Focus on Osteosclerotic Progression in Primary Myelofibrosis

 , , ,
, , ,  ,
,  , , , and
, , , and 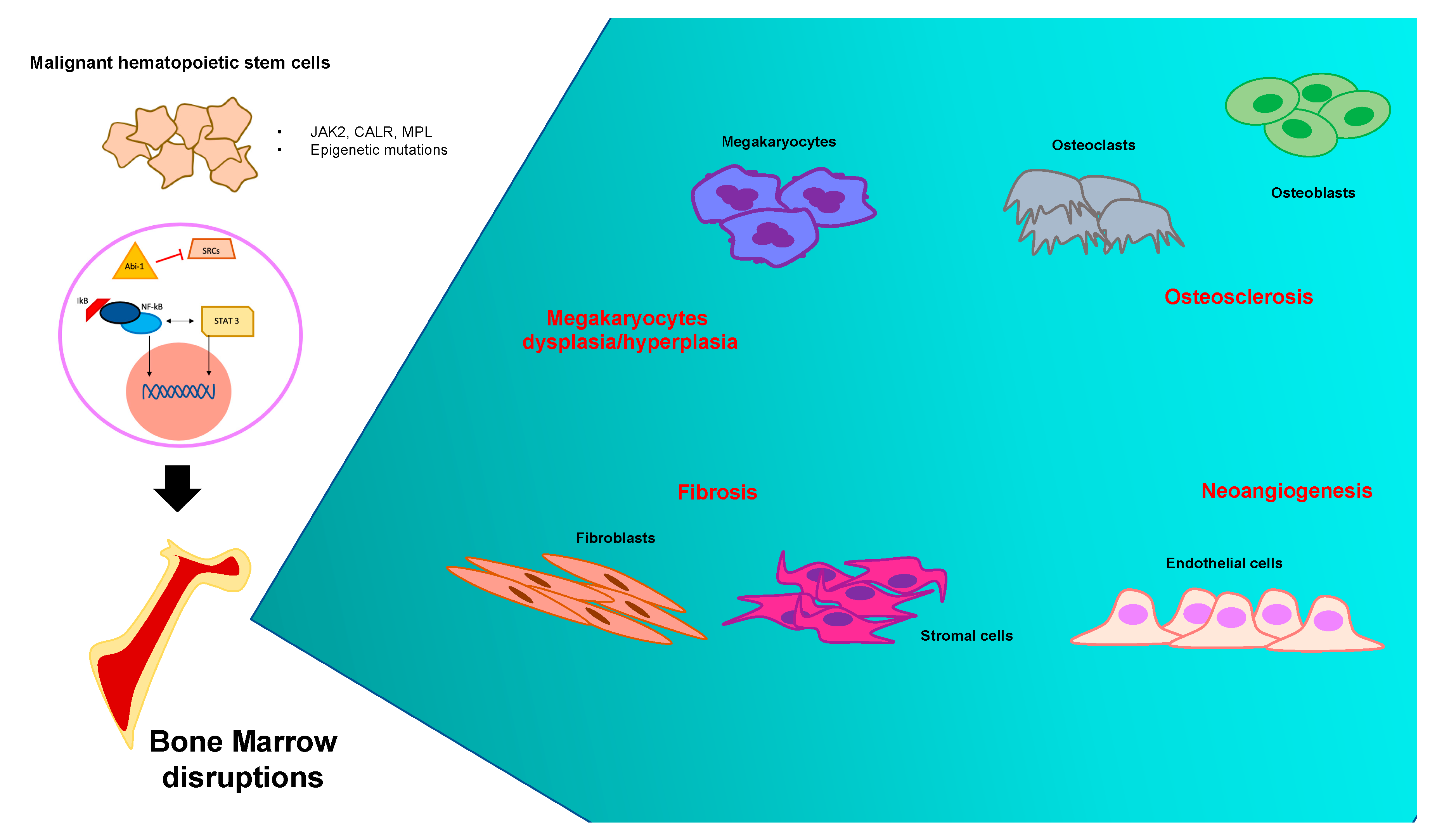{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Clinical Features
Bone Marrow Niche and Microenvironment Disruptions in PMF
3. Fibrosis as PMF Banner
4. Megakaryocytes Role in Bone Marrow Imbalance
5. The Biochemical Network of Osteosclerosis in PMF
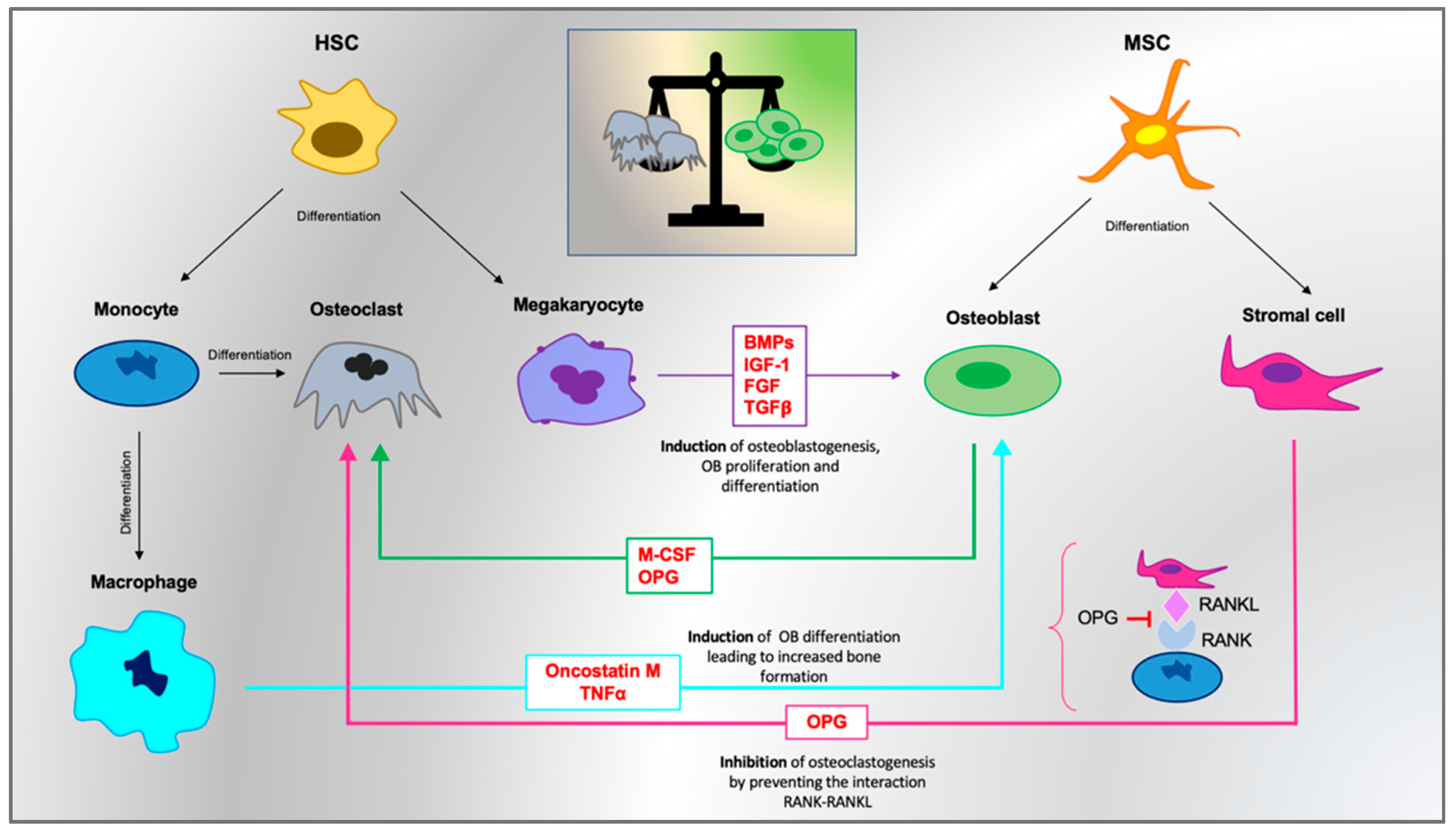
5.1. Bone Marrow as Bone Remodeling “Workshop”
5.2. The OB/OC Ratio
5.3. The Monocytic Line: Role of Osteal Macrophages (OsteoMacs)
5.4. RANKL/OPG Axis and the Wnt/b-Catenin Pathway
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Imai, M.; Araki, M.; Komatsu, N. Somatic mutations of calreticulin in myeloproliferative neoplasms. Int. J. Hematol. 2017, 105, 743–747. [Google Scholar] [CrossRef]
- Di Rosa, M.; Giallongo, C.; Romano, A.; Tibullo, D.; Li Volti, G.; Musumeci, G.; Barbagallo, I.; Imbesi, R.; Castrogiovanni, P.; Palumbo, G.A. Immunoproteasome Genes Are Modulated in CD34 (+) JAK2 (V617F) Mutated Cells from Primary Myelofibrosis Patients. Int. J. Mol. Sci. 2020, 21, 2926. [Google Scholar] [CrossRef] [PubMed]
- Longhitano, L.; Li Volti, G.; Giallongo, C.; Spampinato, M.; Barbagallo, I.; Di Rosa, M.; Romano, A.; Avola, R.; Tibullo, D.; Palumbo, G.A. The Role of Inflammation and Inflammasome in Myeloproliferative Disease. J. Clin. Med. 2020, 9, 2334. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, G.A.; Stella, S.; Pennisi, M.S.; Pirosa, C.; Fermo, E.; Fabris, S.; Cattaneo, D.; Iurlo, A. The Role of New Technologies in Myeloproliferative Neoplasms. Front. Oncol. 2019, 9, 321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pikman, Y.; Lee, B.H.; Mercher, T.; McDowell, E.; Ebert, B.L.; Gozo, M.; Cuker, A.; Wernig, G.; Moore, S.; Galinsky, I.; et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006, 3, e270. [Google Scholar] [CrossRef] [Green Version]
- Vainchenker, W.; Kralovics, R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood 2017, 129, 667–679. [Google Scholar] [CrossRef] [Green Version]
- Gangat, N.; Tefferi, A. Myelofibrosis biology and contemporary management. Br. J. Haematol. 2020, 191, 152–170. [Google Scholar] [CrossRef]
- Wong, W.J.; Baltay, M.; Getz, A.; Fuhrman, K.; Aster, J.C.; Hasserjian, R.P.; Pozdnyakova, O. Gene expression profiling distinguishes prefibrotic from overtly fibrotic myeloproliferative neoplasms and identifies disease subsets with distinct inflammatory signatures. PLoS ONE 2019, 14, e0216810. [Google Scholar] [CrossRef] [Green Version]
- Cokic, V.P.; Mitrovic-Ajtic, O.; Beleslin-Cokic, B.B.; Markovic, D.; Buac, M.; Diklic, M.; Kraguljac-Kurtovic, N.; Damjanovic, S.; Milenkovic, P.; Gotic, M.; et al. Proinflammatory Cytokine IL-6 and JAK-STAT Signaling Pathway in Myeloproliferative Neoplasms. Mediat. Inflamm. 2015, 2015, 453020. [Google Scholar] [CrossRef] [Green Version]
- Woods, B.; Chen, W.; Chiu, S.; Marinaccio, C.; Fu, C.; Gu, L.; Bulic, M.; Yang, Q.; Zouak, A.; Jia, S.; et al. Activation of JAK/STAT Signaling in Megakaryocytes Sustains Myeloproliferation In Vivo. Clin. Cancer Res. 2019, 25, 5901–5912. [Google Scholar] [CrossRef]
- Kleppe, M.; Kwak, M.; Koppikar, P.; Riester, M.; Keller, M.; Bastian, L.; Hricik, T.; Bhagwat, N.; McKenney, A.S.; Papalexi, E.; et al. JAK-STAT pathway activation in malignant and nonmalignant cells contributes to MPN pathogenesis and therapeutic response. Cancer Discov. 2015, 5, 316–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, I.; Huang, Z.; Wen, Q.; Stankiewicz, M.J.; Gilles, L.; Goldenson, B.; Schultz, R.; Diebold, L.; Gurbuxani, S.; Finke, C.M.; et al. AKT is a therapeutic target in myeloproliferative neoplasms. Leukemia 2013, 27, 1882–1890. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.A.C.; Malkova, O.; Engle, E.K.; Miner, C.A.; Fulbright, M.C.; Behbehani, G.K.; Collins, T.B.; Bandyopadhyay, S.; Zhou, A.; Nolan, G.P.; et al. Mass cytometry analysis reveals hyperactive NF Kappa B signaling in myelofibrosis and secondary acute myeloid leukemia. Leukemia 2017, 31, 1962–1974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, B.; Burton, J.H. Myeloproliferative disorders. Emerg. Med. Clin. N. Am. 2014, 32, 597–612. [Google Scholar] [CrossRef]
