
Screening and Molecular Modeling Evaluation of Food Peptides to Inhibit Key Targets of COVID-19 Virus


1
Institute of Food Science and Technology, Chinese Academy of Agricultural Sciences/Key Laboratory of Agricultural Product Processing and Quality Control, Ministry of Agricultural and Rural Affairs, Beijing 100193, China
2
Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA 02115, USA
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Biomolecules 2021, 11(2), 330; https://0-doi-org.brum.beds.ac.uk/10.3390/biom11020330
Submission received: 20 January 2021
/
Revised: 18 February 2021
/
Accepted: 19 February 2021
/
Published: 22 February 2021
(This article belongs to the Section Molecular Medicine)
Abstract
:Peptide drugs, especially food-derived peptides, have a variety of functional activities including antiviral and may also have a therapeutic effect on COVID-19. In this study, comparing with the reported drugs, 79 peptides were found to bind to the key targets of COVID-19 virus with higher non-covalent interaction, while among them, six peptides showed high non-covalent interactions with the three targets, which may inhibit the COVID-19 virus. In the simulation, peptides of nine to 10 amino acids with a hydrophilic amino acid and acidic amino acid in the middle and aromatic amino acids on the side showed higher binding to angiotensin-converting enzyme 2 (ACE2). Peptides of five to six amino acids with a basic amnio acid in the head, acidic amnio acid in the neck, hydrophobicity group in the middle, and basic amino acids in the tail showed higher binding to COVID-19 virus main protease (Mpro), while those with basic amino acids and acidic amino acids in the two sides and aromatic amino acids in the middle might have stronger interaction with COVID-19 virus RNA-dependent RNA polymerase (RdRp).
1. Introduction
Since December 2019, coronavirus disease 2019 (COVID-19) has been raging for a whole year; over 100 million people were infected, and over 2.2 million people died [1]. Although related to the severe acute respiratory syndrome (SARS) and the Middle East respiratory syndrome (MERS), COVID-19 shows some peculiar pathogenetic, epidemiological, and clinical features. At the molecular level, these three viruses were genus β-Coronavirus with nucleic acid similarity of 79.5% (SARS) and 54.9% (MERS), but the gene length was different (COVID-19, 29.9 kb; SARS, 29.7 kb, MERS, 30.1 kb). Moreover, the functional receptors for COVID-19 and SARS were angiotensin-converting enzyme 2 (ACE2), while it was (dipeptidyl peptidase 4) DPP4 for MERS [2]. Although we have the experience to deal with SARS and MERS, there are still many unknowns about COVID-19. This also resulted in the extremely serious epidemic situation, and the prevention and control pressure are still huge. Specific drugs and antibodies for the prevention and control of COVID-19 are still being studied, and there is still uncertainty about the same type of drugs that have been reported to have certain effects [3,4]. Remdesivir, which was developed to target SARS and MERS, has been reported with effects in inhibiting COVID-19 virus, but the effectiveness and safety are still being questioned [5]. Similarly, the antimalarial drugs, chloroquine, and hydroxychloroquine, have also been applied to fight the COVID-19 virus, but there are still concerns about their strong side effects [6]. Safer and more effective drugs to control COVID-19 are urgently needed.
Polypeptide drugs are widely used in the treatment of various human diseases such as pain, arthritis, cancer, hepatitis, and acquired immune deficiency syndrome (AIDS) due to their wide indications, high safety, and significant efficacy [7,8,9,10,11]. As most disease targets are proteins, polypeptide drugs have incomparable advantages and potentials in the binding ability and structural diversity comparing with small molecule drugs [12]. Moreover, their non-covalent binding to the targets has also been shown with the priority to the covalently bound drug in terms of controlling side effects. Nowadays, few covalent drugs are currently designed to treat anything other than cancer [13,14], and this may also explain the side effects of drugs such as Remdesivir and other covalently bound drugs used to treat COVID-19. Therefore, the screening of peptide drugs represents an important direction in the search for specific drugs to inhibit COVID-19.
In nature, peptides are mostly generated from the proteolysis, and food-derived peptides with 2 to 10 amino acids have been proven to have higher absorption efficiency and functional effects, including an antiviral effect [10,11]. In the face of the abundant varieties of peptide sources and structures, computer-aided screening is the first step in drug screening, one of which is the molecular modeling [15,16]. Although the molecular modeling cannot provide a definitive conclusion of the activity, the results may provide important information on initiating drug developments [17].
Based on the in-depth study on the infection mechanism of COVID-19 virus, three major action sites may be key targets for treatment, including COVID-19 virus spike protein receptor binding domain angiotensin-converting enzyme 2 (ACE2) [18,19], COVID-19 virus main protease (COVID-19 virus Mpro) [20], and COVID-19 virus RNA-dependent RNA polymerase (COVID-19 virus RdRp) [21]. The research findings from our group and other investigators indicate that non-specific binding peptides may have similar inhibitory activity to ACE and ACE2 as they show highly homology [22]. Meanwhile, ACE is a type of metal ion-dependent enzyme with “active pocket” structure [23], and its inhibiting mechanism is quite similar to that of COVID-19 Mpro and RdRp; all contain an “active pocket or other molecule like metal ion, nucleotide” [20,21].
In this study, we first widely collected the food-derived peptides and then systematically studied the interaction between peptides and three key targets, ACE2, COVID-19 Mpro, and RdRp. Through calculation and comparison, we evaluated the binding affinity of collected food-derived peptides with those key targets of COVID-19 virus and identify the most potential peptides that may inhibit those three targets and their interaction models, aiming to provide necessary information for the development of more effective drugs against COVID-19.
2. Material and Methods
2.1. Receptor and Ligands Preparation
The structure of human angiotensin-converting enzyme 2 (ACE2, the COVID-19 coronavirus spike protein receptor binding domain), COVID-19 coronavirus main protease (COVID-19 virus Mpro), and RNA-dependent RNA polymerase (COVID-19 virus RdRp) were downloaded from the Protein Data Bank (PDB) website with necessary treatment. The structure of ACE2 was derived from “Crystal structure of SARS-CoV-2 spike receptor-binding domain bound with ACE2, PDB No. 6M0J” with deleting the spike protein [19]. The structure of COVID-19 virus Mpro was derived from “The crystal structure of COVID-19 main protease in complex with an inhibitor N3, PDB No. 6LU7” with deleting the inhibitor N3 (cysteine residues were replenished) [20]. The docking site for ACE2 was defined from the PDB Site Records and the interaction between ACE2 and COVID-19 virus spike protein. The structure of COVID-19 virus RdRp was derived from “Template RNA and Remdesivir bound RdRp complex, PDB No. 7BV2” with deleting the inhibitor Remdesivir (Uracil residues were replenished) [21].
The peptides were collected from the latest reviews on ACE-inhibitory peptides derived from the natural food resources including plant, animal, marine, and mushroom [23,24,25,26,27,28,29]. All the peptides were drawn using Chem3D software (Version 15.0, Cambridge Software Inc., Cambridge, MA, USA, 2018), and then, one step of optimization was employed to get the effective structure.
2.2. Molecular Docking Simulation
The inhibition mechanism of peptides against human angiotensin-converting enzyme 2 (ACE2, the COVID-19 coronavirus spike protein binding receptor), COVID-19 coronavirus main protease (COVID-19 virus Mpro), and RNA-dependent RNA polymerase (RdRp) were conducted through the molecular docking and dynamic test using Discovery studio client software (v16.1.0.15350) (BIOVIA, San Diego, CA, USA) based on our previous methods [15,23,24]. MLN-4760, inhibitor N3, and Remdesivir were set as positive controls [18,20,21]. The main procedures were as follows.
(a) Defining the active site: After loading the structure of receptor into Discovery studio client software (DS 4.0), all water molecules and ligands were removed, and the conforming, hierarchical, adaptive refinement methods (CHARMs) force field was applied using the PREPARE PROTEIN tool in DS 4.0. Then, we chose to define the active sites based on the active cavities of the receptor and PDB site records.
(b) Molecular docking: CDOCKER tools in DS 4.0 were used for the molecular docking. We first loaded the structure of peptides and three targets into DS 4.0 and then chose the active site described in Section 2.1. The CHARMs force field was a default during the whole docking process.
(c) Conformation Scoring. After the docking process, the top 10 conformation poses were generated for each ligand based on docking score. The conformation with the highest CDocker energy (Score, Total Interaction Energy) was obtained (“-” represents inter-attraction). Then, the interaction energy of the ligand–receptor interaction was calculated, and interaction sites were illustrated in 2D and 3D images. Then, the main interaction forces and key amino acids could be identified.
(d) Analysis of peptides overlapping: Based on the calculation of interaction energy (absolute value), we could obtain a series of peptides with high potential inhibitory activities against the receptor. Through the overlapping of these peptides and analysis of surface properties, we could conclude the basic principle for drug searching.
