
Different Roles of Mitochondria in Cell Death and Inflammation: Focusing on Mitochondrial Quality Control in Ischemic Stroke and Reperfusion


 , , and
, , and 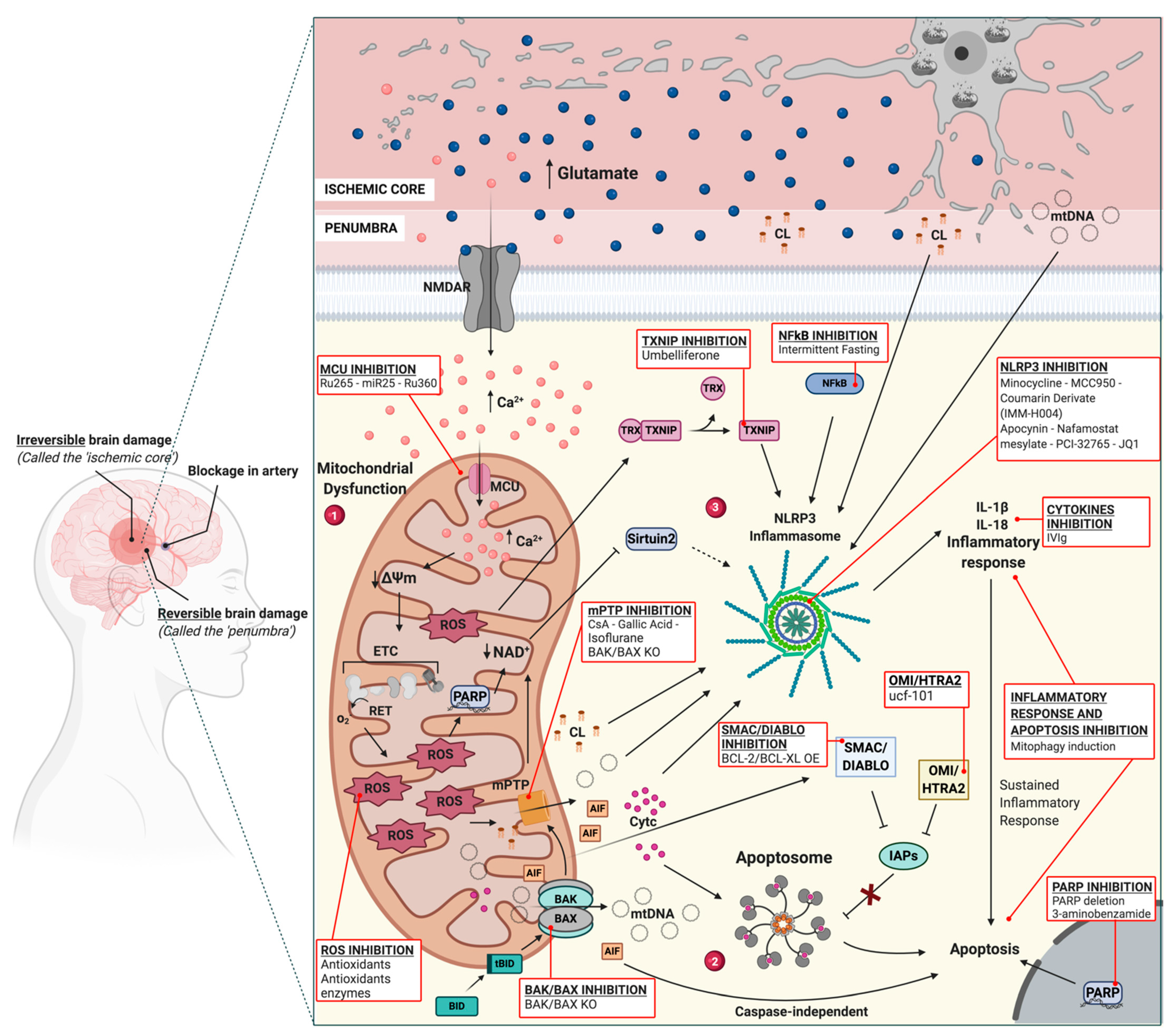{kind=link}
{kind=link}
Abstract
:1. Introduction
2. From OGD to Cell Death and Inflammation: Targeting Mitochondrial Molecular Mechanisms
2.1. Mitochondria, OGD and Ca2+ Release in IS
2.2. Mitochondria and Cell Death in IS
2.3. Mitochondria and ROS Production in IS
2.4. Mitochondria and Inflammation during I/R
3. Mito-Recovery: Regaining Mitochondrial Homeostasis in IS and I/R
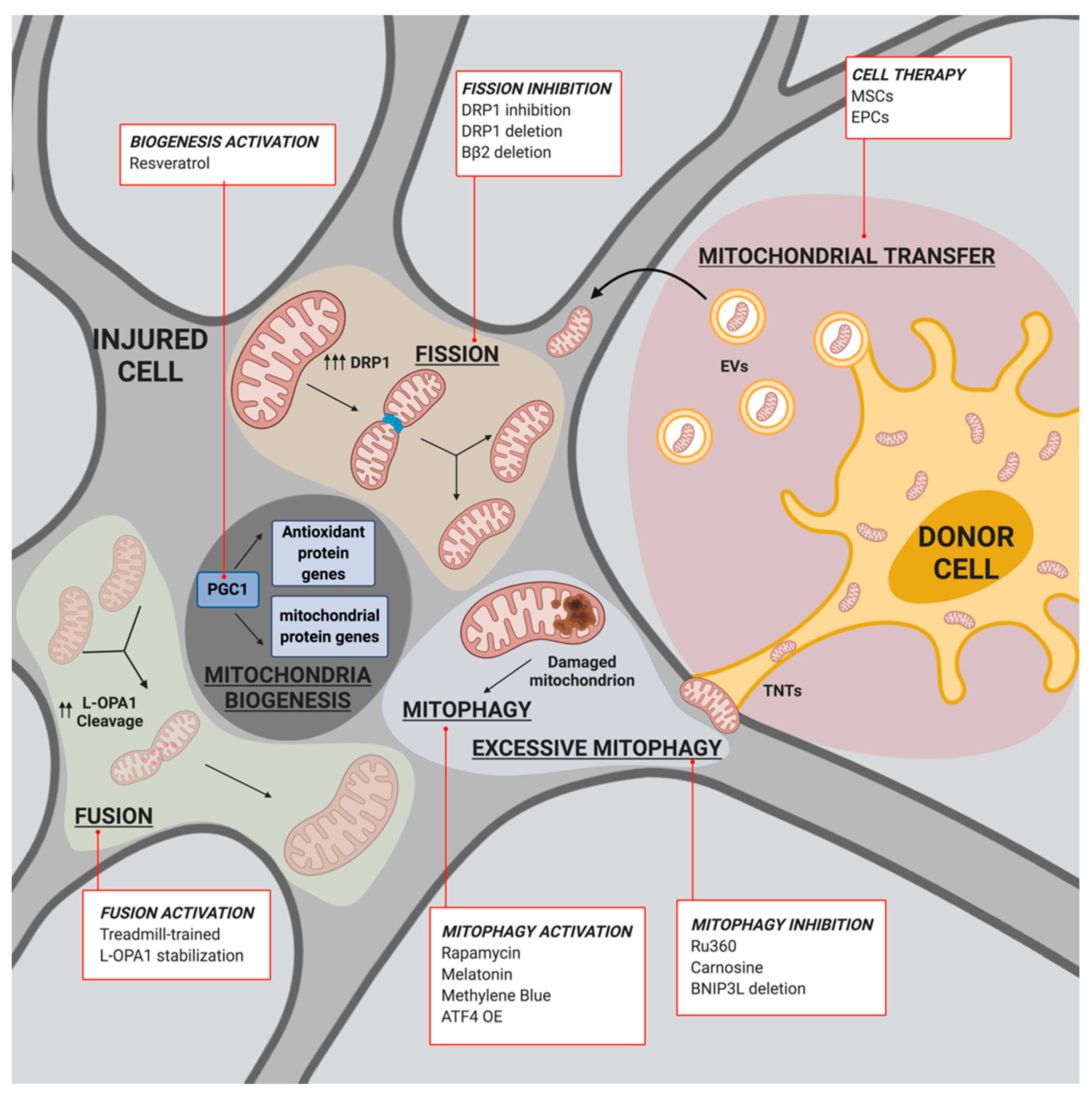
3.1. Mitochondrial Biogenesis and IS
3.2. Mitochondrial Fusion/Fission and IS
3.3. Mitophagy and IS
3.4. Mitochondrial Transfer and IS
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| APAF-1 | apoptotic protease activating factor 1 |
| ATF4 | activating transcription factor 4 |
| bmMSCs | bone marrow derived-MSCs |
| CaMKIα | calcium/calmodulin-dependent protein kinase Iα |
| CDK1 | cyclin dependent kinase 1 |
| CL | cardiolipin |
| CNS | central nervous system |
| Cyt c | cytochrome c |
| DRP1 | dynamin-related protein 1 |
| EPCs | endothelial progenitor cells |
| ERK | extracellular signal-regulated-kinase |
| EVs | extracellular vesicles |
| FIS1 | fission factor 1 |
| FKBP8 | FK506-binding protein 8 |
| FUNCD1 | FUN14 Domain Containing 1 |
| i-AAA | intermembrane space ATPase |
| I/R | ischemic reperfusion |
| IL-18 | interleukin-18 |
| IL-1β | Interleukin-1β |
| IMM | inner mitochondrial membrane |
| IMS | mitochondrial intermembrane space |
| IS | ischemic stroke |
| L-OPA1 | long optic atrophy 1 |
| LCI | local cooling infusion |
| m-AAA | matrix ATPases |
| MAMs | mitochondria associated membranes |
| MCU | mitochondria calcium uniporter |
| MFF | mitochondrial fission factor |
| MFN1 | mitofusin 1 |
| MFN2 | mitofusin 2 |
| MID49 | mitochondrial dynamics protein of 49 KDa |
| MID51 | mitochondrial dynamics protein of 51 KDa |
| mPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| MSCs | mesenchymal stem cells |
| mtDNA | mitochondrial DNA |
| NBR1 | neighbor of Brca1 |
| NDP52 | Nuclear Dot Protein 52 |
| NIX/BNIP3L | NIP3-Like Protein X/BCL2 Interacting Protein 3 Like |
| NLRP3 | nucleotide-binding domain and leucine-rich repeat containing protein 3 |
| NMDA | N-methyl-D-aspartate |
| NRF-1 | Nuclear Respiratory Factor 1 |
| NRF-2 | Nuclear Respiratory Factor 2 |
| OGD | oxygen and glucose deprivation |
| OMA1 | overlapping activity with m-AAA protease |
| OMM | outer mitochondrial membrane |
| OPA1 | optic atrophy 1 |
| OPTN | optineurin |
| OXPHOS | oxidative phosphorylation |
| p62/SQSTM1 | p62/sequestosome 1 |
| PARL | presenilin-associated rhomboid-like |
| PGC-1 | peroxisome proliferator-activated receptor-γ coactivator 1 (known as PPAR-Gamma Coactivator 1) |
| PINK1 | PTEN-induced kinase 1 |
| PKA | cyclic AMP-dependent protein kinase |
| PKCδ | protein kinase C delta |
| ROCK1 | Rho-associated coiled coil-containing protein kinase 1 |
| ROS | reactive oxygen species |
| S-OPA1 | short optic atrophy 1 |
| SIRT1 | Sirtuin 1 |
| SOD2 | superoxide dismutase 2 |
| TAX1BP1 | Tax1 Binding Protein 1 |
| TBK1 | TANK Binding Kinase 1 |
| tMCAO | transient middle cerebral artery occlusion |
| TNFAIP2 | TNF Alpha Induced Protein 2 |
| TNTs | named tunneling nanotubes |
| tPA | tissue plasminogen |
| UCP2 | uncoupling protein 2 |
| Ψm | mitochondrial membrane potential |
References
- Adams, H.P.; Bendixen, B.H.; Kappelle, L.J.; Biller, J.; Love, B.B.; Gordon, D.L.; Marsh, E.E. Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. TOAST. Trial of Org 10172 in Acute Stroke Treatment. Stroke 1993, 24, 35–41. [Google Scholar] [CrossRef] [Green Version]
