
Molecular Process Producing Oncogene Fusion in Lung Cancer Cells by Illegitimate Repair of DNA Double-Strand Breaks

Abstract
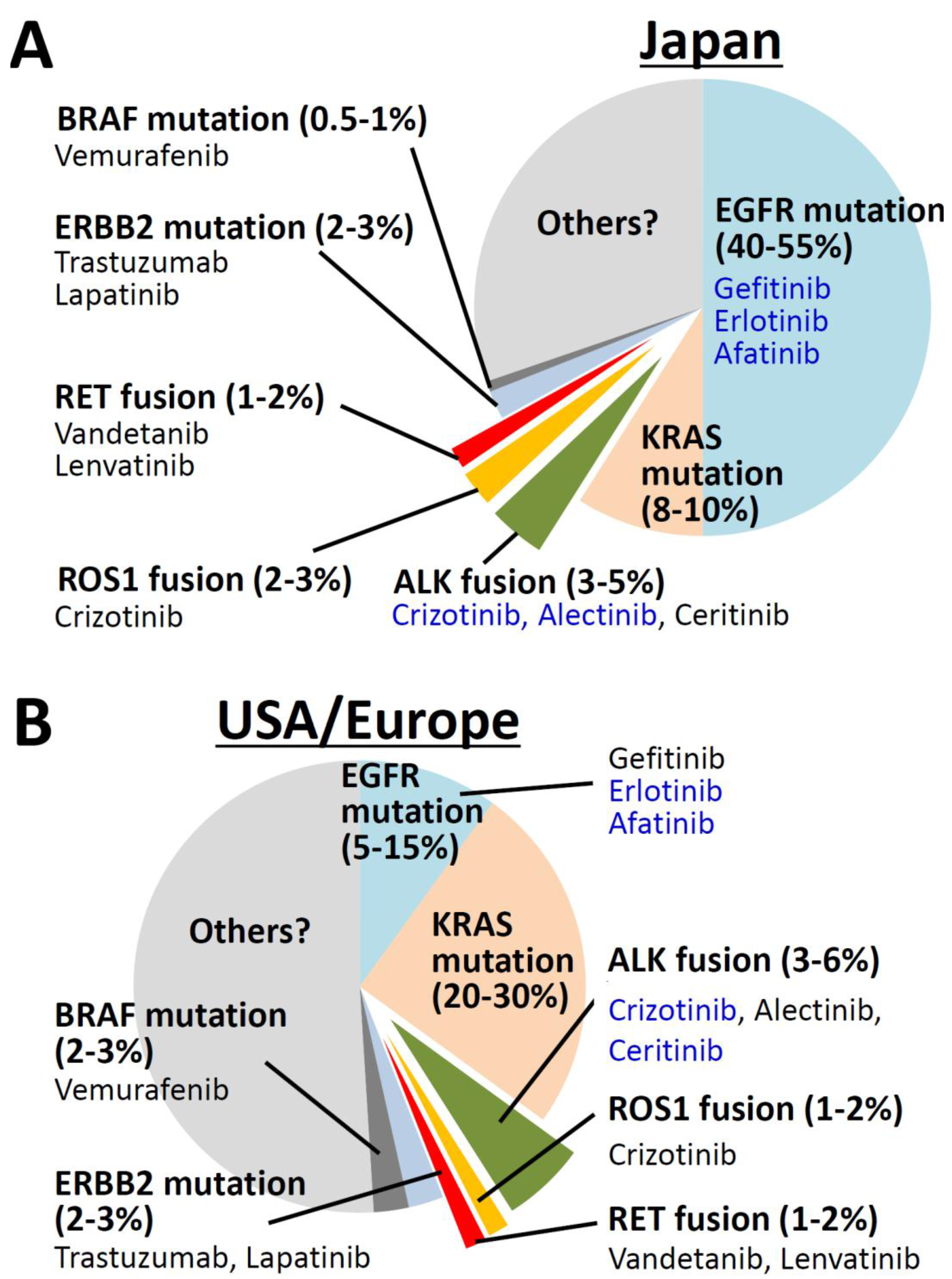
:1. Introduction


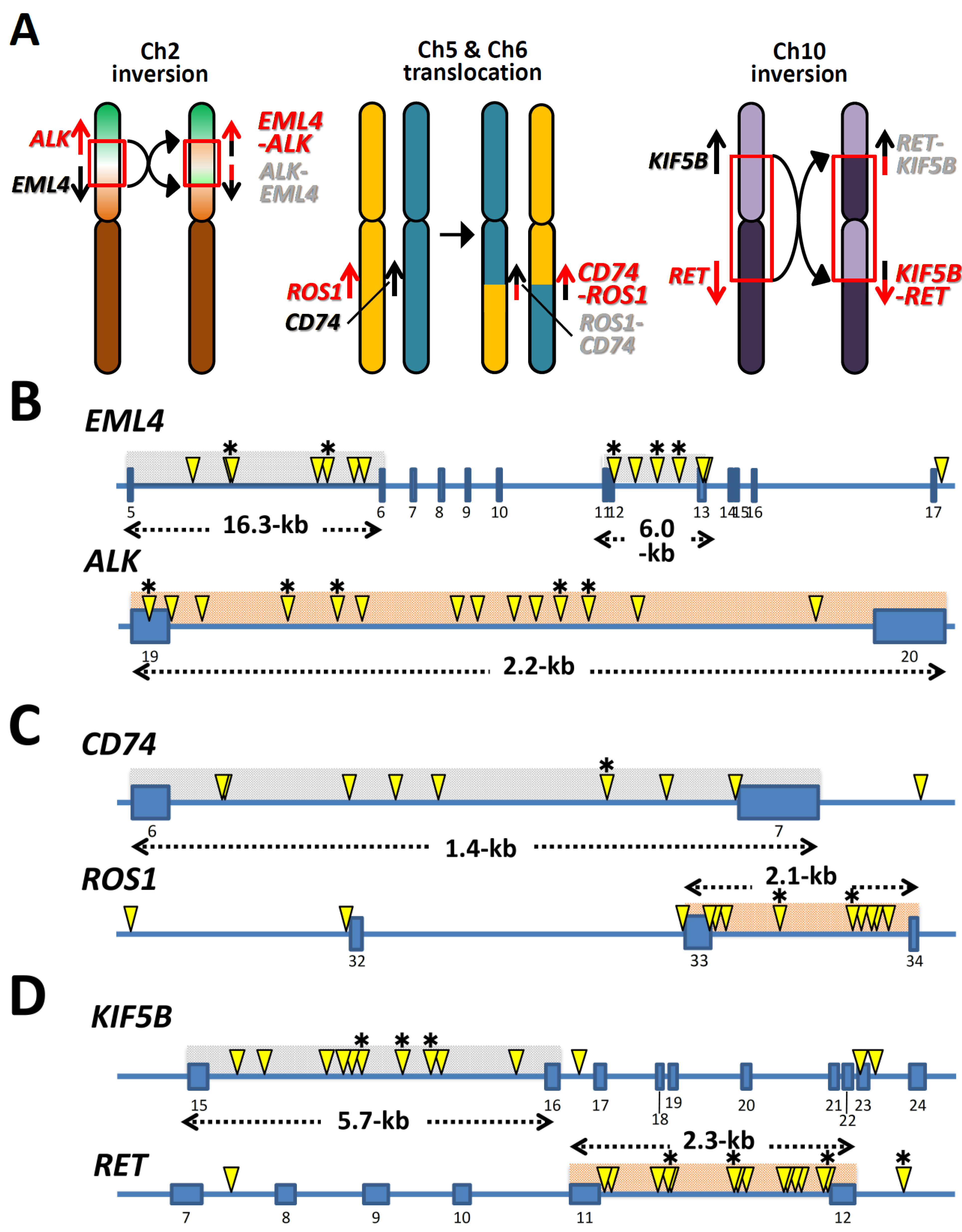
2. Distribution of Breakpoints in Oncogenes and Partner Genes
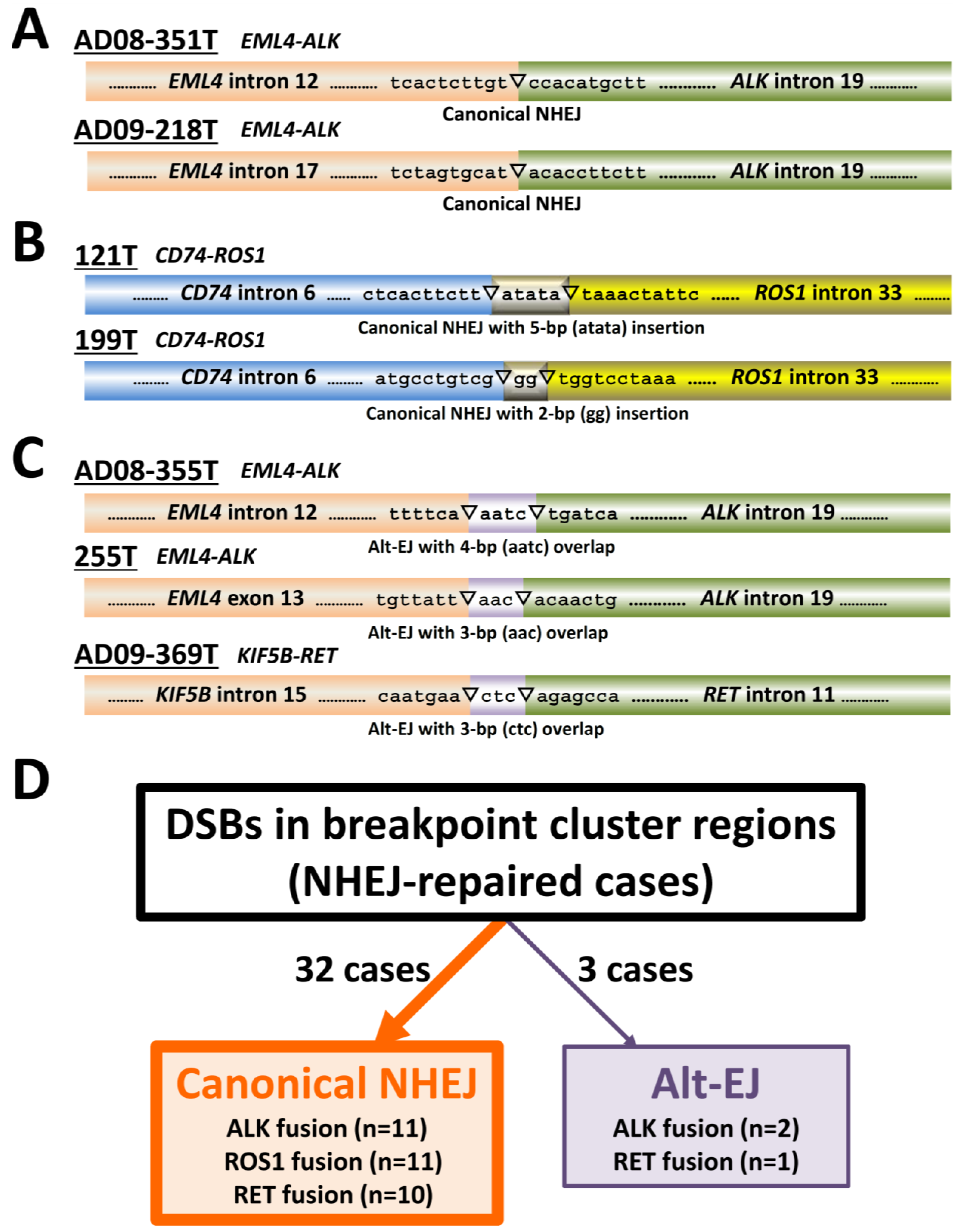
3. Canonical Non-Homologous End-Joining (NHEJ), a Major DSB Repair Pathway for Illegitimate Joining of DNA Ends

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Onco-gene | Sample name | Partner gene | Nucleotide deletion | DNA segment duplication | Mode of DNA end joining | Smoking | ||
|---|---|---|---|---|---|---|---|---|
| Kinase | Partner | Kinase | Partner | |||||
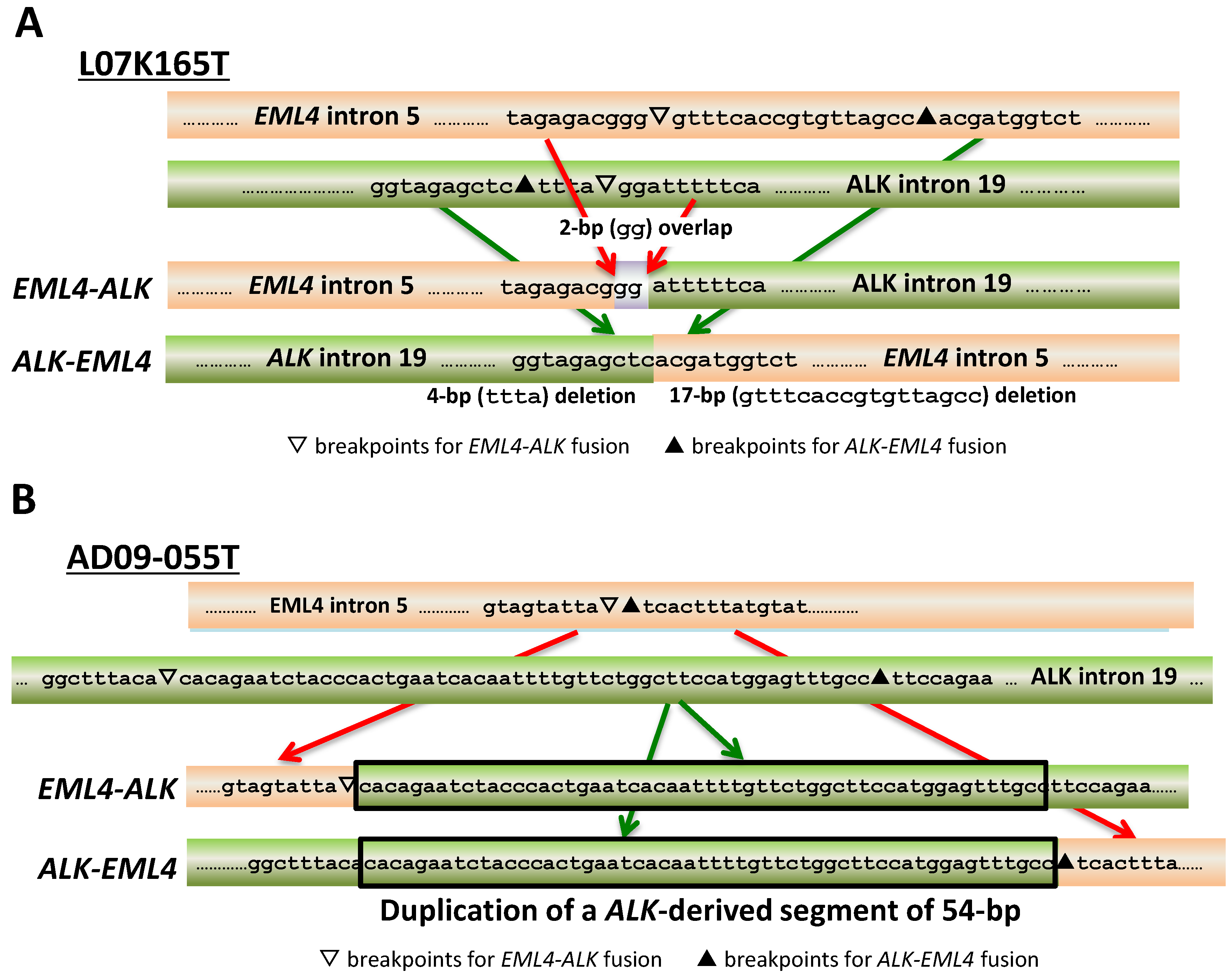
| ALK | L07K165_T | EML4 | 4-bp | 17-bp | - | - | NHEJ | No |
| AD09-357T | EML4 | 4-bp | - | - | - | NHEJ | No | |
| L07K154_T | EML4 | - | - | - | - | NHEJ | No | |
| AD09-055T | EML4 | - | - | 54-bp | - | SDEJ | No | |
| ROS1 | 103T | CD74 | - | 32-bp | 41-bp | - | SDEJ | No |
| RET | BR0020 | KIF5B | - | - | - | - | NHEJ | No |
| L07K201T | KIF5B | 15-bp | 19-bp | - | - | NHEJ | Yes | |
| 349T | KIF5B | 1-bp | 7-bp | - | - | NHEJ | Yes | |
| AD08-341T | KIF5B | 16-bp | 26-bp | - | - | NHEJ | No | |
| RET-024 | CCDC6 | 14-bp | 2-bp | - | - | NHEJ | Yes | |
| RET-030 | CCDC6 | 52-bp | 1021-bp | - | - | NHEJ | No | |
| AD12-106T | KIF5B | - | 573-bp | 490-bp | - | SDEJ | Yes | |