2.3. Molecular Dynamic Simulation
To analyze the structure changes of the receptor including ACE2, COVID-19 virus Mpro, and COVID-19 RdRp caused by molecule docking, the molecular dynamics simulation of the ligand–receptor complex was performed using the SIMULATION tool in the DS 4.0 with CHARMs force field. Specifically, the ligand–receptor complex was first pretreated in the absence of a water molecule, and later, a 7 Å solvation shell was added. At same time, 0.145 M NaCl was used to simulate the human environment. Two minimization cycles (steepest descent and conjugate gradient) were performed until the root mean square (RMS) of energy gradient was <0.1 kcal/mol·Å [15,30]. The steepest descent cycle was performed with 2000 steps (time step: 0.001 ps), while a conjugate gradient was performed with 1000 steps (time step: 0.001 ps). The SHAKE algorithm was applied throughout the molecular dynamics (MD) simulation to hold all the bonds involving hydrogen atoms. The long-range electrostatic forces were treated with the PME method. After minimization, the sample was gradually heated to a target temperature from 50 to 300 K over an interval of 5 ps. After this heating process, a 5000 steps-long (time step: 0.002 ps) equilibration phase was applied. The production stage was performed in 50,000 steps using a time step of 0.002 ps using NPT canonical assembly. The decay time for the temperature coupling was 5.0 ps. The whole complex structure changes were illustrated by highlighting the ligand and docking pocket comparing with the complex structure before molecular dynamic simulation. After the molecular dynamic simulation, 50 conformation poses were produced, and we could obtain the pose with the lowest total energy. The structure changes before MD and after MD could be analyzed, while the interaction between the ligand and receptor could also be illustrated in 2D and 3D images.
3. Results
3.1. Candidate Peptides That Target ACE2
From the latest research studies, the COVID-19 virus could bind to ACE2 on the surface of host cells. By binding to ACE2, the virus starts the first step in cell invasion. Inhibition of virus binding to ACE2 may be an important strategy for preventing viral infections. Based on the crystalline structure of the COVID-19 virus spike protein receptor binding domain ACE2 complex, the main binding sites could be illustrated (Supplementary Materials, Figure S1A–C). By docking the peptides to the COVID-19 virus spike protein RBD of ACE2, we found 187 peptides that could bind to ACE2 with the -CDocker energy (Score, Total Interaction Energy) varying from 14.592 to 225.598 kcal/mol (Supplementary Materials, Table S1). For the reported inhibitor of ACE2, including MLN-4760 and DX600 (Supplementary Materials, Figure S1D,E), we also found that the MLN-4760 could bind to ACE2 at the COVID-19 virus spike receptor binding domain with the -CDocker energy (Score) of 21.028 kcal/mol (Supplementary Materials, Table S2; Figure S2A–E). The DX600 could not bind to ACE2 at this specific domain.
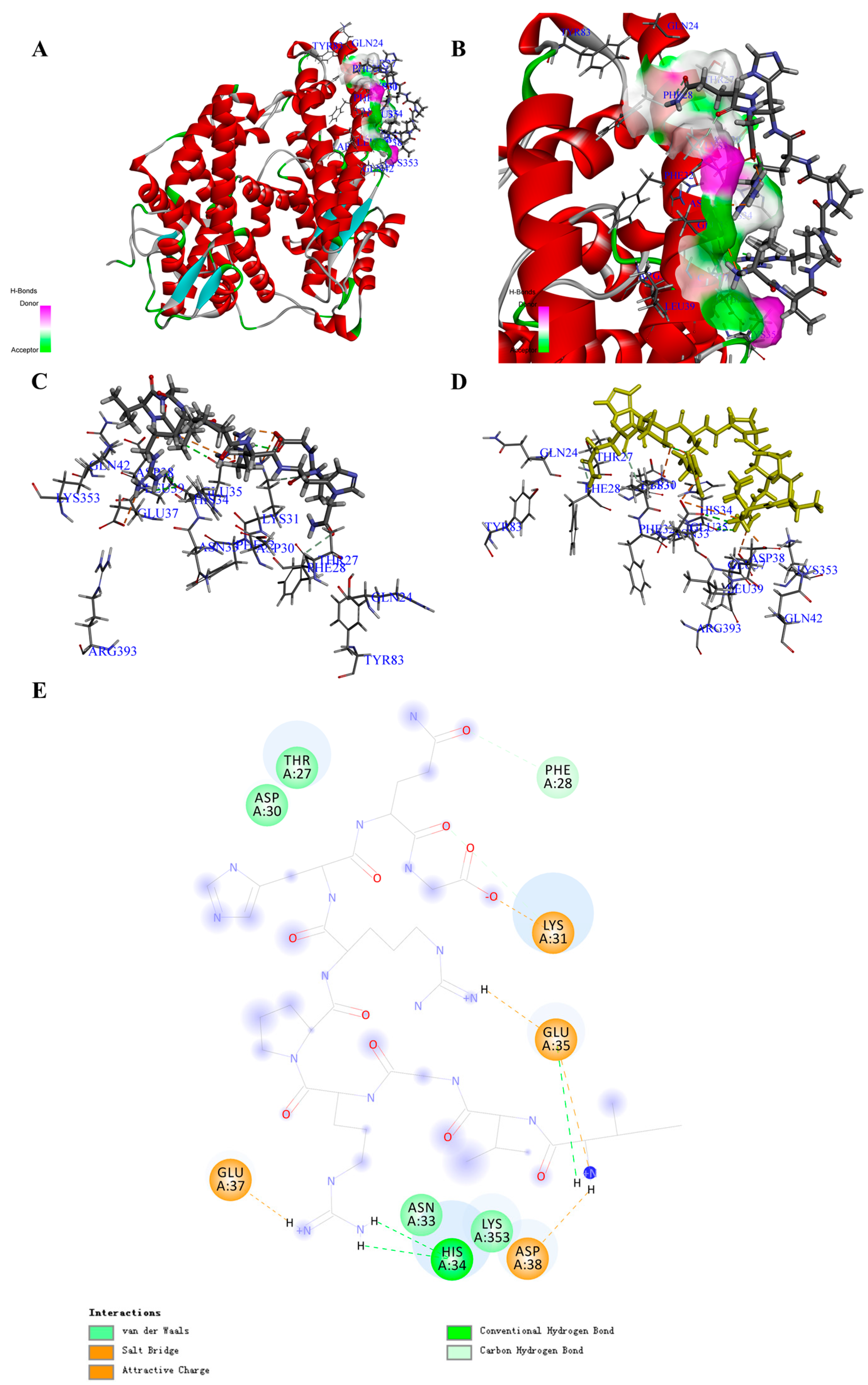
Based on the screening by -CDocker energy (Score), we chose 30 peptides with the highest scores for the interaction analysis (Supplementary Materials, Table S2). Finally, we obtained the interaction counts, residues, and energy between peptides and ACE2, and the peptide having the highest total interaction energy was PA1 with an amino acids sequence of Ile-Val-Gly-Arg-Pro-Arg-His-Gln-Gly. The total interaction energy (absolute value) between PA1 and ACE2 was 330.556 kcal/mol, which was made up of 10 non-covalent bonds (Table 1; Supplementary Materials, Table S8). Specifically, five electrostatic (including five attractive charges and three salt bridges) and five H-bonds were formed between peptide and amino acid residues including GLU35, ASP38, GLU37, ASP30, HIS34, THR27, PHE32, GLN24, and ASN33 (Figure 1A–E; Supplementary Materials, Table S8). Comparing with the reported drugs, the total interaction energy (absolute value) for MLN-4760 was 86.407 kcal/mol, and the main interaction amino acid residues were GLU37, LYS31, ASP38, LYS353, THR27, GLU35, ASP30, and ASN33 (Table 2). The main forces for MLN-4760 were H-bond and hydrophobic (Supplementary Materials, Table S5). Through the calculation of interaction between the COVID-19 spike protein and ACE2, the total interaction energy (absolute value) was 158.342 kcal/mol, which was contributed by GLN24, ASP30, LYS31, HIS34, GLU35, ASP38, TYR41, GLN42, MET82, TYR83, LYS353, and GLY354 (Table 2 and Supplementary Materials, Figure S5). The main interaction forces were H-bond and hydrophobic interaction (Supplementary Materials, Table S5) [19]. The identified peptides that could bind to the receptor-binding domain of ACE2 with higher interaction energy are listed in Table 1 and Table 2.
The docking poses in Figure S5 clearly show how the MLN-4760 and virus spike protein bind to ACE2. The bonding region was the surface of two helical chains of ACE2, the middle region was highly hydrophilic, and both sides were highly hydrophobic. For the COVID-19 virus spike protein, it was like “handle” bound to ACE2, where lots of H-bond appeared in the middle region and hydrophobic interaction appeared on both sides.
Through overlapping the peptides of higher interaction energy bound to ACE2, we obtained the “portrayal” of the peptides that might be potential for the development of the natural inhibitory drug (Figure S3A–D). The peptide commonly had 9–10 amino acids (Ave. MW ≈ 1.14 KDa) (Table 3), with hydrophilic amino acid and acidic amino acid in the middle and aromatic amino acids on the side (Supplementary Materials, Figure S3E,F). The overlapping results could also show that the α-helix at SER19 to TYR50 was the main binding site, which was half covered by the peptides such as “tweezer” (Supplementary Materials, Figure S3G,H). This kind of site occupation and spatial obstruction might give the possibility for the peptides inhibiting the interaction of COVID-19 spike protein and ACE2 [31].