- Zafar, A.; Al-Khamis, F.A.; Al-Bakr, A.I.; Alsulaiman, A.A.; Msmar, A.H. Risk factors and subtypes of acute ischemic stroke. Neurosciences 2016, 21, 246–251. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.-Y.; Murphy, B.D.; Aviv, R.I.; Fox, A.J.; Black, S.E.; Sahlas, D.J.; Symons, S.; Lee, D.H.; Pelz, D.; Gulka, I.B.; et al. Cerebral Blood Flow Threshold of Ischemic Penumbra and Infarct Core in Acute Ischemic Stroke: A Systematic Review. Stroke 2006, 37, 2201. [Google Scholar] [CrossRef] [Green Version]
- Gomez, C.R. Time Is Brain: The Stroke Theory of Relativity. J. Stroke Cerebrovasc. Dis. 2018, 27, 2214–2227. [Google Scholar] [CrossRef]
- Murphy, B.D.; Fox, A.J.; Lee, D.H.; Sahlas, D.J.; Black, S.E.; Hogan, M.J.; Coutts, S.B.; Demchuk, A.; Goyal, M.; Aviv, R.; et al. White Matter Thresholds for Ischemic Penumbra and Infarct Core in Patients with Acute Stroke: CT Perfusion Study 1. Radiology 2008, 247, 818–825. [Google Scholar] [CrossRef] [PubMed]
- Bandera, E.; Botteri, M.; Minelli, C.; Sutton, A.; Abrams, K.R.; Latronico, N. Cerebral blood flow threshold of ischemic penumbra and infarct core in acute ischemic stroke: A systematic review. Stroke 2006, 37, 1334–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Institute of Neurological Disorders; Stroke rt-PA Stroke Study Group. Tissue plasminogen activator for acute ischemic stroke. N. Engl. J. Med. 1995, 333, 1581–1587. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Campbell, B.C.; Parsons, M.W.; Churilov, L.; Levi, C.R.; Hsu, C.; Kleinig, T.J.; Wijeratne, T.; Curtze, S.; Dewey, H.M.; et al. Thrombolysis Guided by Perfusion Imaging up to 9 Hours after Onset of Stroke. N. Engl. J. Med. 2019, 380, 1795–1803. [Google Scholar] [CrossRef] [PubMed]
- Berkhemer, O.A.; Fransen, P.S.S.; Beumer, D.; Berg, L.A.V.D.; Lingsma, H.F.; Yoo, A.J.; Schonewille, W.J.; Vos, J.A.; Nederkoorn, P.J.; Wermer, M.J.H.; et al. A Randomized Trial of Intraarterial Treatment for Acute Ischemic Stroke. N. Engl. J. Med. 2015, 372, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Goyal, M.; Demchuk, A.M.; Menon, B.K.; Eesa, M.; Rempel, J.L.; Thornton, J.; Roy, D.; Jovin, T.G.; Willinsky, R.A.; Sapkota, B.L.; et al. Randomized Assessment of Rapid Endovascular Treatment of Ischemic Stroke. N. Engl. J. Med. 2015, 372, 1019–1030. [Google Scholar] [CrossRef]
- Saver, J.L.; Goyal, M.; Bonafe, A.; Diener, H.-C.; Levy, E.I.; Pereira, V.M.; Albers, G.W.; Cognard, C.; Cohen, D.J.; Hacke, W.; et al. Stent-Retriever Thrombectomy after Intravenous t-PA vs. t-PA Alone in Stroke. N. Engl. J. Med. 2015, 372, 2285–2295. [Google Scholar] [CrossRef] [Green Version]
- Campbell, B.C.; Mitchell, P.J.; Kleinig, T.J.; Dewey, H.M.; Churilov, L.; Yassi, N.; Yan, B.; Dowling, R.J.; Parsons, M.W.; Oxley, T.J.; et al. Endovascular Therapy for Ischemic Stroke with Perfusion-Imaging Selection. N. Engl. J. Med. 2015, 372, 1009–1018. [Google Scholar] [CrossRef] [Green Version]
- Jovin, T.G.; Chamorro, A.; Cobo, E.; De Miquel, M.A.; Molina, C.A.; Rovira, A.; Román, L.S.; Serena, J.; Abilleira, S.; Ribó, M.; et al. Thrombectomy within 8 Hours after Symptom Onset in Ischemic Stroke. N. Engl. J. Med. 2015, 372, 2296–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyal, M.; Menon, B.K.; Van Zwam, W.H.; Dippel, D.W.; Mitchell, P.J.; Demchuk, A.M.; Dávalos, A.; Majoie, C.B.; Van Der Lugt, A.; De Miquel, M.A.; et al. Endovascular thrombectomy after large-vessel ischaemic stroke: A meta-analysis of individual patient data from five randomised trials. Lancet 2016, 387, 1723–1731. [Google Scholar] [CrossRef]
- Ragoschke-Schumm, A.; Walter, S. DAWN and DEFUSE-3 trials: Is time still important? Radiologe 2018, 58 (Suppl. S1), 20–23. [Google Scholar] [CrossRef]
- Nogueira, R.G.; Jadhav, A.P.; Haussen, D.C.; Bonafe, A.; Budzik, R.F.; Bhuva, P.; Yavagal, D.R.; Ribo, M.; Cognard, C.; Hanel, R.A.; et al. Thrombectomy 6 to 24 Hours after Stroke with a Mismatch between Deficit and Infarct. N. Engl. J. Med. 2018, 378, 11–21. [Google Scholar] [CrossRef]
