8-O-Debutanoylthapsigargin" Biomolecules 10, no. 12: 1640. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10121640