
Neuroprotective Effects of Pharmacological Hypothermia on Hyperglycolysis and Gluconeogenesis in Rats after Ischemic Stroke

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
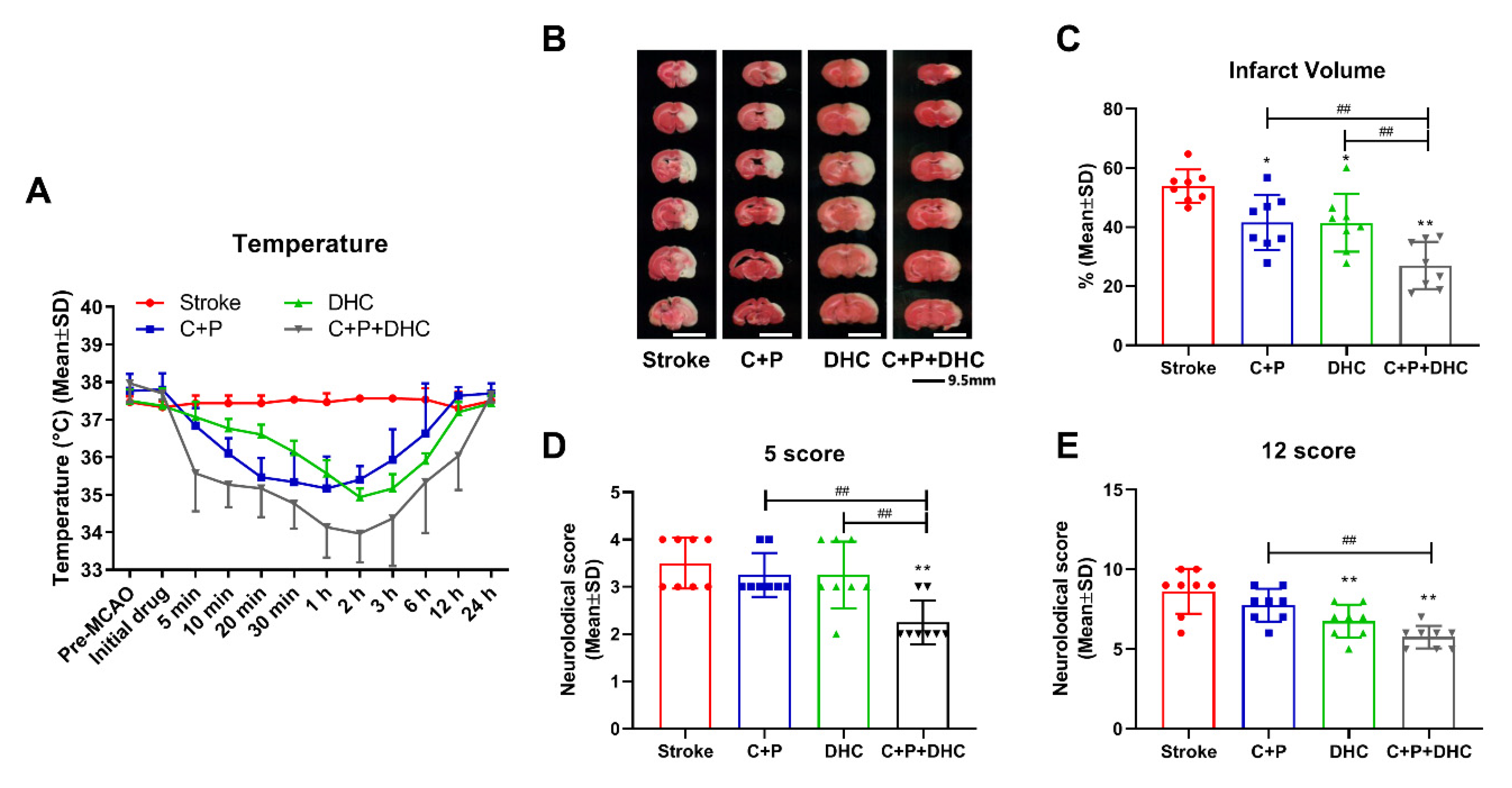
2.1. Physiological Parameters
2.2. Induction of Pharmacological Hypothermia
2.3. Infarct Volume and Neurological Deficits
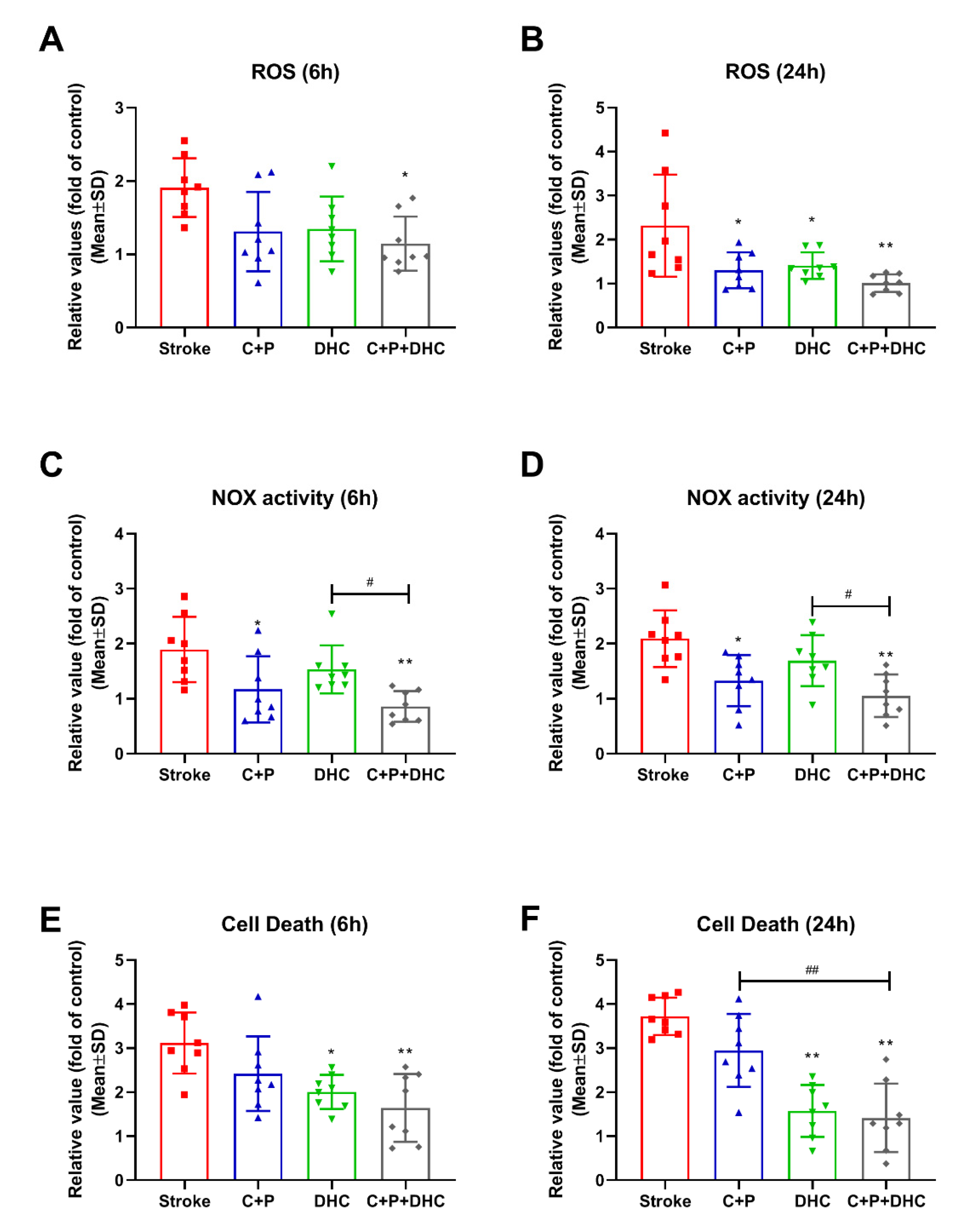
2.4. ROS Levels
2.5. NOX Activity
2.6. Apoptotic Cell Death
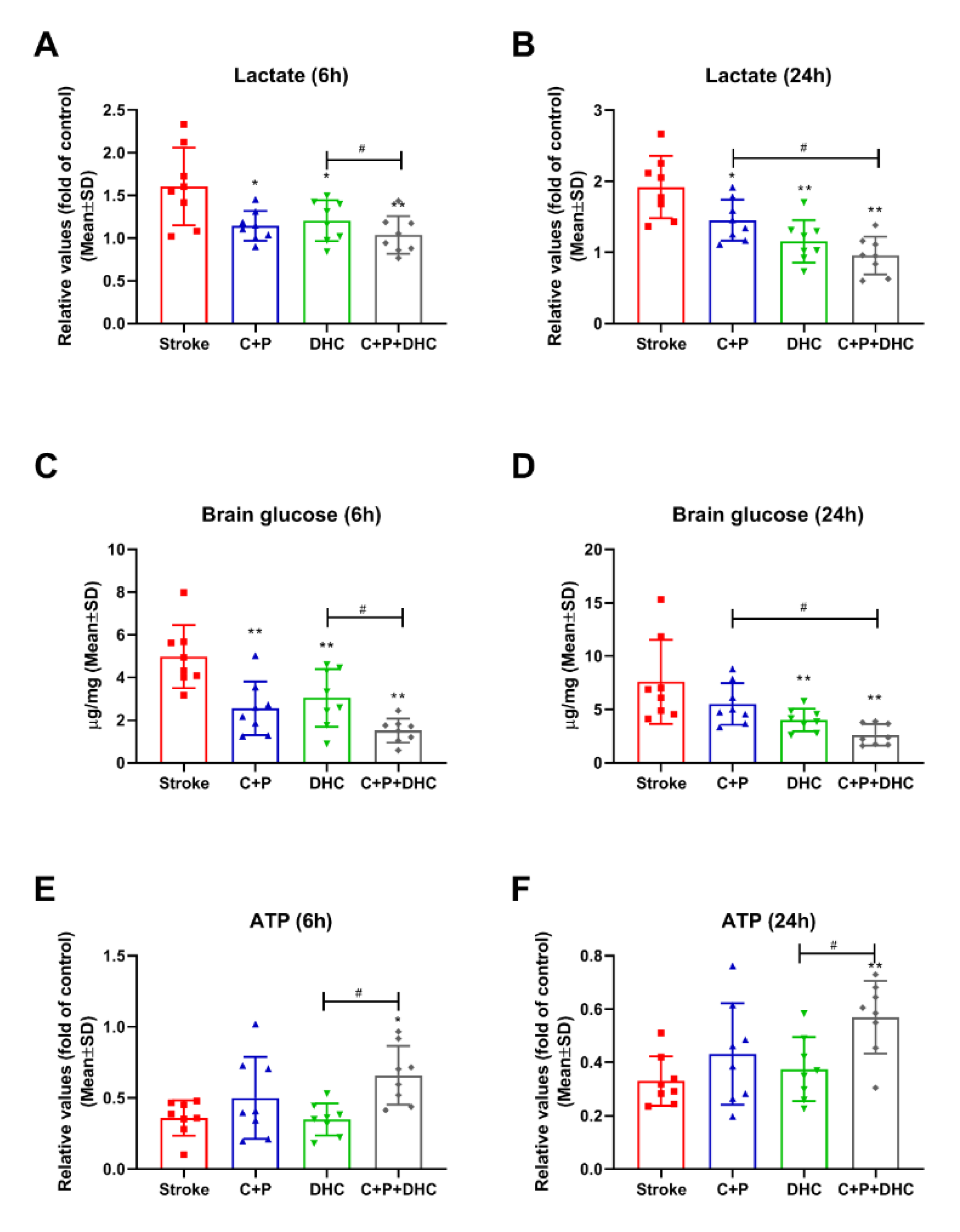
2.7. Lactate Level
2.8. Cerebral Glucose Concentration
2.9. ATP Level
2.10. Hypothermia Induced Neuroprotection Is Associated with Glucose Metabolism
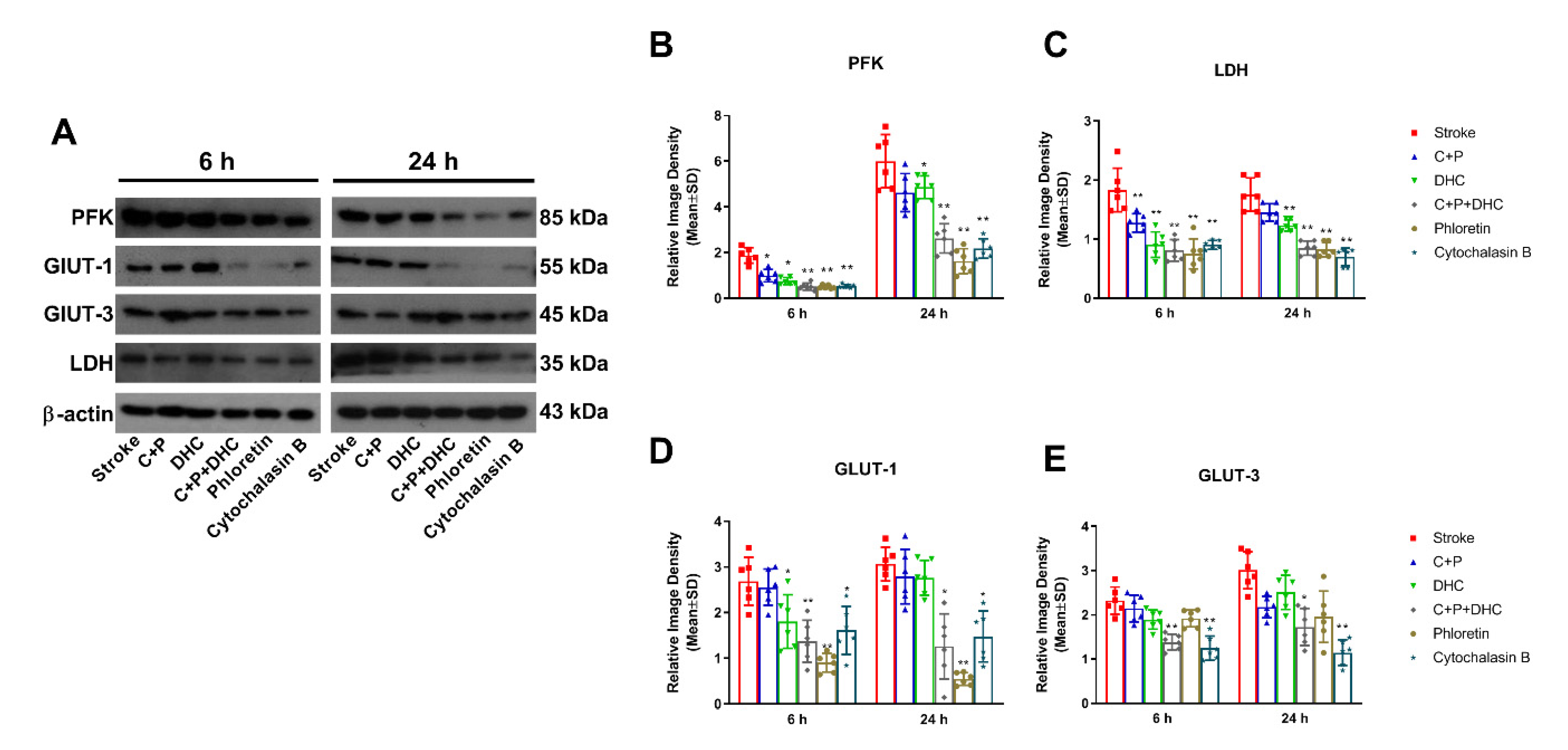
2.11. Pharmacological Hypothermia Reduced Expression of Glycolytic Enzyme
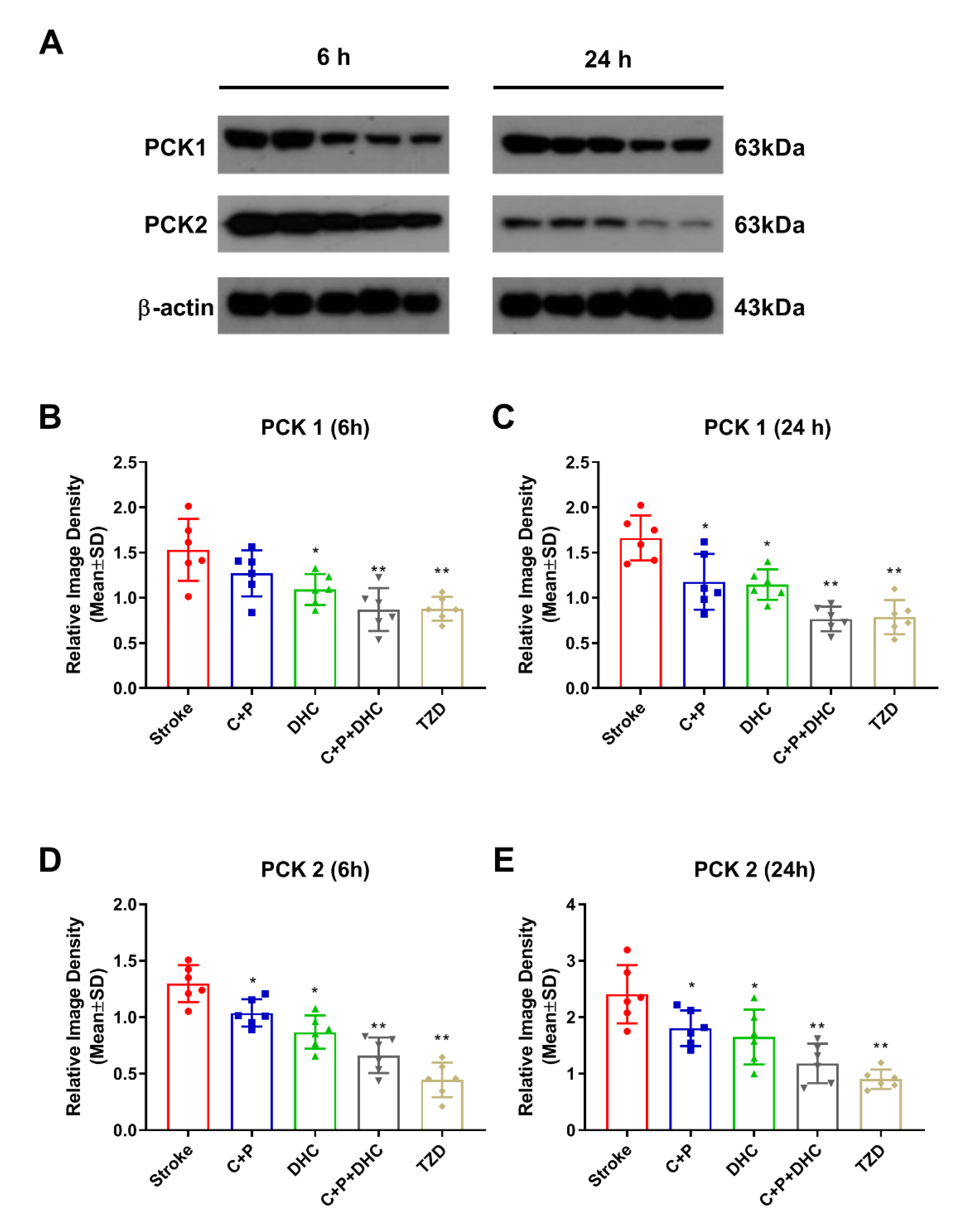
2.12. Pharmacological Hypothermia Reduced Expression of Gluconeogenic Enzymes
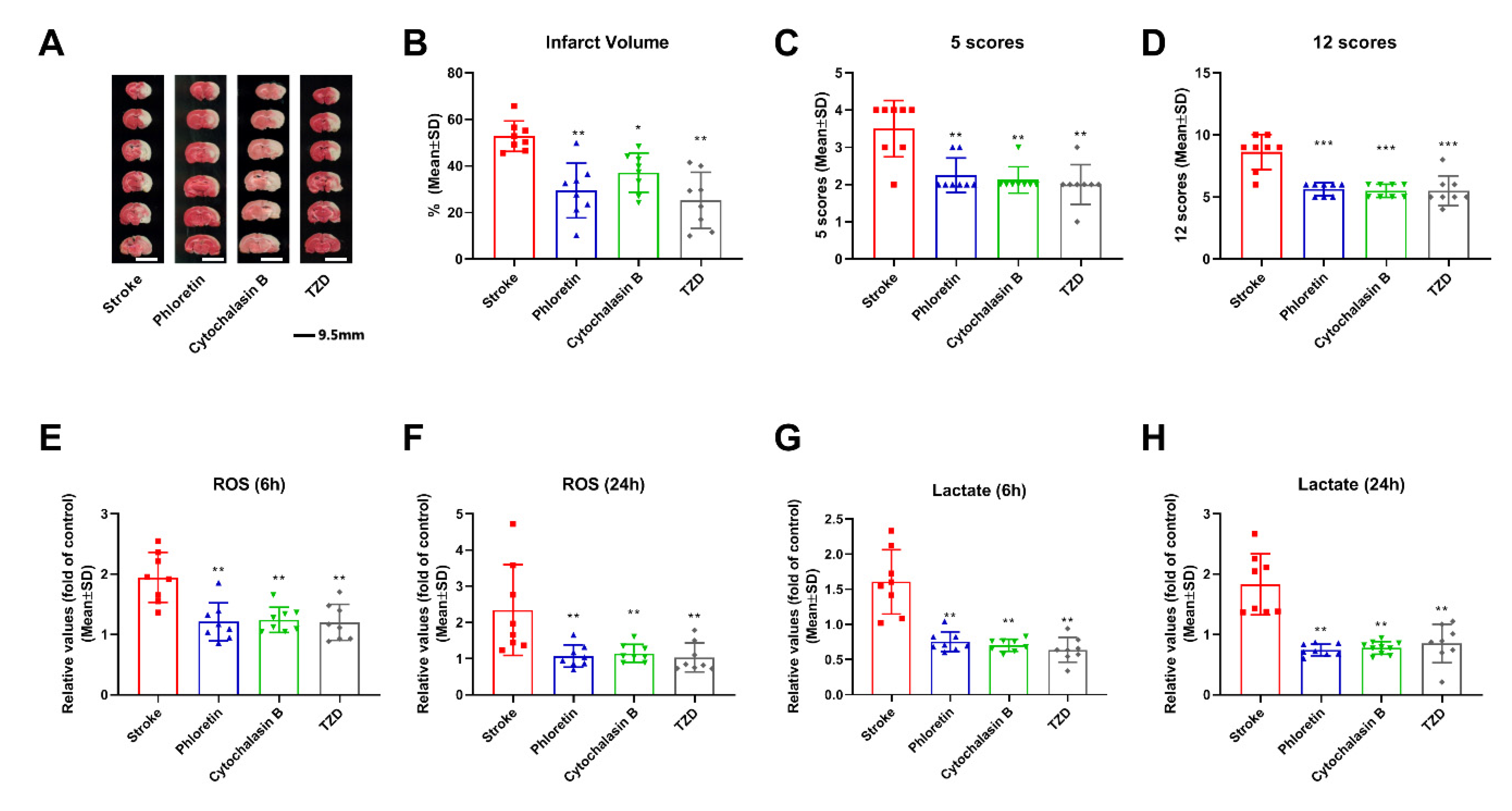
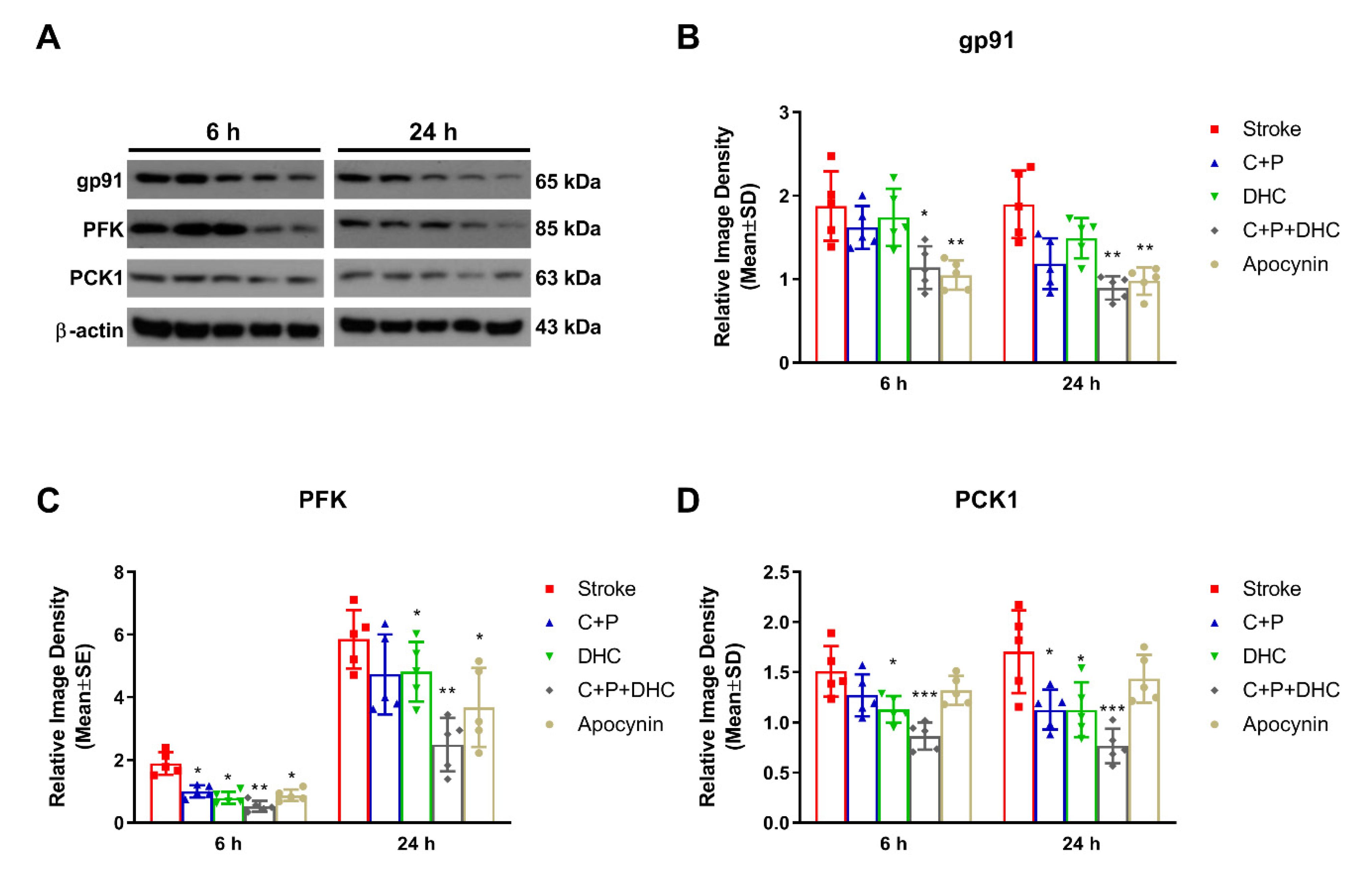
2.13. Effect of NOX on the Neuroprotection Induced by Pharmacological Hypothermia
3. Discussion
4. Materials and Methods
4.1. Subject
4.2. Focal Cerebral Ischemia and Reperfusion
4.3. Pharmacological Hypothermia with C + P and DHC, and Inhibitor Administration
4.4. Infarct Volume
4.5. Neurological Deficits
4.6. Cerebral Glucose, Lactate, and ATP Production
4.7. ROS Production
4.8. NADPH Oxidase (NOX) Activity
4.9. Cell Death
4.10. Protein Expression
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Campbell, B.C.V.; Khatri, P. Stroke. Lancet 2020, 396, 129–142. [Google Scholar] [CrossRef]
- Deng, X.-X.; Li, S.-S.; Sun, F.-Y. Necrostatin-1 Prevents Necroptosis in Brains after Ischemic Stroke via Inhibition of RIPK1-Mediated RIPK3/MLKL Signaling. Aging Dis. 2019, 10, 807–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, B.C.V.; Silva, D.A.D.; Macleod, M.R.; Coutts, S.B.; Schwamm, L.H.; Davis, S.M.; Donnan, G.A. Ischaemic stroke. Nat. Reviews. Dis. Primers 2019, 5, 70. [Google Scholar] [CrossRef] [PubMed]
- Rabinstein, A.A. Update on Treatment of Acute Ischemic Stroke. Continuum 2020, 26, 268–286. [Google Scholar] [CrossRef]