- Moulard, O.; Mehta, J.; Fryzek, J.; Olivares, R.; Iqbal, U.; Mesa, R.A. Epidemiology of myelofibrosis, essential thrombocythemia, and polycythemia vera in the European Union. Eur. J. Haematol. 2014, 92, 289–297. [Google Scholar] [CrossRef]
- Harrison, C.N.; Schaap, N.; Mesa, R.A. Management of myelofibrosis after ruxolitinib failure. Ann. Hematol. 2020, 99, 1177–1191. [Google Scholar] [CrossRef] [Green Version]
- Polverelli, N.; Palumbo, G.A.; Binotto, G.; Abruzzese, E.; Benevolo, G.; Bergamaschi, M.; Tieghi, A.; Bonifacio, M.; Breccia, M.; Catani, L.; et al. Epidemiology, outcome, and risk factors for infectious complications in myelofibrosis patients receiving ruxolitinib: A multicenter study on 446 patients. Hematol. Oncol. 2018, 36, 561–569. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Jimma, T.; Finke, C.M.; Gangat, N.; Vaidya, R.; Begna, K.H.; Al-Kali, A.; Ketterling, R.P.; Hanson, C.A.; et al. One thousand patients with primary myelofibrosis: The mayo clinic experience. Mayo Clin. Proc. 2012, 87, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A. Primary myelofibrosis: 2019 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2018, 93, 1551–1560. [Google Scholar] [CrossRef] [Green Version]
- Polverelli, N.; Elli, E.M.; Abruzzese, E.; Palumbo, G.A.; Benevolo, G.; Tiribelli, M.; Bonifacio, M.; Tieghi, A.; Caocci, G.; D’Adda, M.; et al. Second primary malignancy in myelofibrosis patients treated with ruxolitinib. Br. J. Haematol. 2020. [Google Scholar] [CrossRef]
- Koschmieder, S.; Mughal, T.I.; Hasselbalch, H.C.; Barosi, G.; Valent, P.; Kiladjian, J.J.; Jeryczynski, G.; Gisslinger, H.; Jutzi, J.S.; Pahl, H.L.; et al. Myeloproliferative neoplasms and inflammation: Whether to target the malignant clone or the inflammatory process or both. Leukemia 2016, 30, 1018–1024. [Google Scholar] [CrossRef] [PubMed]
- Elli, E.M.; Barate, C.; Mendicino, F.; Palandri, F.; Palumbo, G.A. Mechanisms Underlying the Anti-inflammatory and Immunosuppressive Activity of Ruxolitinib. Front. Oncol. 2019, 9, 1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, M.K.; Park, B.B.; Uhm, J.E. Understanding Splenomegaly in Myelofibrosis: Association with Molecular Pathogenesis. Int. J. Mol. Sci. 2018, 19, 898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A. The forgotten myeloproliferative disorder: Myeloid metaplasia. Oncologist 2003, 8, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Cervantes, F. How I treat splenomegaly in myelofibrosis. Blood Cancer J. 2011, 1, e37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palandri, F.; Palumbo, G.A.; Abruzzese, E.; Iurlo, A.; Polverelli, N.; Elli, E.; Bonifacio, M.; Bergamaschi, M.; Martino, B.; Tiribelli, M.; et al. Impact of 2016 WHO diagnosis of early and overt primary myelofibrosis on presentation and outcome of 232 patients treated with ruxolitinib. Hematol. Oncol. 2019, 37, 418–423. [Google Scholar] [CrossRef]
- Palandri, F.; Palumbo, G.A.; Bonifacio, M.; Breccia, M.; Latagliata, R.; Martino, B.; Polverelli, N.; Abruzzese, E.; Tiribelli, M.; Nicolosi, M.; et al. Durability of spleen response affects the outcome of ruxolitinib-treated patients with myelofibrosis: Results from a multicentre study on 284 patients. Leuk. Res. 2018, 74, 86–88. [Google Scholar] [CrossRef]
- Calvi, L.M.; Adams, G.B.; Weibrecht, K.W.; Weber, J.M.; Olson, D.P.; Knight, M.C.; Martin, R.P.; Schipani, E.; Divieti, P.; Bringhurst, F.R.; et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 2003, 425, 841–846. [Google Scholar] [CrossRef]
- Hooper, A.T.; Butler, J.M.; Nolan, D.J.; Kranz, A.; Iida, K.; Kobayashi, M.; Kopp, H.G.; Shido, K.; Petit, I.; Yanger, K.; et al. Engraftment and reconstitution of hematopoiesis is dependent on VEGFR2-mediated regeneration of sinusoidal endothelial cells. Cell Stem Cell 2009, 4, 263–274. [Google Scholar] [CrossRef] [Green Version]
- Yin, T.; Li, L. The stem cell niches in bone. J. Clin. Invest. 2006, 116, 1195–1201. [Google Scholar] [CrossRef]
- O’Sullivan, J.M.; Harrison, C.N. Myelofibrosis: Clinicopathologic features, prognosis, and management. Clin. Adv. Hematol. Oncol. 2018, 16, 121–131. [Google Scholar] [PubMed]