- Kamal, N.; Majmundar, N.; Damadora, N.; El-Ghanem, M.; Nuoman, R.; Keller, I.A.; Schonfeld, S.; Rybinnik, I.; Gupta, G.; Roychowdry, S.; et al. Mechanical thrombectomy—Is time still brain? The DAWN of a new era. Br. J. Neurosurg. 2018, 32, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Albers, G.W.; Marks, M.P.; Kemp, S.; Christensen, S.; Tsai, J.P.; Ortega-Gutierrez, S.; McTaggart, R.A.; Torbey, M.T.; Kim-Tenser, M.; Leslie-Mazwi, T.; et al. Thrombectomy for Stroke at 6 to 16 Hours with Selection by Perfusion Imaging. N. Engl. J. Med. 2018, 378, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, A.P.; Desai, S.M.; Kenmuir, C.L.; Rocha, M.; Starr, M.T.; Molyneaux, B.J.; Gross, B.A.; Jankowitz, B.T.; Jovin, T.G. Eligibility for Endovascular Trial Enrollment in the 6- to 24-Hour Time Window: Analysis of a Single Comprehensive Stroke Center. Stroke 2018, 49, 1015–1017. [Google Scholar] [CrossRef] [PubMed]
- Gauberti, M.; Lapergue, B.; Martinez de Lizarrondo, S.; Vivien, D.; Richard, S.; Bracard, S.; Piotin, M.; Gory, B. Ischemia-Reperfusion Injury After Endovascular Thrombectomy for Ischemic Stroke. Stroke 2018, 49, 3071–3074. [Google Scholar] [CrossRef] [Green Version]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell Biology of Ischemia/Reperfusion Injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.; Wang, X.; Yu, Z. Ischemia-reperfusion Injury in the Brain: Mechanisms and Potential Therapeutic Strategies. Biochem. Pharmacol. Open Access 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Andrabi, S.S.; Parvez, S.; Tabassum, H. Ischemic stroke and mitochondria: Mechanisms and targets. Protoplasma 2020, 257, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Huang, J.-L.; Manaenko, A.; Ye, Z.-H.; Sun, X.-J. Hypoxia therapy--a new hope for the treatment of mitochondrial dysfunctions. Med. Gas Res. 2016, 6, 174–176. [Google Scholar] [CrossRef] [Green Version]
- Tucker, D.; Lu, Y.; Zhang, Q. From Mitochondrial Function to Neuroprotection—An Emerging Role for Methylene Blue. Mol. Neurobiol. 2018, 55, 5137–5153. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [Green Version]
- Macdonald, R.; Barnes, K.; Hastings, C.; Mortiboys, H. Mitochondrial abnormalities in Parkinson’s disease and Alzheimer’s disease: Can mitochondria be targeted therapeutically? Biochem. Soc. Trans. 2018, 46, 891–909. [Google Scholar] [CrossRef]
- Grunewald, A.; Kumar, K.R.; Sue, C.M. New insights into the complex role of mitochondria in Parkinson’s disease. Prog. Neurobiol. 2019, 177, 73–93. [Google Scholar] [CrossRef]
- Ramos-Cejudo, J.; Wisniewski, T.; Marmar, C.; Zetterberg, H.; Blennow, K.; de Leon, M.J.; Fossati, S. Traumatic Brain Injury and Alzheimer’s Disease: The Cerebrovascular Link. EBioMedicine 2018, 28, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Vezzani, B.; Carinci, M.; Patergnani, S.; Pasquin, M.P.; Guarino, A.; Aziz, N.; Pinton, P.; Simonato, M.; Giorgi, C. The Dichotomous Role of Inflammation in the CNS: A Mitochondrial Point of View. Biomolecules 2020, 10, 1437. [Google Scholar] [CrossRef] [PubMed]
- Palade, G.E. An electron microscope study of the mitochondrial structure. J. Histochem. Cytochem. 1953, 1, 188–211. [Google Scholar] [CrossRef] [PubMed]
- Schonfeld, P.; Reiser, G. Why does brain metabolism not favor burning of fatty acids to provide energy? Reflections on disadvantages of the use of free fatty acids as fuel for brain. J. Cereb. Blood Flow Metab. 2013, 33, 1493–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatefi, Y. The mitochondrial electron transport and oxidative phosphorylation system. Annu. Rev. Biochem. 1985, 54, 1015–1069. [Google Scholar] [CrossRef] [PubMed]
- Sazanov, L.A. A giant molecular proton pump: Structure and mechanism of respiratory complex I. Nat. Rev. Mol. Cell Biol. 2015, 16, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Attwell, D.; Laughlin, S.B. An Energy Budget for Signaling in the Grey Matter of the Brain. Br. J. Pharmacol. 2001, 21, 1133–1145. [Google Scholar] [CrossRef]