Moreover, after molecular dynamic simulation, we could obtain the most stable conformation with the lowest total energy. For instance, the PA1 could also show a much stronger interaction to ACE2. Comparing with the complex before molecular dynamic simulation, the total interaction energy (absolute value) between PA1 and amino acid residues increased from 330.556 to 440.463 kcal/mol (Supplementary Materials, Figure S4A–D), and the interaction bond counts increased from 10 to 15 with more H-bonds formed (Supplementary Materials, Tables S5 and S11; Figure S4E). This result suggests that PA1 may be a potential candidate bound to ACE2 for inhibiting the interaction of the COVID-19 virus, although further experimental verification is required.
Docking results between 187 peptides and ACE2 could be found in Supplementary Materials, Table S1. The interaction analysis results of the 30 peptides with the highest -CDocker energy (Score) could be found in Supplementary Materials, Table S2.
3.2. Candidate Peptides That Target COVID-19 Virus Mpro
The COVID-19 virus Mpro is another target that can directly stop the replication and construction of the enzyme and protein for the new virus. By docking the peptides to COVID-19 virus Mpro (Supplementary Materials, Figure S6A–D), a total of 167 peptides could successfully bind to the COVID-19 virus Mpro, and the -CDocker energy (Score) varied from 21.009 kcal/mol to 131.774 kcal/mol (Supplementary Materials, Table S1).
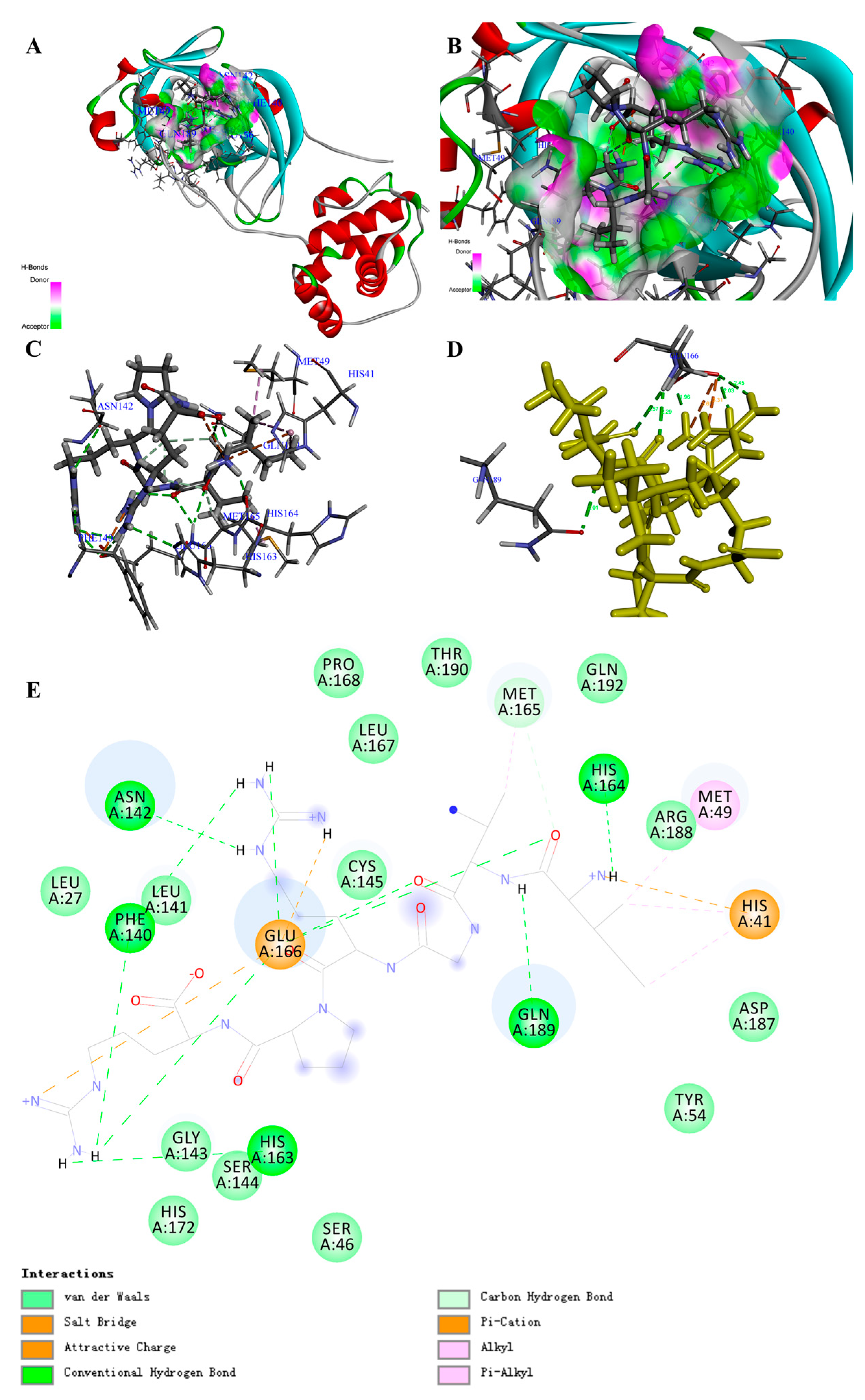
Based on the screening by -CDocker energy (Score), we chose 30 peptides with the highest score for the interaction analysis and obtained the interaction counts, residues, and energy between peptides and COVID-19 virus Mpro (Supplementary Materials, Table S3). For instance, the highest total interaction energy (absolute value) between peptide (Ile-Val-Gly-Arg-Pro-Arg) and COVID-19 virus Mpro was -366.214 kcal/mol, which was made up of 24 bonds (Table 1; Supplementary Materials, Table S9). Specifically, 2 attractive charges, 12 H-bonds, 1 Pi-Cation, 2 Alkyl, and 2 Pi-Alkyls were formed between the peptide and amino acid residues including HIS41, MET49, ASN142, GLY143, CYS145(SG), HIS164, MET165, GLU166, GLN189, and THR190 (Figure 2A–E; Supplementary Materials, Table S9). Comparing with the reported drugs, the total interaction energy (absolute value) for Ritonavir was 87.46983 kcal/mol, while it was -102 kcal/mol for Lopinavir (Table 2; Figure S6E,F). This result is in accordance with the clinical report of Lopinavir [32]. With regard to the interaction residues, GLY143, CYS145, MET165, GLU166, and GLN189 were the common targets to inhibit COVID-19 virus Mpro, and the main interaction forces for Ritonavir and Lopinavir were H-bond and Pi bond (Supplementary Materials, Table S6). Through the in-depth review of the reported inhibitor N3 (Supplementary Materials, Figure S6D), we could also find the similar interaction sites. The main difference was that the interaction between inhibitor N3 and COVID-19 virus Mpro contained a covalent bond (C-S), which was the main interaction force (Supplementary Materials, Figure S7A–E; Table S6) [20], since the interaction energy (absolute value) of no-covalent bonds for inhibitor N3 was just 37.435 kcal/mol (Table 2).
The docking poses in Figure S10 clearly illustrated that the active site of COVID-19 virus Mpro was similar to a deep “pocket”, and two hydrophobicity residues were guarded similar to a “door switch” (Supplementary Materials, Figure S8A–D). All the drugs and peptides (top 3 of total interaction energy (absolute value)) were inserted into the pocket site of the COVID-19 virus Mpro.
Through overlapping the peptides of higher interaction energy, we obtained the “portrayal” of the peptides that might have potential for the development of natural inhibitory drugs to target COVID-19 virus Mpro. The peptide commonly had five to six amino acids (Ave. MW ≈ 0.67 KDa) (Table 3), with a basic amino acid of the “head” section, an acidic amino acid of the “neck” section, a hydrophobicity group of the mid-section, and basic amino acids of the “tail” section (Supplementary Materials, Figure S8E–H).
Moreover, after molecular dynamic simulation, we could obtain the most stable conformation with the lowest total energy. For instance, the PM1 could also show stronger interaction to COVID-19 virus Mpro (Supplementary Materials, Figure S9A–D). Comparing with the complex before molecular dynamic simulation, the interaction bond counts between PM1 and amino acid residues increased from 19 to 29, although the total interaction energy (absolute value) varied from −366.21436 to −341.462362 kcal/mol (Supplementary Materials, Tables S6 and S11; Figure S9E). This result suggests that PM1 may be a potential candidate for inhibiting COVID-19 virus Mpro. Certainly, further experimental verification and clinical trials are needed in the future.
Docking results between 187 peptides and COVID-19 virus Mpro are shown in Supplementary Materials, Table S1. The interaction analysis results of the 30 peptides with the highest -CDocker energy (Score) can be found in Supplementary Materials, Table S3.