| BR0030 | KIF5B | - | - | - | 211-bp | SDEJ | No | |
| 442T | KIF5B | 269-bp | - | - | 235-bp | SDEJ | No | |
| AD08-144T | KIF5B | 7-bp | - | - | 2576-bp | SDEJ | No | |
| Onco-gene | Sample name | Partner gene | Reciprocal | Nucleotide overlap at junction | Nucleotide insertion at junction | Mode of NHEJ | Smoking |
|---|---|---|---|---|---|---|---|
| ALK | L07K165_T | EML4 | yes | GG | - | C-NHEJ | No |
| AD09-357T | EML4 | yes | T | - | C-NHEJ | No | |
| L07K154_T | EML4 | yes | - | AC | C-NHEJ | No | |
| 43T | EML4 | no | T | - | C-NHEJ | Yes | |
| 137T | EML4 | no | CT | - | C-NHEJ | Yes | |
| 169T | EML4 | no | - | - | C-NHEJ | Yes | |
| 236T | EML4 | no | TA | - | C-NHEJ | No | |
| 255T | EML4 | no | AAC | - | Alt-EJ | No | |
| L07K098_T | EML4 | no | A | - | C-NHEJ | No | |
| AD08_351T | EML4 | no | - | - | C-NHEJ | Yes | |
| AD08_355T | EML4 | no | AATC | - | Alt-EJ | No | |
| AD09-218T | EML4 | no | - | - | C-NHEJ | No | |
| AD09-352T | EML4 | no | - | - | C-NHEJ | Yes | |
| ROS1 | 121T | CD74 | no | - | ATATA | C-NHEJ | No |
| 199T | CD74 | no | - | GG | C-NHEJ | No | |
| L07K147_T | EZR | no | - | - | C-NHEJ | No | |
| AD08_009T | CD74 | no | - | - | C-NHEJ | No | |
| AD08_034T | EZR | no | T | - | C-NHEJ | No | |
| AD08_047T | CD74 | no | CA | - | C-NHEJ | No | |
| AD09-074T | CD74 | no | - | - | C-NHEJ | No | |
| AD09-224T | CD74 | no | - | - | C-NHEJ | No | |