- Ames, A. CNS energy metabolism as related to function. Brain Res. Rev. 2000, 34, 42–68. [Google Scholar] [CrossRef]
- Hall, C.N.; Klein-Flügge, M.C.; Howarth, C.; Attwell, D. Oxidative Phosphorylation, Not Glycolysis, Powers Presynaptic and Postsynaptic Mechanisms Underlying Brain Information Processing. J. Neurosci. 2012, 32, 8940–8951. [Google Scholar] [CrossRef]
- Mongin, A.A. Disruption of ionic and cell volume homeostasis in cerebral ischemia: The perfect storm. Pathophysiology 2007, 14, 183–193. [Google Scholar] [CrossRef] [Green Version]
- Fann, D.Y.-W.; Lee, S.-Y.; Manzanero, S.; Chunduri, P.; Sobey, C.G.; Arumugam, T.V. Pathogenesis of acute stroke and the role of inflammasomes. Ageing Res. Rev. 2013, 12, 941–966. [Google Scholar] [CrossRef]
- Yuan, J. Neuroprotective strategies targeting apoptotic and necrotic cell death for stroke. Apoptosis 2009, 14, 469–477. [Google Scholar] [CrossRef] [Green Version]
- Epstein, F.H.; Lipton, S.A.; Rosenberg, P.A. Excitatory Amino Acids as a Final Common Pathway for Neurologic Disorders. N. Engl. J. Med. 1994, 330, 613–622. [Google Scholar] [CrossRef]
- Vandenberg, R.J.; Ryan, R.M. Mechanisms of Glutamate Transport. Physiol. Rev. 2013, 93, 1621–1657. [Google Scholar] [CrossRef] [Green Version]
- Lai, T.W.; Zhang, S.; Wang, Y.T. Excitotoxicity and stroke: Identifying novel targets for neuroprotection. Prog. Neurobiol. 2014, 115, 157–188. [Google Scholar] [CrossRef] [Green Version]
- Schipke, C.G.; Ohlemeyer, C.; Matyash, M.; Nolte, C.; Kettenmann, H.; Kirchhoff, F. Astrocytes of the mouse neocortex express functional N-methyl-D-aspartate receptors. FASEB J. 2001, 15, 1270–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaindl, A.M.; Degos, V.; Peineau, S.; Gouadon, E.; Chhor, V.; Loron, G.; Le Charpentier, T.; Josserand, J.; Ali, C.; Vivien, D.; et al. Activation of microglial N-methyl-D-aspartate receptors triggers inflammation and neuronal cell death in the developing and mature brain. Ann. Neurol. 2012, 72, 536–549. [Google Scholar] [CrossRef] [PubMed]
- White, R.J.; Reynolds, I.J. Mitochondrial Depolarization in Glutamate-Stimulated Neurons: An Early Signal Specific to Excitotoxin Exposure. J. Neurosci. 1996, 16, 5688–5697. [Google Scholar] [CrossRef]
- Mody, I.; MacDonald, J.F. NMDA receptor-dependent excitotoxicity: The role of intracellular Ca2+ release. Trends Pharmacol. Sci. 1995, 16, 356–359. [Google Scholar] [CrossRef]
- Smith, J.P.; Cunningham, L.A.; Partridge, L.D. Coupling of AMPA receptors with the Na(+)/Ca(2+) exchanger in cultured rat astrocytes. Brain Res. 2000, 887, 98–109. [Google Scholar] [CrossRef]
- Demaurex, N.; Distelhorst, C. Cell biology. Apoptosis—The calcium connection. Science 2003, 300, 65–67. [Google Scholar] [CrossRef] [Green Version]
- Giorgi, C.; Danese, A.; Missiroli, S.; Patergnani, S.; Pinton, P. Calcium Dynamics as a Machine for Decoding Signals. Trends Cell Biol. 2018, 28, 258–273. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Retta, S.F.; Pinton, P. Publisher Correction: The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 746. [Google Scholar] [CrossRef]
- Patergnani, S.; Danese, A.; Bouhamida, E.; Aguiari, G.; Previati, M.; Pinton, P.; Giorgi, C. Various Aspects of Calcium Signaling in the Regulation of Apoptosis, Autophagy, Cell Proliferation, and Cancer. Int. J. Mol. Sci. 2020, 21, 8323. [Google Scholar] [CrossRef] [PubMed]
- Danese, A.; Patergnani, S.; Bonora, M.; Wieckowski, M.R.; Previati, M.; Giorgi, C.; Pinton, P. Calcium regulates cell death in cancer: Roles of the mitochondria and mitochondria-associated membranes (MAMs). Biochim. Biophys. Acta (BBA) Bioenerg. 2017, 1858, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G. A role for the mitochondrion in the protection of cells against calcium overload? Prog. Brain Res. 1985, 63, 97–106. [Google Scholar] [PubMed]
- Chinopoulos, C.; Adam-Vizi, V. Mitochondrial Ca2+ sequestration and precipitation revisited. FEBS J. 2010, 277, 3637–3651. [Google Scholar] [CrossRef]