- Nazha, A.; Khoury, J.D.; Rampal, R.K.; Daver, N. Fibrogenesis in Primary Myelofibrosis: Diagnostic, Clinical, and Therapeutic Implications. Oncologist 2015, 20, 1154–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciurea, S.O.; Merchant, D.; Mahmud, N.; Ishii, T.; Zhao, Y.; Hu, W.; Bruno, E.; Barosi, G.; Xu, M.; Hoffman, R. Pivotal contributions of megakaryocytes to the biology of idiopathic myelofibrosis. Blood 2007, 110, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C.; Novetsky, A.; Chen, C.; Novetsky, A.D. Plasma matrix metalloproteinase and tissue inhibitor of metalloproteinase in patients with agnogenic myeloid metaplasia or idiopathic primary myelofibrosis. Br. J. Haematol. 2002, 119, 709–712. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Bianchi, L.; Cellai, C.; Paoletti, F.; Rana, R.A.; Lorenzini, R.; Migliaccio, G.; Migliaccio, A.R. Development of myelofibrosis in mice genetically impaired for GATA-1 expression (GATA-1 (low) mice). Blood 2002, 100, 1123–1132. [Google Scholar] [CrossRef]
- Abbonante, V.; Di Buduo, C.A.; Gruppi, C.; Malara, A.; Gianelli, U.; Celesti, G.; Anselmo, A.; Laghi, L.; Vercellino, M.; Visai, L.; et al. Thrombopoietin/TGF-beta1 Loop Regulates Megakaryocyte Extracellular Matrix Component Synthesis. Stem Cells 2016, 34, 1123–1133. [Google Scholar] [CrossRef]
- Jacobson, R.J.; Salo, A.; Fialkow, P.J. Agnogenic myeloid metaplasia: A clonal proliferation of hematopoietic stem cells with secondary myelofibrosis. Blood 1978, 51, 189–194. [Google Scholar] [CrossRef] [Green Version]
- Reeder, T.L.; Bailey, R.J.; Dewald, G.W.; Tefferi, A. Both B and T lymphocytes may be clonally involved in myelofibrosis with myeloid metaplasia. Blood 2003, 101, 1981–1983. [Google Scholar] [CrossRef]
- Reilly, J.T. Idiopathic myelofibrosis: Pathogenesis, natural history and management. Blood Rev. 1997, 11, 233–242. [Google Scholar] [CrossRef]
- Dong, M.; Blobe, G.C. Role of transforming growth factor-beta in hematologic malignancies. Blood 2006, 107, 4589–4596. [Google Scholar] [CrossRef] [Green Version]
- Martyre, M.C.; Romquin, N.; Le Bousse-Kerdiles, M.C.; Chevillard, S.; Benyahia, B.; Dupriez, B.; Demory, J.L.; Bauters, F. Transforming growth factor-beta and megakaryocytes in the pathogenesis of idiopathic myelofibrosis. Br. J. Haematol. 1994, 88, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Yanagida, M.; Ide, Y.; Imai, A.; Toriyama, M.; Aoki, T.; Harada, K.; Izumi, H.; Uzumaki, H.; Kusaka, M.; Tokiwa, T. The role of transforming growth factor-beta in PEG-rHuMGDF-induced reversible myelofibrosis in rats. Br. J. Haematol. 1997, 99, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Farmer, S.; Vestergaard, H.; Hansen, S.; Shanbhogue, V.V.; Stahlberg, C.I.; Hermann, A.P.; Frederiksen, H. Bone geometry, bone mineral density, and micro-architecture in patients with myelofibrosis: A cross-sectional study using DXA, HR-pQCT, and bone turnover markers. Int J. Hematol. 2015, 102, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Wagner-Ballon, O.; Chagraoui, H.; Prina, E.; Tulliez, M.; Milon, G.; Raslova, H.; Villeval, J.L.; Vainchenker, W.; Giraudier, S. Monocyte/macrophage dysfunctions do not impair the promotion of myelofibrosis by high levels of thrombopoietin. J. Immunol. 2006, 176, 6425–6433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasselbalch, H.C. Chronic inflammation as a promotor of mutagenesis in essential thrombocythemia, polycythemia vera and myelofibrosis. A human inflammation model for cancer development? Leuk. Res. 2013, 37, 214–220. [Google Scholar] [CrossRef]
- Desterke, C.; Martinaud, C.; Ruzehaji, N.; Le Bousse-Kerdiles, M.C. Inflammation as a Keystone of Bone Marrow Stroma Alterations in Primary Myelofibrosis. Mediat. Inflamm. 2015, 2015, 415024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leiva, O.; Ng, S.K.; Chitalia, S.; Balduini, A.; Matsuura, S.; Ravid, K. The role of the extracellular matrix in primary myelofibrosis. Blood Cancer J. 2017, 7, e525. [Google Scholar] [CrossRef]