| AD09-230T | CD74 | no | AT | - | C-NHEJ | No | |
| AD09-254T | CD74 | no | AC | - | C-NHEJ | Yes | |
| AD09-466T | EZR | no | - | T | C-NHEJ | Yes | |
| RET | BR0020 | KIF5B | yes | - | - | C-NHEJ | No |
| L07K201T | KIF5B | yes | C | ATA | C-NHEJ | Yes | |
| 349T | KIF5B | yes | - | A | C-NHEJ | Yes | |
| AD08-341T | KIF5B | yes | - | - | C-NHEJ | No | |
| RET-024 | CCDC6 | yes | - | - | C-NHEJ | Yes | |
| RET-030 | CCDC6 | yes | - | - | C-NHEJ | No | |
| BR1001 | KIF5B | no | - | AGT | C-NHEJ | No | |
| BR1002 | KIF5B | no | A | - | C-NHEJ | No | |
| BR1003 | KIF5B | no | - | CTTT | C-NHEJ | No | |
| AD09-369T | KIF5B | no | CTC | - | Alt-EJ | No | |
| AD12-001T | KIF5B | no | - | - | C-NHEJ | Yes |



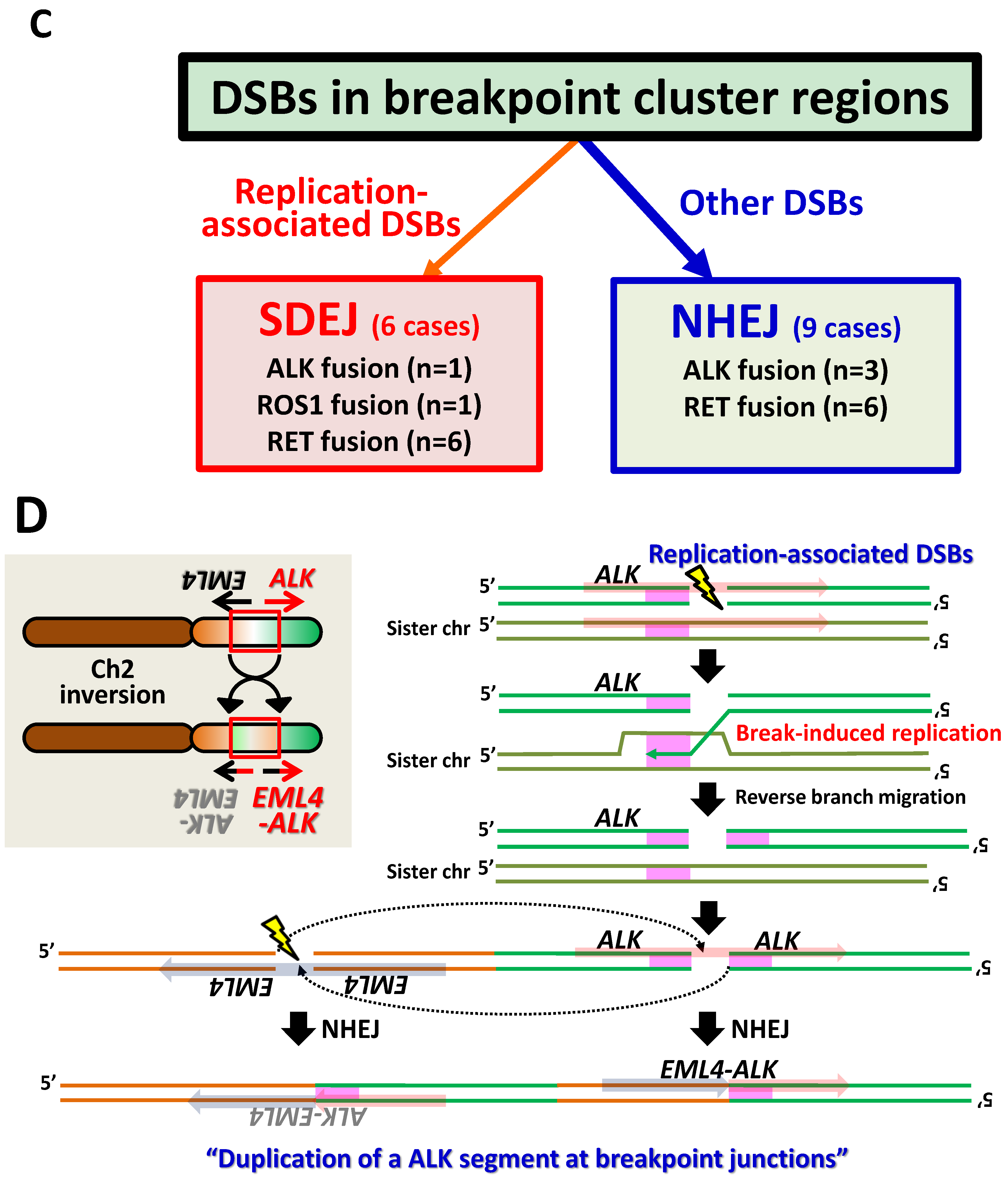
4. Synthesis-Dependent End-Joining (SDEJ), another DSB Repair Pathway for Illegitimate Joining of DNA Ends
5. Molecular Process for Chromosome Rearrangements Producing Gene Fusion
6. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contribution
Conflicts of Interests
References
- Kohno, T.; Nakaoku, T.; Tsuta, K.; Tsuchihara, K.; Matsumoto, S.; Yoh, K.; Goto, K. Beyond ALK-RET, ROS1 and other oncogene fusions in lung cancer. Transl. Lung Cancer Res. 2015, 4, 156–164. [Google Scholar] [PubMed]
- Shaw, A.T.; Hsu, P.P.; Awad, M.M.; Engelman, J.A. Tyrosine kinase gene rearrangements in epithelial malignancies. Nat. Rev. Cancer 2013, 13, 772–787. [Google Scholar] [CrossRef] [PubMed]