- Liao, Y.; Dong, Y.; Cheng, J. The Function of the Mitochondrial Calcium Uniporter in Neurodegenerative Disorders. Int. J. Mol. Sci. 2017, 18, 248. [Google Scholar] [CrossRef]
- Kirichok, Y.; Krapivinsky, G.; Clapham, D.E. The mitochondrial calcium uniporter is a highly selective ion channel. Nat. Cell Biol. 2004, 427, 360–364. [Google Scholar] [CrossRef]
- Yu, N.; Wang, S.; Wang, P.; Li, Y.; Li, S.; Wang, L.; Chen, H.; Wang, Y. The calcium uniporter regulates the permeability transition pore in isolated cortical mitochondria. Neural Regen. Res. 2012, 7, 109–113. [Google Scholar]
- Morciano, G.; Bonora, M.; Campo, G.; Aquila, G.; Rizzo, P.; Giorgi, C.; Wieckowski, M.R.; Pinton, P. Mechanistic Role of mPTP in Ischemia-Reperfusion Injury. Adv. Exp. Med. Biol. 2017, 982, 169–189. [Google Scholar] [CrossRef]
- Zhao, Q.; Wang, S.; Li, Y.; Wang, P.; Li, S.; Guo, Y.; Yao, R. The role of the mitochondrial calcium uniporter in cerebral ischemia/reperfusion injury in rats involves regulation of mitochondrial energy metabolism. Mol. Med. Rep. 2013, 7, 1073–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morciano, G.; Giorgi, C.; Bonora, M.; Punzetti, S.; Pavasini, R.; Wieckowski, M.R.; Campo, G.; Pinton, P. Molecular identity of the mitochondrial permeability transition pore and its role in ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 2015, 78, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, P.G.; Rabchevsky, A.; Waldmeier, P.; Springer, J. Mitochondrial permeability transition in CNS trauma: Cause or effect of neuronal cell death? J. Neurosci. Res. 2005, 79, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Patergnani, S.; Ramaccini, D.; Morciano, G.; Pedriali, G.; Kahsay, A.E.; Bouhamida, E.; Giorgi, C.; Wieckowski, M.R.; Pinton, P. Physiopathology of the Permeability Transition Pore: Molecular Mechanisms in Human Pathology. Biomolecules 2020, 10, 998. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Huang, B.-J.; Ma, X.-E.; Wang, S.-Y.; Feng, J.; Lv, F.; Liu, Y.; Liu, Y.; Li, C.-M.; Liang, D.; et al. MiR-25 Protects Cardiomyocytes against Oxidative Damage by Targeting the Mitochondrial Calcium Uniporter. Int. J. Mol. Sci. 2015, 16, 5420–5433. [Google Scholar] [CrossRef] [Green Version]
- Novorolsky, R.J.; Nichols, M.; Kim, J.S.; Pavlov, E.V.; Woods, J.J.; Wilson, J.J.; Robertson, G.S. The cell-permeable mitochondrial calcium uniporter inhibitor Ru265 preserves cortical neuron respiration after lethal oxygen glucose deprivation and reduces hypoxic/ischemic brain injury. Br. J. Pharmacol. 2020, 40, 1172–1181. [Google Scholar] [CrossRef]
- Yao, H.; Takasawa, R.; Fukuda, K.; Shiokawa, D.; Sadanaga-Akiyoshi, F.; Ibayashi, S.; Tanuma, S.-I.; Uchimura, H. DNA fragmentation in ischemic core and penumbra in focal cerebral ischemia in rats. Mol. Brain Res. 2001, 91, 112–118. [Google Scholar] [CrossRef]
- Leist, M.; Single, B.; Castoldi, A.F.; Kühnle, S.; Nicotera, P. Intracellular Adenosine Triphosphate (ATP) Concentration: A Switch in the Decision Between Apoptosis and Necrosis. J. Exp. Med. 1997, 185, 1481–1486. [Google Scholar] [CrossRef]
- Bano, D.; Nicotera, P. Ca2+ Signals and Neuronal Death in Brain Ischemia. Stroke 2007, 38, 674–676. [Google Scholar] [CrossRef] [Green Version]
- Uzdensky, A.B. Regulation of apoptosis in the ischemic penumbra in the first day post-stroke. Neural Regen. Res. 2020, 15, 253–254. [Google Scholar] [CrossRef]