- Cheng, Z.; Geng, X.; Gao, J.; Hussain, M.; Moon, S.-J.; Du, H.; Ding, Y. Intravenous Administration of Standard Dose Tirofiban after Mechanical Arterial Recanalization is Safe and Relatively Effective in Acute Ischemic Stroke. Aging Dis. 2019, 10, 1049–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albers, G.W.; Marks, M.P.; Kemp, S.; Christensen, S.; Tsai, J.P.; Ortega-Gutierrez, S.; McTaggart, R.A.; Torbey, M.T.; Kim-Tenser, M.; Leslie-Mazwi, T.; et al. Thrombectomy for Stroke at 6 to 16 Hours with Selection by Perfusion Imaging. N. Engl. J. Med. 2018, 378, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Knecht, T.; Borlongan, C.; Dela Peña, I. Combination therapy for ischemic stroke: Novel approaches to lengthen therapeutic window of tissue plasminogen activator. Brain Circ. 2018, 4, 99–108. [Google Scholar] [CrossRef]
- Katan, M.; Luft, A. Global Burden of Stroke. Semin. Neurol. 2018, 38, 208–211. [Google Scholar] [CrossRef] [Green Version]
- Savitz, S.I.; Baron, J.C.; Fisher, M. Stroke Treatment Academic Industry Roundtable X: Brain Cytoprotection Therapies in the Reperfusion Era. Stroke 2019, 50, 1026–1031. [Google Scholar] [CrossRef]
- Lyden, P.; Buchan, A.; Boltze, J.; Fisher, M. Top Priorities for Cerebroprotective Studies: A Paradigm Shift. Stroke 2021, 52, 3063–3071. [Google Scholar] [CrossRef]
- Kuczynski, A.M.; Demchuk, A.M.; Almekhlafi, M.A. Therapeutic hypothermia: Applications in adults with acute ischemic stroke. Brain Circ. 2019, 5, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Wu, D.; Yang, T.; Xu, J.; Chen, J.; Wang, L.; Xu, S.; Zhao, W.; Wu, C.; Ji, X. Hypothermic neuroprotection against acute ischemic stroke: The 2019 update. J. Cereb. Blood Flow Metab. 2020, 40, 461–481. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Liu, L.; Zhang, H.; Geng, X.; Jiao, L.; Li, G.; Coutinho, J.M.; Ding, Y.; Liebeskind, D.S.; Ji, X. Endovascular Hypothermia in Acute Ischemic Stroke: Pilot Study of Selective Intra-Arterial Cold Saline Infusion. Stroke 2016, 47, 1933–1935. [Google Scholar] [CrossRef] [PubMed]