- Kohno, T.; Ichikawa, H.; Totoki, Y.; Yasuda, K.; Hiramoto, M.; Nammo, T.; Sakamoto, H.; Tsuta, K.; Furuta, K. KIF5B-RET fusions in lung adenocarcinoma. Nat. Med. 2012, 18, 375–377. [Google Scholar] [CrossRef] [PubMed]
- Dogan, S.; Shen, R.; Ang, D.C.; Johnson, M.L.; D’Angelo, S.P.; Paik, P.K.; Brzostowski, E.B.; Riely, G.J.; Kris, M.G.; Zakowski, M.F. Molecular epidemiology of EGFR and KRAS mutations in 3026 lung adenocarcinomas: Higher susceptibility of women to smoking-related KRAS-mutant cancers. Clin. Cancer Res. 2012, 18, 6169–6177. [Google Scholar] [CrossRef] [PubMed]
- Couraud, S.; Zalcman, G.; Milleron, B.; Morin, F.; Souquet, P.J. Lung cancer in never smokers—A review. Eur. J. Cancer 2012, 48, 1299–1311. [Google Scholar] [PubMed]
- Yang, L.; Luquette, L.J.; Gehlenborg, N.; Xi, R.; Haseley, P.S.; Hsieh, C.H.; Zhang, C.; Ren, X.; Protopopov, A.; Chin, L. Diverse mechanisms of somatic structural variations in human cancer genomes. Cell 2013, 153, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Kohno, T.; Yokota, J. Molecular processes of chromosome 9p21 deletions causing inactivation of the p16 tumor suppressor gene in human cancer: Deduction from structural analysis of breakpoints for deletions. DNA Repair (Amst) 2006, 5, 1273–1281. [Google Scholar] [CrossRef]