- Manabat, C.; Han, B.H.; Wendland, M.; Derugin, N.; Fox, C.K.; Choi, J.; Holtzman, D.M.; Ferriero, D.M.; Vexler, Z.S. Reperfusion Differentially Induces Caspase-3 Activation in Ischemic Core and Penumbra After Stroke in Immature Brain. Stroke 2003, 34, 207–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niizuma, K.; Yoshioka, H.; Chen, H.; Kim, G.S.; Jung, J.E.; Katsu, M.; Okami, N.; Chan, P.H. Mitochondrial and apoptotic neuronal death signaling pathways in cerebral ischemia. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2010, 1802, 92–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engel, T.; Plesnila, N.; Prehn, J.H.M.; Henshall, D.C. In vivo Contributions of BH3-Only Proteins to Neuronal Death Following Seizures, Ischemia, and Traumatic Brain Injury. Br. J. Pharmacol. 2011, 31, 1196–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karch, J.; Kwong, J.Q.; Burr, A.R.; Sargent, M.A.; Elrod, J.W.; Peixoto, P.M.; Martínez-Caballero, S.; Osinska, H.; Cheng, E.H.-Y.; Robbins, J.; et al. Bax and Bak function as the outer membrane component of the mitochondrial permeability pore in regulating necrotic cell death in mice. eLife 2013, 2, e00772. [Google Scholar] [CrossRef]
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta (BBA) Bioenerg. 2013, 1833, 3448–3459. [Google Scholar] [CrossRef] [Green Version]
- Chandra, D.; Liu, J.-W.; Tang, D.G.; Canela, N.; Rodriguez-Vilarrupla, A.; Estanyol, J.M.; Díaz, C.; Pujol, M.J.; Agell, N.; Bachs, O. Early Mitochondrial Activation and Cytochrome c Up-regulation during Apoptosis. J. Biol. Chem. 2002, 277, 50842–50854. [Google Scholar] [CrossRef] [Green Version]
- Riley, J.S.; Quarato, G.; Cloix, C.; Lopez, J.; O’Prey, J.; Pearson, M.; Chapman, J.; Sesaki, H.; Carlin, L.M.; Passos, J.F.; et al. Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Yin, X.-M.; Luo, Y.; Cao, G.; Bai, L.; Pei, W.; Kuharsky, D.K.; Chen, J. Bid-mediated Mitochondrial Pathway Is Critical to Ischemic Neuronal Apoptosis and Focal Cerebral Ischemia. J. Biol. Chem. 2002, 277, 42074–42081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Omori, N.; Sato, K.; Jin, G.; Nagano, I.; Manabe, Y.; Shoji, M.; Abe, K. Coordinate expression of survival p-ERK and proapoptotic cytochrome c signals in rat brain neurons after transient MCAO. Brain Res. 2002, 958, 83–88. [Google Scholar] [CrossRef]
- Fujimura, M.; Morita-Fujimura, Y.; Murakami, K.; Kawase, M.; Chan, P.H. Cytosolic Redistribution of Cytochrome C after Transient Focal Cerebral Ischemia in Rats. Br. J. Pharmacol. 1998, 18, 1239–1247. [Google Scholar] [CrossRef]
- Yu, S.W.; Wang, H.; Poitras, M.F.; Coombs, C.; Bowers, W.J.; Federoff, H.J.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 2002, 297, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Minami, M.; Pei, W.; Yan, C.; Chen, D.; O’Horo, C.; Graham, S.H.; Chen, J. Intracellular Bax Translocation after Transient Cerebral Ischemia: Implications for a Role of the Mitochondrial Apoptotic Signaling Pathway in Ischemic Neuronal Death. Br. J. Pharmacol. 2001, 21, 321–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Culmsee, C.; Zhu, C.; Landshamer, S.; Becattini, B.; Wagner, E.; Pellecchia, M.; Blomgren, K.; Plesnila, N. Apoptosis-Inducing Factor Triggered by Poly(ADP-Ribose) Polymerase and Bid Mediates Neuronal Cell Death after Oxygen-Glucose Deprivation and Focal Cerebral Ischemia. J. Neurosci. 2005, 25, 10262–10272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schinzel, A.C.; Takeuchi, O.; Huang, Z.; Fisher, J.K.; Zhou, Z.; Rubens, J.; Hetz, C.; Danial, N.N.; Moskowitz, M.A.; Korsmeyer, S.J. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc. Natl. Acad. Sci. USA 2005, 102, 12005–12010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, T.-H.; Aguettaz, P.; Campuzano, O.; Charriaut-Marlangue, C.; Riou, A.; Berthezène, Y.; Nighoghossian, N.; Ovize, M.; Wiart, M.; Chauveau, F. Pre- and Post-treatment with Cyclosporine a in a Rat Model of Transient Focal Cerebral Ischaemia with Multimodal MRI Screening. Int. J. Stroke 2012, 8, 669–674. [Google Scholar] [CrossRef]