- Kurisu, K.; Kim, J.Y.; You, J.; Yenari, M.A. Therapeutic Hypothermia and Neuroprotection in Acute Neurological Disease. Curr. Med. Chem. 2019, 26, 5430–5455. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Ding, Y. Temporal limits of therapeutic hypothermia onset in clinical trials for acute ischemic stroke: How early is early enough? Brain Circ. 2020, 6, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Khan, H.; Geng, X.; Zhang, J.; Ding, Y. Pharmacological hypothermia: A potential for future stroke therapy? Neurol. Res. 2016, 38, 478–490. [Google Scholar] [CrossRef]
- Han, Y.; Geng, X.-k.; Lee, H.; Li, F.; Ding, Y. Neuroprotective Effects of Early Hypothermia Induced by Phenothiazines and DHC in Ischemic Stroke. Evid.-Based Complement. Altern. Med. 2021, 2021, 1207092. [Google Scholar] [CrossRef]
- Cao, Z.; Balasubramanian, A.; Marrelli, S.P. Pharmacologically induced hypothermia via TRPV1 channel agonism provides neuroprotection following ischemic stroke when initiated 90 min after reperfusion. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 306, R149–R156. [Google Scholar] [CrossRef] [Green Version]
- Mohammed, M.; Madden, C.J. Activation of TRPV1 in nucleus tractus solitarius reduces brown adipose tissue thermogenesis, arterial pressure, and heart rate. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R134–R143. [Google Scholar] [CrossRef]
- Geng, X.; Parmar, S.; Li, X.; Peng, C.; Ji, X.; Chakraborty, T.; Li, W.A.; Du, H.; Tan, X.; Ling, F.; et al. Reduced apoptosis by combining normobaric oxygenation with ethanol in transient ischemic stroke. Brain Res. 2013, 1531, 17–24. [Google Scholar] [CrossRef]
- Kurisu, K.; Yenari, M.A. Therapeutic hypothermia for ischemic stroke; pathophysiology and future promise. Neuropharmacology 2018, 134, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Li, W.A.; Moore-Langston, S.; Chakraborty, T.; Rafols, J.A.; Conti, A.C.; Ding, Y. Hyperglycemia in stroke and possible treatments. Neurol. Res. 2013, 35, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Yip, J.; Geng, X.; Shen, J.; Ding, Y. Cerebral Gluconeogenesis and Diseases. Front. Pharmacol. 2016, 7, 521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, X.; Shen, J.; Li, F.; Yip, J.; Guan, L.; Rajah, G.; Peng, C.; DeGracia, D.; Ding, Y. Phosphoenolpyruvate Carboxykinase (PCK) in the Brain Gluconeogenic Pathway Contributes to Oxidative and Lactic Injury After Stroke. Mol. Neurobiol. 2021, 58, 2309–2321. [Google Scholar] [CrossRef] [PubMed]