- Saito, M.; Shimada, Y.; Shiraishi, K.; Sakamoto, H.; Tsuta, K.; Totsuka, H.; Chiku, S.; Ichikawa, H.; Kato, M.; Watamabe, S.; et al. Development of lung adenocarcinomas with exclusive dependence on oncogene fusions. Cancer Res. 2015, 75, 2264–2271. [Google Scholar] [CrossRef] [PubMed]
- Mizukami, T.; Shiraishi, K.; Shimada, Y.; Ogiwara, H.; Tsuta, K.; Ichikawa, H.; Sakamoto, H.; Kato, M.; Shibata, T.; Nakano, T.; et al. Molecular mechanisms underlying oncogenic RET fusion in lung adenocarcinoma. J. Thorac. Oncol. 2014, 9, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Tsuta, K.; Kohno, T.; Yoshida, A.; Shimada, Y.; Asamura, H.; Furuta, K.; Kushima, R. RET-rearranged non-small-cell lung carcinoma: A clinicopathological and molecular analysis. Br. J. Cancer 2014, 110, 1571–1578. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, A.; Kohno, T.; Tsuta, K.; Wakai, S.; Arai, Y.; Shimada, Y.; Asamura, H.; Furuta, K.; Shibata, T.; Tsuda, H. ROS1-rearranged lung cancer: A clinicopathologic and molecular study of 15 surgical cases. Am. J. Surg. Pathol. 2013, 37, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, A.; Tsuta, K.; Nakamura, H.; Kohno, T.; Takahashi, F.; Asamura, H.; Sekine, I.; Fukayama, M.; Shibata, T.; Furuta, K.; et al. Comprehensive histologic analysis of ALK-rearranged lung carcinomas. Am. J. Surg. Pathol. 2011, 35, 1226–1234. [Google Scholar] [CrossRef] [PubMed]