3.3. Candidate Peptides That Target COVID-19 Virus RdRp
During the replication and transcription of viral genomes, the RNA-dependent RNA polymerase (RdRp) plays an important role that might be another target to prevent the transmission and infection of COVID-19 virus. By docking the peptides to COVID-19 virus RdRp (Supplementary Materials, Figure S11A–D), in total, we found 173 peptides that could bind to RdRp, and the -CDocker energy (Score) varied from −320.607 to 186.305 kcal/mol (Supplementary Materials, Table S1).
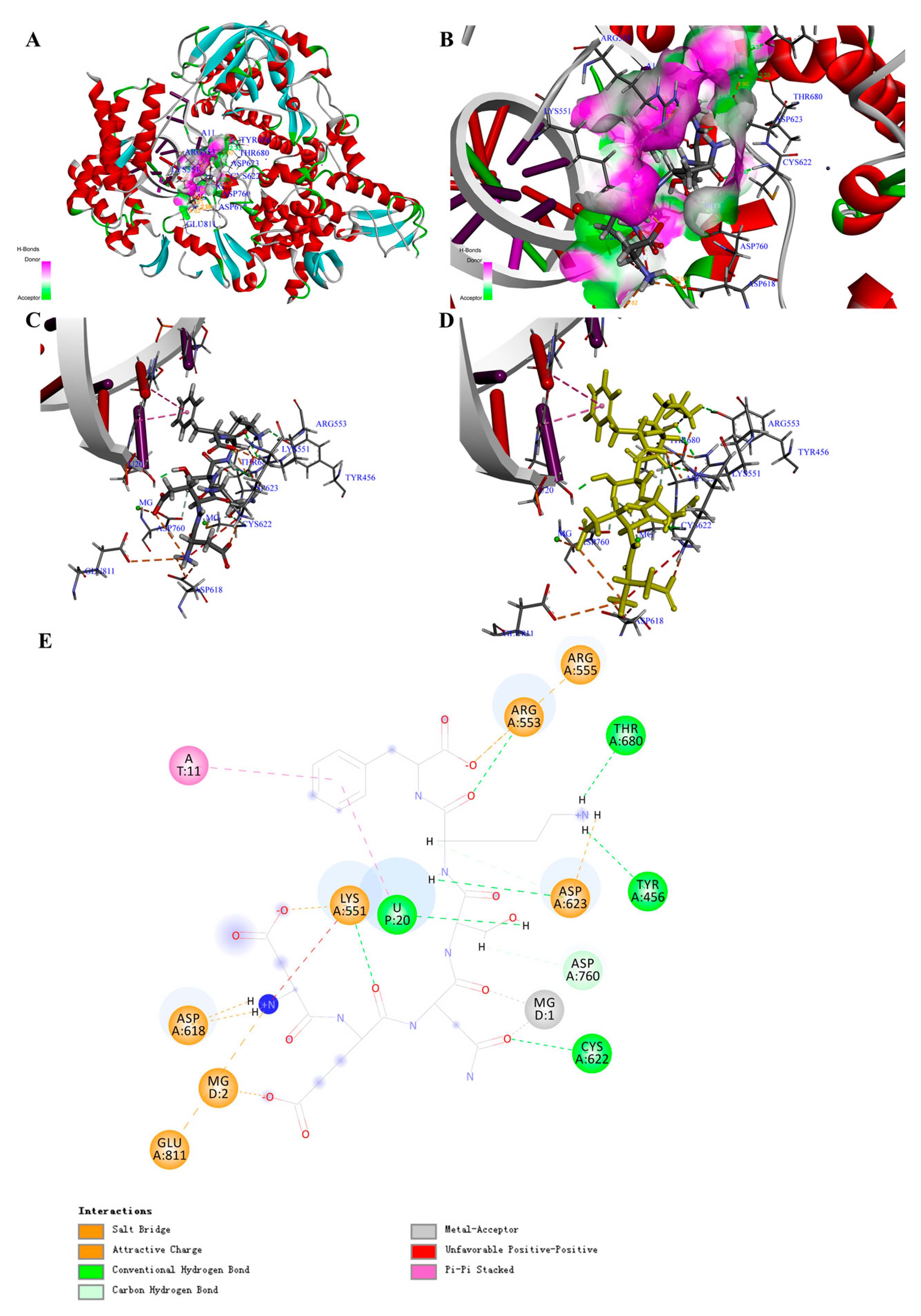
By comparing the -CDocker energy (Score), we obtained 30 peptides with the highest scores, which might show higher binding energy and inhibitory activity. The interaction counts, residues, and energy between the peptides and COVID-19 virus RdRp were listed in Table S4. Peptide Asp-Glu-Asn-Ser-Lys-Phe (PR1) showed the highest total interaction energy (absolute value): 870.287 kcal/mol (Table 1), and the main interaction residues for COVID-19 virus RdRp were MG1, MG2, A11, U10, U20, ASP452, LYS545, LYS551, ARG553, ARG555, LYS621, ASP623, and LYS798 (Figure 3A–E). In total, 23 non-covalent bonds were formed between PR1 and COVID-19 virus RdRp. Specifically, eight electrostatic interactions, nine H-bonds, two metal acceptors, and four Pi-Pi stacked were the main interaction forces (Supplementary Materials, Table S10). The non-covalent interaction energy (absolute value) between the drug Remdesivir (Supplementary Materials, Figure S11D) and COVID-19 virus RdRp was 223.131 kcal/mol (Table 2; Supplementary Materials, Figure S11E,F), which was contributed by two electrostatic interactions, three H-bonds, and four hydrophobic interactions (Supplementary Materials, Figure S12A–E; Table S7). The O-P covalent bond between Remdesivir and RNA was thought to be a key mechanism for inhibiting RNA replication (Supplementary Materials, Figure S12D,E; Table S7) [21], and the main interaction amino acid residues for Remdesivir were MG1, MG2, U20, A11, LYS545, ARG555, CYS622, SER682, THR687, ASN691, and SER759, which were quite similar to PR1 (Table 2).
The results of docking poses in Figure S15 could clearly illustrate that the active site for COVID-19 virus RdRp was combined with RNA and the key domain was formed similar to a “pore canal” (Supplementary Materials, Figure S13A–D). All the drugs and peptides (top three of the total interaction energy (absolute value)) were inserted into the pocket site of the COVID-19 virus RdRp. Furthermore, the region away from RNA showed higher hydrophily, which was used to deliver the substrate for RNA polymerization.
Through overlapping the peptides of higher interaction energy, we obtained the “portrayal” of the peptides, which might indicate potential for the development of natural inhibitory drugs that target the COVID-19 virus RdRp. The peptide commonly had five to six amino acids (Ave. MW ≈ 0.67 KDa) (Table 3), with a basic amino acid and an acidic amino acid in the two sides and aromatic amino acids in the middle (Figure S13E–H).
Moreover, after molecular dynamic simulation, we could obtain the most stable conformation with the lowest total energy. For instance, the PR1 could show stronger interaction to COVID-19 virus RdRp (Supplementary Materials, Figure S14A–D). Comparing with the complex before molecular dynamic simulation, the interaction bond counts between PR1 and amino acid residues increased from 23 to 30, while the total interaction energy (absolute value) increased from 870.287 to 971.613 kcal/mol (Supplementary Materials, Tables S7 and S11; Figure S14E). This result suggests that PR1 may serve as a potential candidate for inhibiting COVID-19 virus RdRp.
Docking results between 187 peptides and COVID-19 virus RdRp could be found in Supplementary Materials, Table S1. The interaction analysis results of the 30 peptides with highest -CDocker energy (Score) could be found in Supplementary Materials, Table S4.