- Balança, B.; Meiller, A.; Bezin, L.; Dreier, J.P.; Marinesco, S.; Lieutaud, T. Altered hypermetabolic response to cortical spreading depolarizations after traumatic brain injury in rats. J. Cereb. Blood Flow Metab. 2017, 37, 1670–1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amorini, A.M.; Lazzarino, G.; Di Pietro, V.; Signoretti, S.; Lazzarino, G.; Belli, A.; Tavazzi, B. Metabolic, enzymatic and gene involvement in cerebral glucose dysmetabolism after traumatic brain injury. Biochim. Biophys. Acta 2016, 1862, 679–687. [Google Scholar] [CrossRef]
- Xu, X.J.; Yang, M.S.; Zhang, B.; Niu, F.; Dong, J.Q.; Liu, B.Y. Glucose metabolism: A link between traumatic brain injury and Alzheimer’s disease. Chin. J. Traumatol. 2021, 24, 5–10. [Google Scholar] [CrossRef]
- Abbasi, Y.; Shabani, R.; Mousavizadeh, K.; Soleimani, M.; Mehdizadeh, M. Neuroprotective effect of ethanol and Modafinil on focal cerebral ischemia in rats. Metab. Brain Dis. 2019, 34, 805–819. [Google Scholar] [CrossRef]
- Cheng, Z.; Li, F.W.; Stone, C.R.; Elkin, K.; Peng, C.Y.; Bardhi, R.; Geng, X.K.; Ding, Y.C. Normobaric oxygen therapy attenuates hyperglycolysis in ischemic stroke. Neural Regen. Res. 2021, 16, 1017–1023. [Google Scholar] [CrossRef]
- Guo, S.; Cosky, E.; Li, F.; Guan, L.; Ji, Y.; Wei, W.; Peng, C.; Geng, X.; Ding, Y. An inhibitory and beneficial effect of chlorpromazine and promethazine (C + P) on hyperglycolysis through HIF-1α regulation in ischemic stroke. Brain Res. 2021, 1763, 147463. [Google Scholar] [CrossRef]
- Haelewyn, B.; Yvon, A.; Hanouz, J.L.; MacKenzie, E.T.; Ducouret, P.; Gérard, J.L.; Roussel, S. Desflurane affords greater protection than halothane against focal cerebral ischaemia in the rat. Br. J. Anaesth. 2003, 91, 390–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Zhao, W.; An, H.; Wu, L.; Chen, J.; Hussain, M.; Ding, Y.; Li, C.; Wei, W.; Duan, J.; et al. Safety, feasibility, and potential efficacy of intraarterial selective cooling infusion for stroke patients treated with mechanical thrombectomy. J. Cereb. Blood Flow Metab. 