- Orkin, S.H.; Shivdasani, R.A.; Fujiwara, Y.; McDevitt, M.A. Transcription factor GATA-1 in megakaryocyte development. Stem Cells 1998, 16 (Suppl. 2), 79–83. [Google Scholar] [CrossRef]
- Villeval, J.L.; Cohen-Solal, K.; Tulliez, M.; Giraudier, S.; Guichard, J.; Burstein, S.A.; Cramer, E.M.; Vainchenker, W.; Wendling, F. High thrombopoietin production by hematopoietic cells induces a fatal myeloproliferative syndrome in mice. Blood 1997, 90, 4369–4383. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Lavu, S.; Mudireddy, M.; Lasho, T.L.; Finke, C.M.; Gangat, N.; Pardanani, A.; Hanson, C.A.; Mannarelli, C.; Guglielmelli, P.; et al. JAK2 exon 12 mutated polycythemia vera: Mayo-Careggi MPN Alliance study of 33 consecutive cases and comparison with JAK2V617F mutated disease. Am. J. Hematol. 2018, 93, E93–E96. [Google Scholar] [CrossRef] [Green Version]
- Emadi, S.; Clay, D.; Desterke, C.; Guerton, B.; Maquarre, E.; Charpentier, A.; Jasmin, C.; Le Bousse-Kerdiles, M.C.; French INSERM Research Network on MMM. IL-8 and its CXCR1 and CXCR2 receptors participate in the control of megakaryocytic proliferation, differentiation, and ploidy in myeloid metaplasia with myelofibrosis. Blood 2005, 105, 464–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsushima, K.; Oppenheim, J.J. Interleukin 8 and MCAF: Novel inflammatory cytokines inducible by IL 1 and TNF. Cytokine 1989, 1, 2–13. [Google Scholar] [CrossRef]
- Hashimoto, S.; Yoda, M.; Yamada, M.; Yanai, N.; Kawashima, T.; Motoyoshi, K. Macrophage colony-stimulating factor induces interleukin-8 production in human monocytes. Exp. Hematol. 1996, 24, 123–128. [Google Scholar] [PubMed]
- Takeuchi, K.; Higuchi, T.; Yamashita, T.; Koike, K. Chemokine production by human megakaryocytes derived from CD34-positive cord blood cells. Cytokine 1999, 11, 424–434. [Google Scholar] [CrossRef]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef] [Green Version]
- Burstein, S.A.; Malpass, T.W.; Yee, E.; Kadin, M.; Brigden, M.; Adamson, J.W.; Harker, L.A. Platelet factor-4 excretion in myeloproliferative disease: Implications for the aetiology of myelofibrosis. Br. J. Haematol. 1984, 57, 383–392. [Google Scholar] [CrossRef]
- Gleitz, H.F.E.; Dugourd, A.J.F.; Leimkuhler, N.B.; Snoeren, I.A.M.; Fuchs, S.N.R.; Menzel, S.; Ziegler, S.; Kroger, N.; Triviai, I.; Busche, G.; et al. Increased CXCL4 expression in hematopoietic cells links inflammation and progression of bone marrow fibrosis in MPN. Blood 2020, 136, 2051–2064. [Google Scholar] [CrossRef]
- Bucelli, C.; Cattaneo, D.; Valli, V.B.; Levati, G.V.; Lonati, S.; Gianelli, U.; Iurlo, A. Osteolytic Lesions in Primary Myelofibrosis and Effect of Ruxolitinib Therapy: Report of a Case and Literature Review. Chemotherapy 2018, 63, 340–344. [Google Scholar] [CrossRef]
- Roberts, B.E.; Woods, C.G.; Miles, D.W.; Paterson, C.R. Bone changes in polycythaemia vera and myelosclerosis. J. Clin. Pathol. 1969, 22, 696–700. [Google Scholar] [CrossRef] [Green Version]
- Merry, G.M.; Aronowitz, P.B. Myelofibrosis with massive hepatosplenomegaly and osteolytic bone lesions. J. Hosp. Med. 2010, 5, E27–E28. [Google Scholar] [CrossRef]
- Levesque, J.P.; Helwani, F.M.; Winkler, I.G. The endosteal ‘osteoblastic’ niche and its role in hematopoietic stem cell homing and mobilization. Leukemia 2010, 24, 1979–1992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Fattore, A.; Capannolo, M.; Rucci, N. Bone and bone marrow: The same organ. Arch. Biochem. Biophys. 2010, 503, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabrese, G.; Giuffrida, R.; Forte, S.; Salvatorelli, L.; Fabbi, C.; Figallo, E.; Gulisano, M.; Parenti, R.; Magro, G.; Colarossi, C.; et al. Bone augmentation after ectopic implantation of a cell-free collagen-hydroxyapatite scaffold in the mouse. Sci. Rep. 2016, 6, 36399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabrese, G.; Forte, S.; Gulino, R.; Cefali, F.; Figallo, E.; Salvatorelli, L.; Maniscalchi, E.T.; Angelico, G.; Parenti, R.; Gulisano, M.; et al. Combination of Collagen-Based Scaffold and Bioactive Factors Induces Adipose-Derived Mesenchymal Stem Cells Chondrogenic Differentiation In Vitro. Front. Physiol. 2017, 8, 50. [Google Scholar] [CrossRef] [Green Version]