4. Discussion
In this study, the results from molecular simulation demonstrate that certain naturally food-derived peptides may have inhibitory activities to the potential targets of COVID-19 virus with PA1 as a potential inhibitor for ACE2, PM1 as a potential inhibitor for Mpro, and PR1 as a potential inhibitor for RdRp. All these peptides could be obtained from plants and animals such as soybean, peanut, corn, walnut, pork, chicken, salmon, sardine, etc. (Supplementary Materials, Table S1)
In the processing of infection, COVID-19 virus directly binds to ACE2 with the help of the spike protein and then enters the cell through receptor-mediated endocytosis [18]. ACE2 has high homology with ACE, and in some reports, the ACE inhibitor has been shown to have the effect of slowing down the infection and damage of pneumonia risk [33]. Although it is unclear if the ACE inhibitor really works on COVID-19, the consensus is that inhibiting the interaction between ACE2 and the spike protein of COVID-19 could stop the infection. After docking 187 natural peptides to ACE2 in the area of the COVID-19 virus spike receptor binding domain, we found a series of peptides that could interact with ACE2. Specifically, for peptide PA1, the total interaction energy (absolute value) was more than twice that of the spike protein residues (158.34175 kcal/mol). For MLN-4760, we could indicate a certain interaction with ACE2 at the area of COVID-19 virus spike receptor binding domain, and for the ACE inhibitory peptides, results were similar. These results strongly supported our hypothesis that the ACE inhibitor could also bind to ACE2 and might show a potential effect to stop the interaction between ACE2 and the spike protein of COVID-19 virus. From our results, the α-helix at SER19 to TYR50 of ACE2 was the main active site interacting with the COVID-19 virus spike protein. Through the overlapping of the peptides with high total interaction energy, it suggested that the peptides with nine to 10 amino acids (Ave. MW ≈ 1.14KDa) might be more suitable for taking up the active sites. At the conformation level, peptides with hydrophilic amino acids, acidic amino acids in the middle, and aromatic amino acids on the two sides might cover the whole α-helix and reduce the interaction between hydrophilic groups of ACE2 and the spike protein. Moreover, after the molecular dynamic simulation, in the lowest energy conformation of the PA1–ACE2 complex, their interaction energy (absolute value) has increased from 330.556 to 440.463 kcal/mol and interaction counts have changed from 10 to 15 with more H-bonds formed. Meanwhile, the binding of peptides has also significantly changed the conformation of ACE2, especially in the area of the COVID-19 virus spike receptor binding domain. This also suggests that a peptide with certain sequences, structure, and conformation might be helpful for preventing the infection of COVID-19 virus at the initial stage through taking up the active sites and blocking their interaction. Moreover, comparing with the conventional ACE-inhibitory drugs, these peptides are of natural origin and do not produce toxic side effects when overused, which might help reduce the risk of traditional drugs when treating COVID-19 [34].
The proliferation of COVID-19 virus relies on a variety of non-structural proteins, including Main protease (Mpro), Papain-like protease, Helicase (Nsp13), RNA-dependent RNA polymerase (RdRp), N-terminal exoribonuclease and C-terminal guanine-N7 methyl transferase (Nsp14), Uridylate-specific endoribonuclease (Nsp15), 2’-O-methyltransferase (Nsp16), and Nsp10 [21,35]. Among them, Mpro and RdRp are the main inhibitory targets reported. Nowadays, lots of drugs such as Remdesivir have been reported at the clinical level with a potential therapeutic effect against the COVID-19 virus. However, there are also many reports on their side effects. According to the latest research, the main interaction for Remdesivir was the covalent bond between Redesivir monophosphate (RMP) and uracil in RdRp [21,36]. Although the covalent bond may largely determine its inhibitory effect, the non-covalent interaction between the inhibitor and receptor also plays an important role. Actually, it is worth noting that the drug and target are permanently bound by a covalent bond, and this irreversibility will bring about a series of side effects [13]. For this reason, more and more researchers have confirmed that the non-covalent bond drug might be more useful.
Here, our results showed that 163 peptides could bind to Mpro, and 30 of them with the highest -CDocker energy (Score) had a total interaction energy (absolute value) over 64.279 kcal/mol. Peptides PM1 could bind to Mpro with the total interaction energy of 366.214 kcal/mol, while Ritonavir and Lopinavir showed the total interaction energy (absolute value) of 87.470 kcal/mol and 102.500 kcal/mol, respectively. This is consistent with the existing reports that Ritonavir and Lopinavir can be used to treat COVID-19. Meanwhile, from the latest research, inhibitor N3 also showed high interaction with Mpro, which was mainly contributed by the C-S covalent bond between CYS145 and N3 and 23 non-covalent bonds [20]. From our results, peptide PM1 formed 19 non-covalent bonds, which is higher than Ritonavir and Lopinavir. Although there is no covalent bond formed between PM1 and Mpro, the total non-covalent interaction energy is ten times comparing with inhibitor N3, and their interaction residues are mainly concreted at GLU166, GLN189, MET165, CYS145, HIS41, ASN142, and HIS164. This suggests that PM1 and other food peptides could have the potential for inhibiting COVID-19 virus Mpro. In addition, it has been reported that the key inhibitory sites were located at the random coil that ranged from THR25 to ALA191 [20]. Our results showed all the peptides and reported drugs were well inserted into the “pocket” and the amino acid residues at GLN189 and ASN142 formed two conventional hydrogen bonds with peptides that function as a “door switch”, which results in the full occupancy of the key active sites. Moreover, from the overlapping of peptides, we can also find out that the peptides with five to six amino acids (Ave. MW ≈ 0.67 KDa) could bind to Mpro, and at the conformation level, peptides with basic amino acids, aliphatic amino acids, and aromatic amino acids might well interact with the residues inside the pocket and also bind tightly to the “door switch”. In addition, after molecular dynamic simulation, the peptide PA1 was still inside the pocket, and more interaction bonds were formed, even though the total interaction energy (absolute value) was slightly reduced. However, the absolute value of the interaction energy was still higher than that of Ritonavir and Lopinavir and also higher than the non-covalent interaction energy of inhibitor N3. Therefore, our results support that certain peptides, especially PM1, might be good candidates for the drug development targeting the COVID-19 virus Mpro.
Our screening found that 172 peptides could bind to RdRp, and 30 of them with the highest scores had the total interaction energy (absolute value) ranging from 213.13233 to 870.28747 kcal/mol. Peptide PR1 had the highest interaction energy with 23 formed bonds and the metal ion (MG) and the nucleotide as the main interaction residues. It has been reported that Remdesivir was metabolized into phosphates (RMP) and then interacted with the uracil (U20). The O-P covalent bond formed and the heterocyclic nitrogen of RMP was bound to the uracil (U10) through two H-bonds [21,36]. Through the calculation, its total non-covalent interaction energy (absolute value) is 233.131 kcal/mol, which is lower than PR1 (870.287 kcal/mol). Although there is no covalent bond formed between PR1 and RdRp, the total non-covalent interaction energy is four times that of Remdesivir. The main interaction residues are similar, including MG1, MG2, U20, A11, LYS545, and ARG555. This suggests that PR1 also could have potential for inhibiting COVID-19 virus RdRp. Our results also showed that the active sites have formed similar to a “pore canal” with higher hydrophilicity, which is used for the transporting of nucleotide substrates. At the end, the RNA chain binds to RdRp. It has been reported that the RdRp-inhibitory mechanism for Remdesivir was via the formation of covalent bonds to prevent the polymerization of RNA [21]. Our results also suggest that the non-covalent interaction between RMP and RdRp amino acid residues might block the “pore canal” and reduce the binding probability of the substrate to the active site. The overlapping results of 30 peptides show that they mostly have five to six amino acids (Ave. MW ≈ 0.67 KDa), show their tight interaction with RNA and MG, and give the brief structure predication of peptides with high interaction energy. Specifically, peptides show more basic charges close to RNA, while there are more acidic groups near the magnesium ions. Peptides with a higher proportion of basic amino acid in the end (close to RNA) and acidic groups in the middle (close to MG) may show higher interaction energy with RdRp. Through the molecular dynamic simulation, the ligand–receptor complex with the lowest energy has been obtained, and the interaction energy between peptides and RdRp has been strengthened. For PR1, the interaction energy has increased to 971.613 kcal/mol with the interaction counts of 30. This can be explained by the introduction of the third magnesium ion. It has been reported that the RdRp strongly relies on the magnesium ion, and comparing with the reported drugs, our results suggest that except for the interaction with RNA, a strong interaction between peptides and MG could make it potential candidate for inhibiting COVID-19 virus RdRp.