- Matsumoto, S.; Friberg, H.; Ferrand-Drake, M.; Wieloch, T. Blockade of the mitochondrial permeability transition pore diminishes infarct size in the rat after transient middle cerebral artery occlusion. J. Cereb. Blood Flow Metab. 1999, 19, 736–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Ren, D.D.; Wan, J.Y.; Chen, C.; Chen, D.; Yang, H.; Feng, C.L.; Gao, J. Desensitizing Mitochondrial Permeability Transition by ERK-Cyclophilin D Axis Contributes to the Neuroprotective Effect of Gallic Acid against Cerebral Ischemia/Reperfusion Injury. Front. Pharmacol. 2017, 8, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, M.; Yang, Q.; Li, G.; Sun, S.; Chen, Y.; Tian, L.; Dong, H. Activation of cannabinoid receptor 1 is involved in protection against mitochondrial dysfunction and cerebral ischaemic tolerance induced by isoflurane preconditioning. Br. J. Anaesth. 2017, 119, 1213–1223. [Google Scholar] [CrossRef] [Green Version]
- Bonora, M.; Morganti, C.; Morciano, G.; Pedriali, G.; Lebiedzinska-Arciszewska, M.; Aquila, G.; Giorgi, C.; Rizzo, P.; Campo, G.; Ferrari, R.; et al. Mitochondrial permeability transition involves dissociation of F1FO ATP synthase dimers and C-ring conformation. EMBO Rep. 2017, 18, 1077–1089. [Google Scholar] [CrossRef]
- Li, Y.; Sun, J.; Wu, R.; Bai, J.; Hou, Y.; Zeng, Y.; Zhang, Y.; Wang, X.; Wang, Z.; Meng, X. Mitochondrial MPTP: A Novel Target of Ethnomedicine for Stroke Treatment by Apoptosis Inhibition. Front. Pharmacol. 2020, 11, 352. [Google Scholar] [CrossRef]
- Broughton, B.R.; Reutens, D.C.; Sobey, C.G. Apoptotic mechanisms after cerebral ischemia. Stroke 2009, 40, e331–e339. [Google Scholar] [CrossRef] [Green Version]
- Culmsee, C.; Landshamer, S. Molecular insights into mechanisms of the cell death program: Role in the progression of neurodegenerative disorders. Curr. Alzheimer Res. 2006, 3, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.C.; Snyder, S.H. Poly(ADP-ribose) polymerase-1 in the nervous system. Neurobiol. Dis. 2000, 7, 225–239. [Google Scholar] [CrossRef] [Green Version]
- Eliasson, M.J.; Sampei, K.; Mandir, A.S.; Hurn, P.D.; Traystman, R.J.; Bao, J.; Pieper, A.; Wang, Z.Q.; Dawson, T.M.; Snyder, S.H.; et al. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat. Med. 1997, 3, 1089–1095. [Google Scholar] [CrossRef]
- Lo, E.H.; Bosque-Hamilton, P.; Meng, W. Inhibition of poly(ADP-ribose) polymerase: Reduction of ischemic injury and attenuation of N-methyl-D-aspartate-induced neurotransmitter dysregulation. Stroke 1998, 29, 830–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Morselli, E.; Kepp, O.; Kroemer, G. Targeting post-mitochondrial effectors of apoptosis for neuroprotection. Biochim. Biophys. Acta (BBA) Bioenerg. 2009, 1787, 402–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasula, S.M.; Hegde, R.; Saleh, A.; Datta, P.; Shiozaki, E.N.; Chai, J.; Lee, R.-A.; Robbins, P.D.; Fernandes-Alnemri, T.; Shi, Y.; et al. A conserved XIAP-interaction motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis. Nat. Cell Biol. 2001, 410, 112–116. [Google Scholar] [CrossRef]
- Althaus, J.; Siegelin, M.; Dehghani, F.; Cilenti, L.; Zervos, A.; Rami, A. The serine protease Omi/HtrA2 is involved in XIAP cleavage and in neuronal cell death following focal cerebral ischemia/reperfusion. Neurochem. Int. 2007, 50, 172–180. [Google Scholar] [CrossRef]
- Saito, A.; Hayashi, T.; Okuno, S.; Ferrand-Drake, M.; Chan, P.H. Interaction between XIAP and Smac/DIABLO in the Mouse Brain after Transient Focal Cerebral Ischemia. Br. J. Pharmacol. 2003, 23, 1010–1019. [Google Scholar] [CrossRef] [Green Version]
- Uzdensky, A.B. Apoptosis regulation in the penumbra after ischemic stroke: Expression of pro- and antiapoptotic proteins. Apoptosis 2019, 24, 687–702. [Google Scholar] [CrossRef]