- Lo Furno, D.; Mannino, G.; Cardile, V.; Parenti, R.; Giuffrida, R. Potential Therapeutic Applications of Adipose-Derived Mesenchymal Stem Cells. Stem Cells Dev. 2016, 25, 1615–1628. [Google Scholar] [CrossRef]
- Morrison, S.J.; Scadden, D.T. The bone marrow niche for haematopoietic stem cells. Nature 2014, 505, 327–334. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, T.; Suda, T. Differentiation and function of osteoclasts. Keio J. Med. 2003, 52, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.C.; Guntur, A.R.; Long, F.; Rosen, C.J. Energy Metabolism of the Osteoblast: Implications for Osteoporosis. Endocr. Rev. 2017, 38, 255–266. [Google Scholar] [CrossRef]
- Dallas, S.L.; Prideaux, M.; Bonewald, L.F. The osteocyte: An endocrine cell … and more. Endocr. Rev. 2013, 34, 658–690. [Google Scholar] [CrossRef] [Green Version]
- Boulais, P.E.; Frenette, P.S. Making sense of hematopoietic stem cell niches. Blood 2015, 125, 2621–2629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, G.B.; Chabner, K.T.; Alley, I.R.; Olson, D.P.; Szczepiorkowski, Z.M.; Poznansky, M.C.; Kos, C.H.; Pollak, M.R.; Brown, E.M.; Scadden, D.T. Stem cell engraftment at the endosteal niche is specified by the calcium-sensing receptor. Nature 2006, 439, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Kiel, M.J.; Yilmaz, O.H.; Iwashita, T.; Yilmaz, O.H.; Terhorst, C.; Morrison, S.J. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 2005, 121, 1109–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacchetti, B.; Funari, A.; Michienzi, S.; Di Cesare, S.; Piersanti, S.; Saggio, I.; Tagliafico, E.; Ferrari, S.; Robey, P.G.; Riminucci, M.; et al. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell 2007, 131, 324–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripodo, C.; Di Bernardo, A.; Ternullo, M.P.; Guarnotta, C.; Porcasi, R.; Ingrao, S.; Gianelli, U.; Boveri, E.; Iannitto, E.; Franco, G.; et al. CD146 (+) bone marrow osteoprogenitors increase in the advanced stages of primary myelofibrosis. Haematologica 2009, 94, 127–130. [Google Scholar] [CrossRef] [Green Version]
- Schepers, K.; Pietras, E.M.; Reynaud, D.; Flach, J.; Binnewies, M.; Garg, T.; Wagers, A.J.; Hsiao, E.C.; Passegue, E. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell 2013, 13, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Centrella, M.; Horowitz, M.C.; Wozney, J.M.; McCarthy, T.L. Transforming growth factor-beta gene family members and bone. Endocr. Rev. 1994, 15, 27–39. [Google Scholar] [CrossRef]
- Veletic, I.; Manshouri, T.; Multani, A.S.; Yin, C.C.; Chen, L.; Verstovsek, S.; Estrov, Z. Myelofibrosis osteoclasts are clonal and functionally impaired. Blood 2019, 133, 2320–2324. [Google Scholar] [CrossRef]
- Thiele, J.; Kvasnicka, H.M. Myelofibrosis in chronic myeloproliferative disorders--dynamics and clinical impact. Histol. Histopathol. 2006, 21, 1367–1378. [Google Scholar] [CrossRef]
- Coindre, J.M.; Reiffers, J.; Goussot, J.F.; De Mascarel, A.; Broustet, A. Histomorphometric analysis of sclerotic bone from idiopathic myeloid metaplasia (nine cases). J. Pathol. 1984, 144, 163–169. [Google Scholar] [CrossRef]
- Krause, D.S.; Scadden, D.T.; Preffer, F.I. The hematopoietic stem cell niche--Home for friend and foe? Cytom. B Clin. Cytom. 2013, 84, 7–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrieux, J.; Roche-Lestienne, C.; Geffroy, S.; Desterke, C.; Grardel, N.; Plantier, I.; Selleslag, D.; Demory, J.L.; Lai, J.L.; Leleu, X.; et al. Bone morphogenetic protein antagonist gene NOG is involved in myeloproliferative disease associated with myelofibrosis. Cancer Genet. Cytogenet. 2007, 178, 11–16. [Google Scholar] [CrossRef]
- Garimella, R.; Kacena, M.A.; Tague, S.E.; Wang, J.; Horowitz, M.C.; Anderson, H.C. Expression of bone morphogenetic proteins and their receptors in the bone marrow megakaryocytes of GATA-1 (low) mice: A possible role in osteosclerosis. J. Histochem. Cytochem. 2007, 55, 745–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pera, M.F.; Andrade, J.; Houssami, S.; Reubinoff, B.; Trounson, A.; Stanley, E.G.; Ward-van Oostwaard, D.; Mummery, C. Regulation of human embryonic stem cell differentiation by BMP-2 and its antagonist noggin. J. Cell Sci. 2004, 117, 1269–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A. Myelofibrosis with myeloid metaplasia. N. Engl. J. Med. 2000, 342, 1255–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramann, R.; Schneider, R.K. The identification of fibrosis-driving myofibroblast precursors reveals new therapeutic avenues in myelofibrosis. Blood 2018, 131, 2111–2119. [Google Scholar] [CrossRef] [Green Version]