2018, 38, 2251–2260. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Li, J.; Rafols, J.A.; Phillis, J.W.; Diaz, F.G. Prereperfusion saline infusion into ischemic territory reduces inflammatory injury after transient middle cerebral artery occlusion in rats. Stroke 2002, 33, 2492–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Z.; Cowan, A.; Tallarida, R.; Rawls, S.M. Capsaicin and nitric oxide synthase inhibitor interact to evoke a hypothermic synergy. Neurosci. Lett. 2006, 409, 41–46. [Google Scholar] [CrossRef]
- Fosgerau, K.; Weber, U.J.; Gotfredsen, J.W.; Jayatissa, M.; Buus, C.; Kristensen, N.B.; Vestergaard, M.; Teschendorf, P.; Schneider, A.; Hansen, P.; et al. Drug-induced mild therapeutic hypothermia obtained by administration of a transient receptor potential vanilloid type 1 agonist. BMC Cardiovasc. Disord. 2010, 10, 51. [Google Scholar] [CrossRef] [Green Version]
- Rawls, S.M.; Benamar, K. Effects of opioids, cannabinoids, and vanilloids on body temperature. Front. Biosci. 2011, 3, 822–845. [Google Scholar] [CrossRef] [Green Version]
- Ota, W.; Nakane, Y.; Kashio, M.; Suzuki, Y.; Nakamura, K.; Mori, Y.; Tominaga, M.; Yoshimura, T. Involvement of TRPM2 and TRPM8 in temperature-dependent masking behavior. Sci. Rep. 2019, 9, 3706. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Wang, H.; Zhao, J.; Chen, C.; Leak, R.K.; Xu, Y.; Vosler, P.; Chen, J.; Gao, Y.; Zhang, F. Drug-induced hypothermia in stroke models: Does it always protect? CNS Neurol. Disord. Drug Targets 2013, 12, 371–380. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, K.; Elmadhoun, O.; Ji, X.; Duan, Y.; Shi, J.; He, X.; Liu, X.; Wu, D.; Che, R.; et al. Synergistically Induced Hypothermia and Enhanced Neuroprotection by Pharmacological and Physical Approaches in Stroke. Aging Dis. 2018, 9, 578–589. [Google Scholar] [CrossRef] [Green Version]
- Ban, T.A. Fifty years chlorpromazine: A historical perspective. Neuropsychiatr. Dis. Treat. 2007, 3, 495–500. [Google Scholar]