- Bunting, S.F.; Nussenzweig, A. End-joining, translocations and cancer. Nat. Rev. Cancer 2013, 13, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Kelly, J.C.; Meloni-Ehrig, A.; Slovak, M.L.; Boles, D.; Christacos, N.C.; Bryke, C.R.; Schonberg, S.A.; Otani-Rosa, J.; Pan, Q.; et al. Incidence and patterns of ALK FISH abnormalities seen in a large unselected series of lung carcinomas. Mol. Cytogenet. 2012, 5. [Google Scholar] [CrossRef]
- Yoshida, A.; Tsuta, K.; Nitta, H.; Hatanaka, Y.; Asamura, H.; Sekine, I.; Grogan, T.M.; Fukayama, M.; Shibata, T.; Furuta, K.; et al. Bright-field dual-color chromogenic in situ hybridization for diagnosing echinoderm microtubule-associated protein-like 4-anaplastic lymphoma kinase-positive lung adenocarcinomas. J. Thorac. Oncol. 2011, 6, 1677–1686. [Google Scholar] [CrossRef] [PubMed]
- Ghezraoui, H.; Piganeau, M.; Renouf, B.; Renaud, J.B.; Sallmyr, A.; Ruis, B.; Oh, S.; Tomkinson, A.E.; Hendrickson, E.A.; Giovannangeli, C.; et al. Chromosomal translocations in human cells are generated by canonical nonhomologous end-joining. Mol. Cell 2014, 55, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Gillert, E.; Leis, T.; Repp, R.; Reichel, M.; Hosch, A.; Breitenlohner, I.; Angermüller, S.; Borkhardt, A.; Harbott, J.; Lampert, F.; et al. A DNA damage repair mechanism is involved in the origin of chromosomal translocations t(4;11) in primary leukemic cells. Oncogene 1999, 18, 4663–4671. [Google Scholar] [CrossRef]