5. Conclusions
Since the outbreak of COVID-19, there is an urgent need for effective prevention and treatment. Although many potential inhibitors have been reported at the clinical and laboratory levels, the development of drugs with natural, less side effects remains one of the top priorities. In this paper, we have proved that natural food-derived peptides could bind to ACE2, COVID-19 virus Mpro, and RdRp with higher non-covalent interaction energy than reported drugs or inhibitors. We also obtained six peptides (Val-Ser-Gly-Ala-Gly-Arg-Tyr, Val-Met-Asp-Lys-Pro-Gln-Gly, Val-Ile-Glu-Lys-Tyr-Pro, Lys-Asp-Tyr-Arg-Leu, Asp-Glu-Asn-Ser-Lys-Phe, Asn-Asn-Asn-Pro-Phe-Lys-Phe) showing the potential inhibitory effect to all these three targets. Through the molecular dynamic simulation, we have also confirmed that the interaction between peptides and COVID-19 could be further strengthened, while the receptor–ligand complex tends to be structurally stable. All these results suggest that natural food peptides might be potential candidates for drug development to prevent and control the infection of COVID-19 virus. Our study results provide critically important evidence to prioritize further experimental research for the development of therapeutic drugs against the COVID-19 virus. The sequence, structure, and conformation of peptides could also benefit from the further optimization of drugs.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2218-273X/11/2/330/s1, Table S1. Docking results of the natural food peptides inhibiting the COVID-19 virus MPro. Table S2 Interaction counts, residues and energy of the peptides inhibiting ACE2. Table S3 Interaction counts, residues and energy of the peptides inhibiting the COVID-19 virus Mpro. Table S4 Interaction counts, residues and energy of the peptides inhibiting the COVID-19 virus RdRp. Table S5 Interaction between reported drugs, Covid-19 virus Spike protein residues and ACE2. Table S6 Interaction mechanism between reported drugs and COVID-19 virus Mpro. Table S7 Interaction mechanism between reported drugs and COVID-19 virus RdRp. Table S8 Interaction mechanism between screening peptides and the ACE2. Table S9 Interaction mechanism between screening peptides and the COVID-19 virus Mpro. Table S10 Interaction mechanism between screening peptides and the COVID-19 virus RdRp. Table S11 Interaction mechanism between PA1, PM1, PR1 and three targets after molecular dynamic simulation. Figure S1 Structure of the ACE2 and reported inhibitory drugs. Figure S2. Structure of ACE2 complex containing Inhibitor MLN-4760 and its binding modes. Figure S3. Structure feature of the docking pocket based on the overlap inhibitors. Figure S4 Structure of the ACE2 complex containing PA1 and its binding modes after molecule dynamic simulation. Figure S5 Docking Poses of the top three peptides inhibiting ACE2 comparing with MLN-4760. Figure S6 Structure of the COVID-19 virus Mpro and reported inhibitory drugs. Figure S7 Structure of the COVID-19 virus Mpro complex containing Inhibitor N3 and its binding modes. Figure S8. Structure feature of the docking pocket based on the overlap inhibitors. Figure S9 Structure of the COVID-19 virus Mpro complex containing PM1 and its binding modes after molecule dynamic simulation. Figure S10 Docking Poses of the top three peptides inhibiting COVID-19 virus Mpro comparing with Inhibitor N3. Figure S11 Structure of the COVID-19 virus RdRp and reported inhibitory drugs. Figure S12 Structure of the COVID-19 virus RdRp complex containing Remdesivir and its binding modes. Figure S13 Structure feature of the docking pocket based on the overlap inhibitors. Figure S14 Structure of the COVID-19 virus RdRp complex containing PR1 and its binding modes after molecular dynamic simulation. Figure S15 Docking Poses of the top three peptides inhibiting COVID-19 virus RdRp comparing with Remdesivir.
Author Contributions
A.-M.S. and Q.W. conceived the project and designed the experiments; A.-M.S. and R.G. performed computational experiments, drew the figures, prepared the supplemental materials, and wrote the draft manuscript. Q.W. and J.-R.Z. helped to revise the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Agricultural Science and Technology Innovation Project (CAAS-ASTIP-201X-IAPPST), Young Elite Scientist Sponsorship program by CAST (2018 QNRC001) and Top Young Talents of Grain Industry in China (LQ2020202). And the APC was funded by Agricultural Science and Technology Innovation Project (CAAS-ASTIP-201X-IAPPST).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The authors confirm that there is no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Abbreviations
| CHARMS | conforming, hierarchical, adaptive refinement methods |
| MD | molecular dynamic |
| MG | magnesium ion |
| NPT | constant-pressure, constant-temperature |
| PME | particle-mesh-ewald |
| POP | phosphonium ion |
| RBD | receptor binding domain |
| RMP | Redesivir monophosphate |
| RMS | root mean square |
References
- World Health Organization (WHO). Weekly operational update on COVID-19—8 February 2021; WHO: Geneva, Switzerland, 2021. [Google Scholar]
- Petrosillo, N.; Viceconte, G.; Ergonul, O.; Ippolito, G.; Petersen, E. COVID-19, SARS and MERS: Are they closely related? Clin. Microbiol. Infect. 2020, 26, 729–734. [Google Scholar] [CrossRef]
- Fihn, S.D.; Perencevich, E.; Bradley, S.M. Caution Needed on the Use of Chloroquine and Hydroxychloroquine for Coronavirus Disease 2019. JAMA Netw. Open 2020, 3, e209035. [Google Scholar] [CrossRef]
- Borba, M.G.S.; Val, F.F.A.; Sampaio, V.S.; Alexandre, M.A.A.; Melo, G.C.; Brito, M.; Mourão, M.P.G.; Brito-Sousa, J.D.; Baía-Da-Silva, D.; Guerra, M.V.F.; et al. Effect of High vs Low Doses of Chloroquine Diphosphate as Adjunctive Therapy for Patients Hospitalized With Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Infection. JAMA Netw. Open 2020, 3, e208857. [Google Scholar] [CrossRef] [Green Version]
- Grein, J.; Ohmagari, N.; Shin, D.; Diaz, G.; Asperges, E.; Castagna, A.; Feldt, T.; Green, G.; Green, M.L.; Lescure, F.-X.; et al. Compassionate Use of Remdesivir for Patients with Severe Covid-19. N. Engl. J. Med. 2020, 382, 2327–2336. [Google Scholar] [CrossRef] [PubMed]
- Kujawski, S.A.; Wong, K.K.; Collins, J.P.; Epstein, L.; Killerby, M.E.; Midgley, C.M.; Abedi, G.R.; Seema Ahmed, N.; Almendares, O.; Alvarez, F.N.; et al. First 12 patients with coronavirus disease 2019 (COVID-19) in the United States. MedRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Mourier, G.; Salinas, M.; Kessler, P.; Stura, E.A.; Leblanc, M.; Tepshi, L.; Besson, T.; Diochot, S.; Baron, A.; Douguet, D.; et al. Mambalgin-1 Pain-relieving Peptide, Stepwise Solid-phase Synthesis, Crystal Structure, and Functional Domain for Acid-sensing Ion Channel 1a Inhibition. J. Biol. Chem. 2016, 291, 2616–2629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sangar, M.L.C.; Girard, E.J.; Hopping, G.; Yin, C.; Pakiam, F.; Brusniak, M.-Y.; Nguyen, E.; Ruff, R.; Gewe, M.M.; Byrnes-Blake, K.; et al. A potent peptide-steroid conjugate accumulates in cartilage and reverses arthritis without evidence of systemic corticosteroid exposure. Sci. Transl. Med. 2020, 12, eaay1041. [Google Scholar] [CrossRef] [PubMed]
- Demeule, M.; Annabi, B.; Currie, J.-C.; Larocque, A.; Charfi, C.; Béliveau, R. New docetaxel-peptide conjugate for the treatment of sortilin-positive triple-negative breast cancer. J. Clin. Oncol. 2019, 37, e12556. [Google Scholar] [CrossRef]
- Anang, S.; Kaushik, N.; Hingane, S.; Kumari, A.; Gupta, J.; Asthana, S.; Shalimar; Nayak, B.; Ranjith-Kumar, C.T.; Surjit, M. Potent Inhibition of Hepatitis E Virus Release by a Cyclic Peptide Inhibitor of the Interaction between Viral Open Reading Frame 3 Protein and Host Tumor Susceptibility Gene 101. J. Virol. 2018, 92, 00684-18. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Jin, H.; Chen, Y.; Li, L.; Zhu, Y.; Chong, H.; He, Y. A Membrane-Anchored Short-Peptide Fusion Inhibitor Fully Protects Target Cells from Infections of Human Immunodeficiency Virus Type 1 (HIV-1), HIV-2, and Simian Immunodeficiency Virus. J. Virol. 2019, 93, 01177-19. [Google Scholar] [CrossRef]
- Maurya, N.S.; Kushwaha, S.; Mani, A. Recent Advances and Computational Approaches in Peptide Drug Discovery. Curr. Pharm. Des. 2019, 25, 3358–3366. [Google Scholar] [CrossRef]
- Tuley, A.; Fast, W. The Taxonomy of Covalent Inhibitors. Biochemistry 2018, 57, 3326–3337. [Google Scholar] [CrossRef]
- Lagoutte, R.; Patouret, R.; Winssinger, N. Covalent inhibitors: An opportunity for rational target selectivity. Curr. Opin. Chem. Biol. 2017, 39, 54–63. [Google Scholar] [CrossRef] [Green Version]
- Stein, R.M.; Kang, H.J.; McCorvy, J.D.; Glatfelter, G.C.; Jones, A.J.; Che, T.; Slocum, S.; Huang, X.-P.; Savych, O.; Moroz, Y.S.; et al. Virtual discovery of melatonin receptor ligands to modulate circadian rhythms. Nat. Cell Biol. 2020, 579, 609–614. [Google Scholar] [CrossRef]
- Cardoso, M.H.; Orozco, R.Q.; Rezende, S.B.; Rodrigues, G.; Oshiro, K.G.N.; Cândido, E.S.; Franco, O.L. Computer-Aided Design of Antimicrobial Peptides: Are We Generating Effective Drug Candidates? Front. Microbiol. 2020, 10, 3097. [Google Scholar] [CrossRef]