- Guan, L.; Guo, S.; Yip, J.; Elkin, K.B.; Li, F.; Peng, C.; Geng, X.; Ding, Y. Artificial Hibernation by Phenothiazines: A Potential Neuroprotective Therapy Against Cerebral Inflammation in Stroke. Curr. Neurovasc. Res. 2019, 16, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Uesono, Y.; Toh-e, A.; Kikuchi, Y.; Araki, T.; Hachiya, T.; Watanabe, C.K.; Noguchi, K.; Terashima, I. Local Anesthetics and Antipsychotic Phenothiazines Interact Nonspecifically with Membranes and Inhibit Hexose Transporters in Yeast. Genetics 2016, 202, 997–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, H.; Duan, Y.; Wu, D.; Yip, J.; Elmadhoun, O.; Wright, J.C.; Shi, W.; Liu, K.; He, X.; Shi, J.; et al. Phenothiazines Enhance Mild Hypothermia-induced Neuroprotection via PI3K/Akt Regulation in Experimental Stroke. Sci. Rep. 2017, 7, 7469. [Google Scholar] [CrossRef] [PubMed]
- Protasoni, M.; Zeviani, M. Mitochondrial Structure and Bioenergetics in Normal and Disease Conditions. Int. J. Mol. Sci. 2021, 22, 586. [Google Scholar] [CrossRef]
- Rich, P.R. The molecular machinery of Keilin’s respiratory chain. Biochem. Soc. Trans. 2003, 31, 1095–1105. [Google Scholar] [CrossRef]
- Kochanski, R.; Peng, C.; Higashida, T.; Geng, X.; Hüttemann, M.; Guthikonda, M.; Ding, Y. Neuroprotection conferred by post-ischemia ethanol therapy in experimental stroke: An inhibitory effect on hyperglycolysis and NADPH oxidase activation. J. Neurochem. 2013, 126, 113–121. [Google Scholar] [CrossRef]
- Zhang, S.; Zuo, W.; Guo, X.F.; He, W.B.; Chen, N.H. Cerebral glucose transporter: The possible therapeutic target for ischemic stroke. Neurochem. Int. 2014, 70, 22–29. [Google Scholar] [CrossRef]
- Yenari, M.A.; Kauppinen, T.M.; Swanson, R.A. Microglial activation in stroke: Therapeutic targets. Neurotherapeutics 2010, 7, 378–391. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.N.; Zheng, Z.; Giffard, R.G.; Yenari, M.A. Significance of marrow-derived nicotinamide adenine dinucleotide phosphate oxidase in experimental ischemic stroke. Ann. Neurol. 2011, 70, 606–615. [Google Scholar] [CrossRef]
- Rastogi, R.; Geng, X.; Li, F.; Ding, Y. NOX Activation by Subunit Interaction and Underlying Mechanisms in Disease. Front. Cell Neurosci. 2016, 10, 301. [Google Scholar] [CrossRef] [Green Version]