- Langer, T.; Metzler, M.; Reinhardt, D.; Viehmann, S.; Borkhardt, A.; Reichel, M.; Stanulla, M.; Schrappe, M.; Creutzig, U.; Ritter, J.; et al. Analysis of t(9;11) chromosomal breakpoint sequences in childhood acute leukemia: Almost identical MLL breakpoints in therapy-related AML after treatment without etoposides. Genes Chromosom. Cancer 2003, 36, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Zucman-Rossi, J.; Legoix, P.; Victor, J.M.; Lopez, B.; Thomas, G. Chromosome translocation based on illegitimate recombination in human tumors. Proc. Natl. Acad. Sci. USA 1998, 95, 11786–11791. [Google Scholar] [CrossRef] [PubMed]
- Mattarucchi, E.; Guerini, V.; Rambaldi, A.; Campiotti, L.; Venco, A.; Pasquali, F.; Lo Curto, F.; Porta, G. Microhomologies and interspersed repeat elements at genomic breakpoints in chronic myeloid leukemia. Genes Chromosom. Cancer 2008, 47, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T. Pathways for mitotic homologous recombination in mammalian cells. Mutat. Res. 2003, 532, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Eshtad, S.; Nik-Zainal, S. Mechanisms underlying mutational signatures in human cancers. Nat. Rev. Genet 2014, 15, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Lawson, A.R.; Hindley, G.F.; Forshew, T.; Tatevossian, R.G.; Jamie, G.A.; Kelly, G.P.; Neale, G.A.; Ma, J.; Jones, T.A.; Ellison, D.W.; et al. RAF gene fusion breakpoints in pediatric brain tumors are characterized by significant enrichment of sequence microhomology. Genome Res. 2011, 21, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Dillon, L.W.; Pierce, L.C.; Lehman, C.E.; Nikiforov, Y.E.; Wang, Y.H. DNA Topoisomerases Participate in Fragility of the Oncogene RET. PLoS ONE 2013, 8, e75741. [Google Scholar] [CrossRef] [Green Version]
- Gorgoulis, V.G.; Vassiliou, L.V.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; Ditullio, R.A., Jr.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.F.; Lawrence, M.S.; Demichelis, F.; Drier, Y.; Cibulskis, K.; Sivachenko, A.Y.; Sboner, A.; Esgueva, R.; Pflueger, D.; Sougnez, C.; et al. The genomic complexity of primary human prostate cancer. Nature 2011, 470, 214–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seki, Y.; Mizukami, T.; Kohno, T. Molecular Process Producing Oncogene Fusion in Lung Cancer Cells by Illegitimate Repair of DNA Double-Strand Breaks. Biomolecules 2015, 5, 2464-2476. https://0-doi-org.brum.beds.ac.uk/10.3390/biom5042464
Seki Y, Mizukami T, Kohno T. Molecular Process Producing Oncogene Fusion in Lung Cancer Cells by Illegitimate Repair of DNA Double-Strand Breaks. Biomolecules. 2015; 5(4):2464-2476. https://0-doi-org.brum.beds.ac.uk/10.3390/biom5042464
Chicago/Turabian StyleSeki, Yoshitaka, Tatsuji Mizukami, and Takashi Kohno. 2015. "Molecular Process Producing Oncogene Fusion in Lung Cancer Cells by Illegitimate Repair of DNA Double-Strand Breaks" Biomolecules 5, no. 4: 2464-2476. https://0-doi-org.brum.beds.ac.uk/10.3390/biom5042464