- Schneider, R.K.; Mullally, A.; Dugourd, A.; Peisker, F.; Hoogenboezem, R.; Van Strien, P.M.H.; Bindels, E.M.; Heckl, D.; Busche, G.; Fleck, D.; et al. Gli1 (+) Mesenchymal Stromal Cells Are a Key Driver of Bone Marrow Fibrosis and an Important Cellular Therapeutic Target. Cell Stem Cell 2017, 20, 785–800.e788. [Google Scholar] [CrossRef] [Green Version]
- Decker, M.; Martinez-Morentin, L.; Wang, G.; Lee, Y.; Liu, Q.; Leslie, J.; Ding, L. Leptin-receptor-expressing bone marrow stromal cells are myofibroblasts in primary myelofibrosis. Nat. Cell Biol. 2017, 19, 677–688. [Google Scholar] [CrossRef]
- Rameshwar, P.; Denny, T.N.; Stein, D.; Gascon, P. Monocyte adhesion in patients with bone marrow fibrosis is required for the production of fibrogenic cytokines. Potential role for interleukin-1 and TGF-beta. J. Immunol. 1994, 153, 2819–2830. [Google Scholar]
- Rameshwar, P.; Narayanan, R.; Qian, J.; Denny, T.N.; Colon, C.; Gascon, P. NF-kappa B as a central mediator in the induction of TGF-beta in monocytes from patients with idiopathic myelofibrosis: An inflammatory response beyond the realm of homeostasis. J. Immunol. 2000, 165, 2271–2277. [Google Scholar] [CrossRef] [Green Version]
- Chang, V.T.; Yook, C.; Rameshwar, P. Synergism between fibronectin and transforming growth factor-beta1 in the production of substance P in monocytes of patients with myelofibrosis. Leuk. Lymphoma 2013, 54, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Chow, A.; Lucas, D.; Hidalgo, A.; Mendez-Ferrer, S.; Hashimoto, D.; Scheiermann, C.; Battista, M.; Leboeuf, M.; Prophete, C.; van Rooijen, N.; et al. Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J. Exp. Med. 2011, 208, 261–271. [Google Scholar] [CrossRef]
- Winkler, I.G.; Sims, N.A.; Pettit, A.R.; Barbier, V.; Nowlan, B.; Helwani, F.; Poulton, I.J.; van Rooijen, N.; Alexander, K.A.; Raggatt, L.J.; et al. Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood 2010, 116, 4815–4828. [Google Scholar] [CrossRef] [Green Version]
- Christopher, M.J.; Rao, M.; Liu, F.; Woloszynek, J.R.; Link, D.C. Expression of the G-CSF receptor in monocytic cells is sufficient to mediate hematopoietic progenitor mobilization by G-CSF in mice. J. Exp. Med. 2011, 208, 251–260. [Google Scholar] [CrossRef]
- Guihard, P.; Danger, Y.; Brounais, B.; David, E.; Brion, R.; Delecrin, J.; Richards, C.D.; Chevalier, S.; Redini, F.; Heymann, D.; et al. Induction of osteogenesis in mesenchymal stem cells by activated monocytes/macrophages depends on oncostatin M signaling. Stem Cells 2012, 30, 762–772. [Google Scholar] [CrossRef] [Green Version]
- Sinder, B.P.; Pettit, A.R.; McCauley, L.K. Macrophages: Their Emerging Roles in Bone. J. Bone Miner. Res. 2015, 30, 2140–2149. [Google Scholar] [CrossRef] [Green Version]
- Kon, T.; Cho, T.J.; Aizawa, T.; Yamazaki, M.; Nooh, N.; Graves, D.; Gerstenfeld, L.C.; Einhorn, T.A. Expression of osteoprotegerin, receptor activator of NF-kappaB ligand (osteoprotegerin ligand) and related proinflammatory cytokines during fracture healing. J. Bone Miner. Res. 2001, 16, 1004–1014. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.K.; Raggatt, L.J.; Alexander, K.A.; Kuliwaba, J.S.; Fazzalari, N.L.; Schroder, K.; Maylin, E.R.; Ripoll, V.M.; Hume, D.A.; Pettit, A.R. Osteal tissue macrophages are intercalated throughout human and mouse bone lining tissues and regulate osteoblast function In Vitro and In Vivo. J. Immunol. 2008, 181, 1232–1244. [Google Scholar] [CrossRef] [Green Version]