- Raad, H.; Mouawia, H.; Hassan, H.; El-Seblani, M.; Arabi-Derkawi, R.; Boussetta, T.; Gougerot-Pocidalo, M.A.; Dang, P.M.; El-Benna, J. The protein kinase A negatively regulates reactive oxygen species production by phosphorylating gp91phox/NOX2 in human neutrophils. Free Radic. Biol. Med. 2020, 160, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Rastogi, R.; Geng, X.; Ding, Y. Nicotinamide adenine dinucleotide phosphate oxidase activation and neuronal death after ischemic stroke. Neural Regen. Res. 2019, 14, 948–953. [Google Scholar] [CrossRef] [PubMed]
- Sumimoto, H.; Minakami, R.; Miyano, K. Soluble Regulatory Proteins for Activation of NOX Family NADPH Oxidases. Methods Mol. Biol. 2019, 1982, 121–137. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.W.; Shin, B.S.; Ma, H.; Van Hoecke, M.; Brennan, A.M.; Yenari, M.A.; Swanson, R.A. Glucose and NADPH oxidase drive neuronal superoxide formation in stroke. Ann. Neurol. 2008, 64, 654–663. [Google Scholar] [CrossRef]
- Tang, X.N.; Cairns, B.; Kim, J.Y.; Yenari, M.A. NADPH oxidase in stroke and cerebrovascular disease. Neurol. Res. 2012, 34, 338–345. [Google Scholar] [CrossRef] [Green Version]
- Si, M.; Wang, Q.; Li, Y.; Lin, H.; Luo, D.; Zhao, W.; Dou, X.; Liu, J.; Zhang, H.; Huang, Y.; et al. Inhibition of hyperglycolysis in mesothelial cells prevents peritoneal fibrosis. Sci. Transl. Med. 2019, 11, eaav5341. [Google Scholar] [CrossRef]
- Cai, L.; Stevenson, J.; Peng, C.; Xin, R.; Rastogi, R.; Liu, K.; Geng, X.; Gao, Z.; Ji, X.; Rafols, J.A.; et al. Adjuvant therapies using normobaric oxygen with hypothermia or ethanol for reducing hyperglycolysis in thromboembolic cerebral ischemia. Neuroscience 2016, 318, 45–57. [Google Scholar] [CrossRef]
- Geng, J.; Zhang, Y.; Li, S.; Li, S.; Wang, J.; Wang, H.; Aa, J.; Wang, G. Metabolomic Profiling Reveals That Reprogramming of Cerebral Glucose Metabolism Is Involved in Ischemic Preconditioning-Induced Neuroprotection in a Rodent Model of Ischemic Stroke. J. Proteome Res. 2019, 18, 57–68. [Google Scholar] [CrossRef]