- Lin, S.; Shen, R.; He, J.; Li, X.; Guo, X. Molecular Modeling Evaluation of the Binding Effect of Ritonavir, Lopinavir and Darunavir to Severe Acute Respiratory Syndrome Coronavirus 2 Proteases. BioRxiv 2020. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, W.; Mao, C.; Luan, X.; Shen, D.D.; Shen, Q.; Su, H.; Wang, X.; Zhou, F.; Zhao, W.; Gao, M.; et al. Structural Basis for the Inhibition of the RNA-Dependent RNA Polymerase from SARS-CoV-2 by Remdesivir. BioRxiv 2020. [Google Scholar] [CrossRef] [PubMed]
- Rice, G.I.; Thomas, D.A.; Grant, P.J.; Turner, A.J.; Hooper, N.M. Evaluation of angiotensin-converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism. Biochem. J. 2004, 383, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Shi, A.; Liu, H.; Liu, L.; Hu, H.; Wang, Q.; Adhikari, B. Isolation, Purification and Molecular Mechanism of a Peanut Protein-Derived ACE-Inhibitory Peptide. PLoS ONE 2014, 9, e111188. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Shi, A.; Wang, Q.; Zhang, G. Multivesicular Liposomes for the Sustained Release of Angiotensin I-Converting Enzyme (ACE) Inhibitory Peptides from Peanuts: Design, Characterization, and In Vitro Evaluation. Molecules 2019, 24, 1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daskaya-Dikmen, C.; Yucetepe, A.; Karbancioglu-Guler, F.; Daskaya, H.; Ozcelik, B. Angiotensin-I-Converting Enzyme (ACE)-Inhibitory Peptides from Plants. Nutrients 2017, 9, 316. [Google Scholar] [CrossRef]
- Guang, C.; Phillips, R.D. Plant Food-Derived Angiotensin I Converting Enzyme Inhibitory Peptides. J. Agric. Food Chem. 2009, 57, 5113–5120. [Google Scholar] [CrossRef]
- He, H.-L.; Liu, D.; Ma, C.-B. Review on the Angiotensin-I-Converting Enzyme (ACE) Inhibitor Peptides from Marine Proteins. Appl. Biochem. Biotechnol. 2012, 169, 738–749. [Google Scholar] [CrossRef] [PubMed]
- Roy, F.; Boye, J.; Simpson, B. Bioactive proteins and peptides in pulse crops: Pea, chickpea and lentil. Food Res. Int. 2010, 43, 432–442. [Google Scholar] [CrossRef]
- Vercruysse, L.; Van Camp, A.J.; Smagghe, G. ACE Inhibitory Peptides Derived from Enzymatic Hydrolysates of Animal Muscle Protein: A Review. J. Agric. Food Chem. 2005, 53, 8106–8115. [Google Scholar] [CrossRef]
- Xu, R.; Banka, A.; Blake, J.F.; Mitchell, I.S.; Wallace, E.M.; Bencsik, J.R.; Kallan, N.C.; Spencer, K.L.; Gloor, S.L.; Martinson, M.; et al. Discovery of spirocyclic sulfonamides as potent Akt inhibitors with exquisite selectivity against PKA. Bioorg. Med. Chem. Lett. 2011, 21, 2335–2340. [Google Scholar] [CrossRef] [PubMed]
- Roller, S.G. Chapter 8 ADME Properties of Peptide Therapeutics in Drug Discovery and Development. In Peptide-based Drug Discovery: Challenges and New Therapeutics; RSC Publishing: Cambridge, UK, 2017. [Google Scholar] [CrossRef]
- Stower, H. Lopinavir–ritonavir in severe COVID-19. Nat. Med. 2020, 26, 465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohkubo, T.; Chapman, N.; Neal, B.; Woodward, M.; Omae, T.; Chalmers, J. Effects of an Angiotensin-converting Enzyme Inhibitor–based Regimen on Pneumonia Risk. Am. J. Respir. Crit. Care Med. 2004, 169, 1041–1045. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; De Clercq, E. Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nat. Rev. Drug Discov. 2020, 19, 149–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, D.-G.; Shin, H.-J.; Kim, M.-H.; Lee, S.; Kim, H.-S.; Myoung, J.; Kim, B.-T.; Kim, S.-J. Current Status of Epidemiology, Diagnosis, Therapeutics, and Vaccines for Novel Coronavirus Disease 2019 (COVID-19). J. Microbiol. Biotechnol. 2020, 30, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of RNA-dependent RNA polymerase from 2019-nCoV, a major antiviral drug target. BioRxiv 2020. [Google Scholar] [CrossRef]
Figure 1.
Structure of the COVID-19 virus spike protein receptor binding domain angiotensin-converting enzyme 2 (ACE2) complex containing potential inhibitory peptide (PA1) and its binding modes. (A) Three-dimensional (3D) structure of the ACE2 complex containing PA1. For clarity, the interface receptor amino acids residues are shown in thin stick style, and PA1 is shown in thick stick style. ACE2 is downloaded from PDB (No. 6M0J) and generated in DS software with deletion of the ligands. The different colors represent the different secondary structure: red is α-helix, blue is β-fold, green is β-turn, white is random coil. PA1 is the peptide, Ile-Val-Gly-Arg-Pro-Arg-His-Gln-Gly, with the highest total interaction energy to ACE2. (B) Enlarged view of the interface in the ACE2 complex containing PA1. (C,D) Binding site between PA1 and ACE2 and the main interaction residues. Dashed line with a different color represents the non-covalent interaction between PA1 and ACE2. (E) 2D representation of interactions between the PA1 and amino acid residues of ACE2.
Figure 1.
Structure of the COVID-19 virus spike protein receptor binding domain angiotensin-converting enzyme 2 (ACE2) complex containing potential inhibitory peptide (PA1) and its binding modes. (A) Three-dimensional (3D) structure of the ACE2 complex containing PA1. For clarity, the interface receptor amino acids residues are shown in thin stick style, and PA1 is shown in thick stick style. ACE2 is downloaded from PDB (No. 6M0J) and generated in DS software with deletion of the ligands. The different colors represent the different secondary structure: red is α-helix, blue is β-fold, green is β-turn, white is random coil. PA1 is the peptide, Ile-Val-Gly-Arg-Pro-Arg-His-Gln-Gly, with the highest total interaction energy to ACE2. (B) Enlarged view of the interface in the ACE2 complex containing PA1. (C,D) Binding site between PA1 and ACE2 and the main interaction residues. Dashed line with a different color represents the non-covalent interaction between PA1 and ACE2. (E) 2D representation of interactions between the PA1 and amino acid residues of ACE2.

Figure 2.
Structure of the COVID-19 virus main protease (COVID-19 virus Mpro) complex containing peptide (PM1) and its binding modes. (A) Three-dimensional (3D) structure of the COVID-19 virus Mpro complex containing PM1. For clarity, the interface receptor amino acids residues are shown in thin stick style and PM1 is shown in thick stick style. COVID-19 virus Mpro is downloaded from PDB (No. 6LU7) and generated in DS software with deleting the ligands. Different color represents different secondary structure, red is α-helix, blue is β-fold, green is β-turn, white is random coil. PM1 is the peptide, Ile-Val-Gly-Arg-Pro-Arg, with the highest total interaction energy to COVID-19 virus Mpro. (B) Enlarged view of the interface in COVID-19 virus Mpro complex containing PM1. (C,D) Binding site between PM1 and COVID-19 virus Mpro and the main interaction residues. The dashed line with the different color represents the non-covalent interaction between PM1 and COVID-19 virus Mpro. (E) 2D representation of interactions between PM1 and amino acid residues of COVID-19 virus Mpro.
Figure 2.
Structure of the COVID-19 virus main protease (COVID-19 virus Mpro) complex containing peptide (PM1) and its binding modes. (A) Three-dimensional (3D) structure of the COVID-19 virus Mpro complex containing PM1. For clarity, the interface receptor amino acids residues are shown in thin stick style and PM1 is shown in thick stick style. COVID-19 virus Mpro is downloaded from PDB (No. 6LU7) and generated in DS software with deleting the ligands. Different color represents different secondary structure, red is α-helix, blue is β-fold, green is β-turn, white is random coil. PM1 is the peptide, Ile-Val-Gly-Arg-Pro-Arg, with the highest total interaction energy to COVID-19 virus Mpro. (B) Enlarged view of the interface in COVID-19 virus Mpro complex containing PM1. (C,D) Binding site between PM1 and COVID-19 virus Mpro and the main interaction residues. The dashed line with the different color represents the non-covalent interaction between PM1 and COVID-19 virus Mpro. (E) 2D representation of interactions between PM1 and amino acid residues of COVID-19 virus Mpro.

Figure 3.
Structure of the COVID-19 virus RNA-dependent RNA polymerase (COVID-19 virus RdRp) complex containing peptide (PR1) and its binding modes. (A) Three-dimensional (3D) structure of the COVID-19 virus RdRp complex containing PR1. For clarity, the interface receptor amino acids residues are shown in thin stick style and PR1 is shown in thick stick style. COVID-19 virus RdRp is downloaded from PDB (No. 7BV2) and generated in DS software with deleting the RMP and POP ligands. Different colors represent different secondary structures: red is α-helix, blue is β-fold, green is β-turn, white is random coil. PR1 is the peptides, Asp-Glu-Asn-Ser-Lys-Phe, with the highest total interaction energy to COVID-19 virus RdRp. (B) Enlarged view of the interface in COVID-19 virus RdRp complex containing PR1. (C,D) Binding site between PR1 and COVID-19 virus RdRp and the main interaction residues. Dashed lines with different colors represent the non-covalent interaction between PR1 and COVID-19 virus RdRp. (E) Two-dimensional (2D) representation of interactions between PR1 and amino acid residues of COVID-19 virus RdRp.