- Nicolaidou, V.; Wong, M.M.; Redpath, A.N.; Ersek, A.; Baban, D.F.; Williams, L.M.; Cope, A.P.; Horwood, N.J. Monocytes induce STAT3 activation in human mesenchymal stem cells to promote osteoblast formation. PLoS ONE 2012, 7, e39871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, K.A.; Chang, M.K.; Maylin, E.R.; Kohler, T.; Muller, R.; Wu, A.C.; Van Rooijen, N.; Sweet, M.J.; Hume, D.A.; Raggatt, L.J.; et al. Osteal macrophages promote in vivo intramembranous bone healing in a mouse tibial injury model. J. Bone Miner. Res. 2011, 26, 1517–1532. [Google Scholar] [CrossRef]
- Wakahashi, K.; Minagawa, K.; Kawano, Y.; Kawano, H.; Suzuki, T.; Ishii, S.; Sada, A.; Asada, N.; Sato, M.; Kato, S.; et al. Vitamin D receptor-mediated skewed differentiation of macrophages initiates myelofibrosis and subsequent osteosclerosis. Blood 2019, 133, 1619–1629. [Google Scholar] [CrossRef]
- Bock, O.; Loch, G.; Schade, U.; Busche, G.; Wasielewski, R.; Wiese, B.; Kreipe, H. Osteosclerosis in advanced chronic idiopathic myelofibrosis is associated with endothelial overexpression of osteoprotegerin. Br. J. Haematol. 2005, 130, 76–82. [Google Scholar] [CrossRef]
- Wang, J.C.; Hemavathy, K.; Charles, W.; Zhang, H.; Dua, P.K.; Novetsky, A.D.; Chang, T.; Wong, C.; Jabara, M. Osteosclerosis in idiopathic myelofibrosis is related to the overproduction of osteoprotegerin (OPG). Exp. Hematol. 2004, 32, 905–910. [Google Scholar] [CrossRef]
- Simonet, W.S.; Lacey, D.L.; Dunstan, C.R.; Kelley, M.; Chang, M.S.; Luthy, R.; Nguyen, H.Q.; Wooden, S.; Bennett, L.; Boone, T.; et al. Osteoprotegerin: A novel secreted protein involved in the regulation of bone density. Cell 1997, 89, 309–319. [Google Scholar] [CrossRef] [Green Version]
- Chagraoui, H.; Tulliez, M.; Smayra, T.; Komura, E.; Giraudier, S.; Yun, T.; Lassau, N.; Vainchenker, W.; Wendling, F. Stimulation of osteoprotegerin production is responsible for osteosclerosis in mice overexpressing TPO. Blood 2003, 101, 2983–2989. [Google Scholar] [CrossRef]
- Peng, Y.; Sheng, X.; Xue, F.; Qian, Y. The genetic association between osteoprotegerin (OPG) gene polymorphisms and bone mineral density (BMD) in postmenopausal women: A meta-analysis. Medicine 2018, 97, e13507. [Google Scholar] [CrossRef]
- De Martinis, M.; Sirufo, M.M.; Suppa, M.; Ginaldi, L. IL-33/IL-31 Axis in Osteoporosis. Int. J. Mol. Sci. 2020, 21, 1239. [Google Scholar] [CrossRef] [Green Version]
- Lacey, D.L.; Timms, E.; Tan, H.L.; Kelley, M.J.; Dunstan, C.R.; Burgess, T.; Elliott, R.; Colombero, A.; Elliott, G.; Scully, S.; et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 1998, 93, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Malyankar, U.M.; Scatena, M.; Suchland, K.L.; Yun, T.J.; Clark, E.A.; Giachelli, C.M. Osteoprotegerin is an alpha vbeta 3-induced, NF-kappa B-dependent survival factor for endothelial cells. J. Biol. Chem. 2000, 275, 20959–20962. [Google Scholar] [CrossRef] [Green Version]
- Mesa, R.A.; Steensma, D.P.; Pardanani, A.; Li, C.Y.; Elliott, M.; Kaufmann, S.H.; Wiseman, G.; Gray, L.A.; Schroeder, G.; Reeder, T.; et al. A phase 2 trial of combination low-dose thalidomide and prednisone for the treatment of myelofibrosis with myeloid metaplasia. Blood 2003, 101, 2534–2541. [Google Scholar] [CrossRef]
- Glass, D.A., 2nd; Bialek, P.; Ahn, J.D.; Starbuck, M.; Patel, M.S.; Clevers, H.; Taketo, M.M.; Long, F.; McMahon, A.P.; Lang, R.A.; et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev. Cell 2005, 8, 751–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, S.; Kopan, R.; Zou, W.; Hilton, M.J.; Ong, C.T.; Long, F.; Ross, F.P.; Teitelbaum, S.L. NOTCH1 regulates osteoclastogenesis directly in osteoclast precursors and indirectly via osteoblast lineage cells. J. Biol. Chem. 2008, 283, 6509–6518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giovos, G.; Yavropoulou, M.P.; Yovos, J.G. The role of cellular senescence in diabetes mellitus and osteoporosis: Molecular pathways and potential interventions. Hormones 2019, 18, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Irelli, A.; Sirufo, M.M.; Scipioni, T.; De Pietro, F.; Pancotti, A.; Ginaldi, L.; De Martinis, M. mTOR Links Tumor Immunity and Bone Metabolism: What are the Clinical Implications? Int. J. Mol. Sci. 2019, 20, 5841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearns, A.E.; Khosla, S.; Kostenuik, P.J. Receptor activator of nuclear factor kappaB ligand and osteoprotegerin regulation of bone remodeling in health and disease. Endocr. Rev. 2008, 29, 155–192. [Google Scholar] [CrossRef]