- Geng, X.; Li, F.; Yip, J.; Peng, C.; Elmadhoun, O.; Shen, J.; Ji, X.; Ding, Y. Neuroprotection by Chlorpromazine and Promethazine in Severe Transient and Permanent Ischemic Stroke. Mol. Neurobiol. 2017, 54, 8140–8150. [Google Scholar] [CrossRef]
- Wang, F.; Wang, Y.; Geng, X.; Asmaro, K.; Peng, C.; Sullivan, J.M.; Ding, J.Y.; Ji, X.; Ding, Y. Neuroprotective effect of acute ethanol administration in a rat with transient cerebral ischemia. Stroke 2012, 43, 205–210. [Google Scholar] [CrossRef] [Green Version]
- Longa, E.Z.; Weinstein, P.R.; Carlson, S.; Cummins, R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1989, 20, 84–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belayev, L.; Alonso, O.F.; Busto, R.; Zhao, W.; Ginsberg, M.D. Middle cerebral artery occlusion in the rat by intraluminal suture. Neurological and pathological evaluation of an improved model. Stroke 1996, 27, 1616–1622; discussion 1623. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Fu, P.; Ji, X.; Peng, C.; Fredrickson, V.; Sy, C.; Meng, R.; Ling, F.; Du, H.; Tan, X.; et al. Synergetic neuroprotection of normobaric oxygenation and ethanol in ischemic stroke through improved oxidative mechanism. Stroke 2013, 44, 1418–1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guan, L.; Lee, H.; Geng, X.; Li, F.; Shen, J.; Ji, Y.; Peng, C.; Du, H.; Ding, Y. Neuroprotective Effects of Pharmacological Hypothermia on Hyperglycolysis and Gluconeogenesis in Rats after Ischemic Stroke. Biomolecules 2022, 12, 851. https://0-doi-org.brum.beds.ac.uk/10.3390/biom12060851
Guan L, Lee H, Geng X, Li F, Shen J, Ji Y, Peng C, Du H, Ding Y. Neuroprotective Effects of Pharmacological Hypothermia on Hyperglycolysis and Gluconeogenesis in Rats after Ischemic Stroke. Biomolecules. 2022; 12(6):851. https://0-doi-org.brum.beds.ac.uk/10.3390/biom12060851
Chicago/Turabian StyleGuan, Longfei, Hangil Lee, Xiaokun Geng, Fengwu Li, Jiamei Shen, Yu Ji, Changya Peng, Huishan Du, and Yuchuan Ding. 2022. "Neuroprotective Effects of Pharmacological Hypothermia on Hyperglycolysis and Gluconeogenesis in Rats after Ischemic Stroke" Biomolecules 12, no. 6: 851. https://0-doi-org.brum.beds.ac.uk/10.3390/biom12060851