Figure 3.
Structure of the COVID-19 virus RNA-dependent RNA polymerase (COVID-19 virus RdRp) complex containing peptide (PR1) and its binding modes. (A) Three-dimensional (3D) structure of the COVID-19 virus RdRp complex containing PR1. For clarity, the interface receptor amino acids residues are shown in thin stick style and PR1 is shown in thick stick style. COVID-19 virus RdRp is downloaded from PDB (No. 7BV2) and generated in DS software with deleting the RMP and POP ligands. Different colors represent different secondary structures: red is α-helix, blue is β-fold, green is β-turn, white is random coil. PR1 is the peptides, Asp-Glu-Asn-Ser-Lys-Phe, with the highest total interaction energy to COVID-19 virus RdRp. (B) Enlarged view of the interface in COVID-19 virus RdRp complex containing PR1. (C,D) Binding site between PR1 and COVID-19 virus RdRp and the main interaction residues. Dashed lines with different colors represent the non-covalent interaction between PR1 and COVID-19 virus RdRp. (E) Two-dimensional (2D) representation of interactions between PR1 and amino acid residues of COVID-19 virus RdRp.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Docking results and binding sites for the potential peptides inhibiting the possible control targets of the COVID-19 virus.
Table 1.
Docking results and binding sites for the potential peptides inhibiting the possible control targets of the COVID-19 virus.
| Target | Drug Information | Main Interaction Residues | Non-Covalent Interaction | ||
|---|---|---|---|---|---|
| Total Interaction Energy (kcal/mol) | Total VDW Interaction Energy (kcal/mol) | Total Electrostatic Interaction Energy (kcal/mol) | |||
| ACE2 | Ile-Val-Gly-Arg-Pro-Arg-His-Gln-Gly (PA1) | GLU35, ASP38, GLU37, ASP30, HIS34, THR27, PHE32, GLN24, ASN33 | −330.55571 | −39.05066 | −291.50505 |
| Phe-Gln-Lys-Pro-Lys-Arg (PA2) | ASP30, GLU35, GLU37, ASP38, HIS34, THR27, PHE28 | −283.75101 | −30.18061 | −253.5704 | |
| Gly-Asp-Leu-Gly-Lys-Thr-Thr-Thr-Val-Ser-Asn-Trp-Ser-Pro-Pro-Lys-Tyr-Lys-Asp-Thr-Pro (PA3) | ASP30, GLU37, GLU35, LEU29, ASP38, THR27, LYS31, ALA386, ALA387, | −272.21947 | −68.03403 | −204.18544 | |
| Mpro | Ile-Val-Gly-Arg-Pro-Arg (PM1) | HIS41, MET49, PHE140, ASN142, HIS163, HIS164, MET165, GLU166, GLN189 | −366.21436 | −52.81027 | −313.40409 |
| Phe-Gln-Lys-Pro-Lys-Arg (PM2) | THR26, PHE140, GLY143, MET165, GLU166, ASP187, GLN189, THR190 | −308.37838 | −59.35911 | −249.01927 | |
| Tyr-Asn-Lys-Leu (PM3) | HIS41, PHE140, LEU141, ASN142, MET165, GLU166, GLN189 | −306.44818 | −54.43838 | −252.00979 | |
| Rdrp | Asp-Glu-Asn-Ser-Lys-Phe (PR1) | MG1, MG2, A11, U10, U20, ASP452, LYS545, LYS551, ARG553, ARG555, LYS621, ASP623, LYS798 | −870.28747 | −44.73179 | −825.55568 |
| Ile-Ala-Glu (PR2) | MG1, MG2, U20, LYS551, ARG553, ARG555, LYS621, CYS622 | −737.55523 | −14.8947 | −722.66053 | |
| Ile-Val-Val-Glu (PR3) | MG1, MG2, A19, U20, LYS551, ARG624, ARG553, LYS621, CYS622, ARG624, SER682 | −613.53135 | −25.86384 | −587.66751 | |
Table 2.
Docking results and binding sites for the reported drugs and ligand residues inhibiting the possible targets of COVID-19 virus.
Table 2.
Docking results and binding sites for the reported drugs and ligand residues inhibiting the possible targets of COVID-19 virus.
| Target | Drug Information | Main Interaction Residues | Covalent Bond | Non-Covalent Interaction | ||
|---|---|---|---|---|---|---|
| Total Interaction Energy (kcal/mol) | Total VDW Interaction Energy (kcal/mol) | Total Electrostatic Interaction Energy (kcal/mol) | ||||
| ACE2 | MLN-4760 C19H23Cl2N3O4 CAS No.: 305335-31-3 | GLU37, LYS31, ASP38, LYS353, THR27, GLU35, ASP30, ASN33 | / | −86.40653 | −26.30758 | −60.09895 |
| COVID-19 virus Spike receptor binding domain | GLN24, ASP30, LYS31, ASP38, TYR41, GLN42, MET82, TYR83, LYS353, GLY354 | / | −158.34175 | 3.79202 | −162.13377 | |
| Mpro | Inhibition N3 C35H48N6O8 CAS:NA | MET49, HIS41, LEU141, GLY143, CYS145, HIS163, HIS164, MET165, GLU166, GLN189, THR190 | C-S | −36.018383 | −6.372228 | −29.646155 |
| Rdrp | Remdesivir C27H35N6O8P CAS No.: 1809249-37-3 | MG1, MG2, U20, A11, LYS545, ARG555, CYS622, SER682, THR687, ASN691, SER759 | P-O | −223.13061 | −24.07386 | −199.05675 |
Table 3.
The summary for the peptides with potential high inhibitory activity comparing with reported drugs (molecular docking simulation level).
Table 3.
The summary for the peptides with potential high inhibitory activity comparing with reported drugs (molecular docking simulation level).
| Target | Content | Peptides (Based on the Data of Peptides with TOP 30 Interaction Energy) | Reported Drugs |
|---|---|---|---|
| ACE2 | Interaction Counts | 14 | 13 |
| Main Interaction Residues | LYS353, LYS31, ASP38, HIS34, GLU35, PHE390, GLU37, ASP30 | GLU37, LYS31, ASP38, LYS353, THR27, GLU35, ASP30, ASN33 | |
| Main Interaction Types | Salt Bridge, Attractive Charge, H-bond, Electrostatic, Hydrophobic | H-bond, Alkyl, Pi-Alkyl | |
| Typical Grops | Hydrophilic amino acid, acidic amino acid, Aromatic amino acids | hydrophilic amino acid, Aromatic amino acids | |
| Mpro | Interaction Counts | 16 | 14 (including 1 Covalent Bond) |
| Main Interaction Residues | GLU166, GLN189, MET165, CYS145, HIS41, ASN142, HIS164 | GLU166, GLY143, GLN189, MET165, CYS145 | |
| Main Interaction Types | H-bond, Salt Bridge, Attractive Charge, Pi-Sigma, Pi-Sulfur, Pi-Alkyl, Pi-Cation, Alkyl | Covalent bond, H-bond Pi-Sigma, Pi-Sulfur, Pi-Pi T-shaped, Alkyl, Pi-Alkyl | |
| Typical Grops | Basic amino acid, Aliphatic amino acid, Aromatic amino acids | Benzene ring, Aromatic five-membered heterocyclic ring | |
| RdRp | Interaction Counts | 18 | 11 (including 1 Covalent Bond) |
| Main Interaction Residues | MG1, MG2, U20, U10, U18, A11, LYS551, LYS621, ASP452, LYS621, ARG553 | MG1, MG2, U20, A11, LYS545, ARG555, CYS622, SER682, THR687, ASN691, SER759 | |
| Main Interaction Types | Salt Bridge; Attractive Charge, H-bond, Metal-Acceptor | Covalent bond, H-bond, Attractive Charge, Pi-Pi Stacked | |
| Typical Grops | basic amino acid and acidic amino acid, Aromatic amino acids | phosphate group, Six-membered azacyclic compounds |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Shi, A.-M.; Guo, R.; Wang, Q.; Zhou, J.-R. Screening and Molecular Modeling Evaluation of Food Peptides to Inhibit Key Targets of COVID-19 Virus. Biomolecules 2021, 11, 330. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11020330
AMA Style
Shi A-M, Guo R, Wang Q, Zhou J-R. Screening and Molecular Modeling Evaluation of Food Peptides to Inhibit Key Targets of COVID-19 Virus. Biomolecules. 2021; 11(2):330. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11020330
Chicago/Turabian StyleShi, Ai-Min, Rui Guo, Qiang Wang, and Jin-Rong Zhou. 2021. "Screening and Molecular Modeling Evaluation of Food Peptides to Inhibit Key Targets of COVID-19 Virus" Biomolecules 11, no. 2: 330. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11020330
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.