- Takayanagi, H. Osteoimmunology: Shared mechanisms and crosstalk between the immune and bone systems. Nat. Rev. Immunol. 2007, 7, 292–304. [Google Scholar] [CrossRef]
- Kollet, O.; Dar, A.; Shivtiel, S.; Kalinkovich, A.; Lapid, K.; Sztainberg, Y.; Tesio, M.; Samstein, R.M.; Goichberg, P.; Spiegel, A.; et al. Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nat. Med. 2006, 12, 657–664. [Google Scholar] [CrossRef]
- Boyce, B.F.; Xing, L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch. Biochem. Biophys. 2008, 473, 139–146. [Google Scholar] [CrossRef] [Green Version]
- Wada, T.; Nakashima, T.; Hiroshi, N.; Penninger, J.M. RANKL-RANK signaling in osteoclastogenesis and bone disease. Trends Mol. Med. 2006, 12, 17–25. [Google Scholar] [CrossRef]
- Murakami, T.; Yamamoto, M.; Ono, K.; Nishikawa, M.; Nagata, N.; Motoyoshi, K.; Akatsu, T. Transforming growth factor-beta1 increases mRNA levels of osteoclastogenesis inhibitory factor in osteoblastic/stromal cells and inhibits the survival of murine osteoclast-like cells. Biochem. Biophys. Res. Commun. 1998, 252, 747–752. [Google Scholar] [CrossRef]
- Thirunavukkarasu, K.; Miles, R.R.; Halladay, D.L.; Yang, X.; Galvin, R.J.; Chandrasekhar, S.; Martin, T.J.; Onyia, J.E. Stimulation of osteoprotegerin (OPG) gene expression by transforming growth factor-beta (TGF-beta). Mapping of the OPG promoter region that mediates TGF-beta effects. J. Biol. Chem. 2001, 276, 36241–36250. [Google Scholar] [CrossRef] [Green Version]
- Grigorie, D.; Lerner, U.H. The Crucial Role of the Wnt System in Bone Remodelling. Acta Endocrinol. 2018, 14, 90–101. [Google Scholar] [CrossRef]
- Tibullo, D.; Longo, A.; Vicario, N.; Romano, A.; Barbato, A.; Di Rosa, M.; Barbagallo, I.; Anfuso, C.D.; Lupo, G.; Gulino, R.; et al. Ixazomib Improves Bone Remodeling and Counteracts sonic Hedgehog signaling Inhibition Mediated by Myeloma Cells. Cancers 2020, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Lucijanic, M.; Livun, A.; Tomasovic-Loncaric, C.; Stoos-Veic, T.; Pejsa, V.; Jaksic, O.; Prka, Z.; Kusec, R. Canonical Wnt/beta-Catenin Signaling Pathway Is Dysregulated in Patients with Primary and Secondary Myelofibrosis. Clin. Lymphoma Myeloma Leuk. 2016, 16, 523–526. [Google Scholar] [CrossRef] [Green Version]
- Vicario, N.; Bernstock, J.D.; Spitale, F.M.; Giallongo, C.; Giunta, M.A.S.; Li Volti, G.; Gulisano, M.; Leanza, G.; Tibullo, D.; Parenti, R.; et al. Clobetasol Modulates Adult Neural Stem Cell Growth via Canonical Hedgehog Pathway Activation. Int. J. Mol. Sci. 2019, 20, 1991. [Google Scholar] [CrossRef] [Green Version]
- Gerds, A.T.; Tauchi, T.; Ritchie, E.; Deininger, M.; Jamieson, C.; Mesa, R.; Heaney, M.; Komatsu, N.; Minami, H.; Su, Y.; et al. Phase 1/2 trial of glasdegib in patients with primary or secondary myelofibrosis previously treated with ruxolitinib. Leuk. Res. 2019, 79, 38–44. [Google Scholar] [CrossRef]
- Yachoui, R.; Kristianto, J.; Sitwala, K.; Blank, R.D. Role of Endothelin-1 in a Syndrome of Myelofibrosis and Osteosclerosis. J. Clin. Endocrinol. Metab. 2015, 100, 3971–3974. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spampinato, M.; Giallongo, C.; Romano, A.; Longhitano, L.; La Spina, E.; Avola, R.; Scandura, G.; Dulcamare, I.; Bramanti, V.; Di Rosa, M.; et al. Focus on Osteosclerotic Progression in Primary Myelofibrosis. Biomolecules 2021, 11, 122. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11010122
Spampinato M, Giallongo C, Romano A, Longhitano L, La Spina E, Avola R, Scandura G, Dulcamare I, Bramanti V, Di Rosa M, et al. Focus on Osteosclerotic Progression in Primary Myelofibrosis. Biomolecules. 2021; 11(1):122. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11010122
Chicago/Turabian StyleSpampinato, Mariarita, Cesarina Giallongo, Alessandra Romano, Lucia Longhitano, Enrico La Spina, Roberto Avola, Grazia Scandura, Ilaria Dulcamare, Vincenzo Bramanti, Michelino Di Rosa, and et al. 2021. "Focus on Osteosclerotic Progression in Primary Myelofibrosis" Biomolecules 11, no. 1: 122. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11010122