- Ferrer, I.; Planas, A.M. Signaling of Cell Death and Cell Survival Following Focal Cerebral Ischemia: Life and Death Struggle in the Penumbra. J. Neuropathol. Exp. Neurol. 2003, 62, 329–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adrain, C.; Creagh, E.M.; Martin, S.J. Apoptosis-associated release of Smac/DIABLO from mitochondria requires active caspases and is blocked by Bcl-2. EMBO J. 2001, 20, 6627–6636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.M.; Bratton, S.B.; Butterworth, M.; MacFarlane, M.; Cohen, G.M. Bcl-2 and Bcl-xL inhibit CD95-mediated apoptosis by preventing mitochondrial release of Smac/DIABLO and subsequent inactivation of X-linked inhibitor-of-apoptosis protein. J. Biol. Chem. 2002, 277, 11345–11351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linnik, M.D.; Zahos, P.; Geschwind, M.D.; Federoff, H.J. Expression of bcl-2 From a Defective Herpes Simplex Virus–1 Vector Limits Neuronal Death in Focal Cerebral Ischemia. Stroke 1995, 26, 1670–1675. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Yenari, M.A.; Cheng, D.; Barreto-Chang, O.L.; Sapolsky, R.M.; Steinberg, G.K. Bcl-2 Transfection via Herpes Simplex Virus Blocks Apoptosis-Inducing Factor Translocation after Focal Ischemia in the Rat. Br. J. Pharmacol. 2004, 24, 681–692. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Xu, Y.; Geng, L. Caspase-3 inhibitor prevents the apoptosis of brain tissue in rats with acute cerebral infarction. Exp. Ther. Med. 2015, 10, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Colbourne, F.; Sun, P.; Zhao, Z.; Buchan, A.M. Caspase Inhibitors Reduce Neuronal Injury After Focal but Not Global Cerebral Ischemia in Rats. Stroke 2000, 31, 176–182. [Google Scholar] [CrossRef] [Green Version]
- Loor, G.; Kondapalli, J.; Iwase, H.; Chandel, N.S.; Waypa, G.B.; Guzy, R.D.; Vanden Hoek, T.L.; Schumacker, P.T. Mito-chondrial oxidant stress triggers cell death in simulated ischemia-reperfusion. Biochim Biophys Acta 2011, 1813, 1382–1394. [Google Scholar] [CrossRef] [Green Version]
- Sanderson, T.H.; Reynolds, C.A.; Kumar, R.; Przyklenk, K.; Hüttemann, M. Molecular Mechanisms of Ischemia–Reperfusion Injury in Brain: Pivotal Role of the Mitochondrial Membrane Potential in Reactive Oxygen Species Generation. Mol. Neurobiol. 2013, 47, 9–23. [Google Scholar] [CrossRef] [Green Version]
- Cruz, C.M.; Rinna, A.; Forman, H.J.; Ventura, A.L.; Persechini, P.M.; Ojcius, D.M. ATP activates a reactive oxygen spe-cies-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J. Biol. Chem. 2007, 282, 2871–2879. [Google Scholar]
- Muller, F.L.; Liu, Y.; Van Remmen, H. Complex III Releases Superoxide to Both Sides of the Inner Mitochondrial Membrane. J. Biol. Chem. 2004, 279, 49064–49073. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Kussmaul, L.; Hirst, J. The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc. Natl. Acad. Sci. USA 2006, 103, 7607–7612. [Google Scholar] [CrossRef] [Green Version]
- Bleier, L.; Dröse, S. Superoxide generation by complex III: From mechanistic rationales to functional consequences. Biochim. Biophys. Acta (BBA) Bioenerg. 2013, 1827, 1320–1331. [Google Scholar] [CrossRef] [Green Version]
- Prag, H.A.; Gruszczyk, A.V.; Huang, M.M.; Beach, T.E.; Young, T.; Tronci, L.; Nikitopoulou, E.; Mulvey, J.F.; Ascione, R.; Hadjihambi, A.; et al. Mechanism of succinate efflux upon reperfusion of the ischemic heart. Cardiovasc. Res. 2020. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; James, A.M.; Work, L.M.; Saeb-Parsy, K.; Frezza, C.; Krieg, T.; Murphy, M.P. A Unifying Mechanism for Mitochondrial Superoxide Production during Ischemia-Reperfusion Injury. Cell Metab. 2016, 23, 254–263. [Google Scholar] [CrossRef] [Green Version]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Kishida, K.T.; Klann, E. Sources and Targets of Reactive Oxygen Species in Synaptic Plasticity and Memory. Antioxidants Redox Signal. 2007, 9, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Gasparovic, A.C.; Žarković, N.; Zarkovic, K.; Semen, K.; Kaminskyy, D.; Yelisyeyevab, O.; Bottari, S.P. Biomarkers of oxidative and nitro-oxidative stress: Conventional and novel approaches. Br. J. Pharmacol. 2017, 174, 1771–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, R.; Tresini, M. Oxidative stress and gene regulation. Free. Radic. Biol. Med. 2000, 28, 463–499. [Google Scholar] [CrossRef]
- Chan, P.H. Oxygen Radicals in Focal Cerebral Ischemia. Brain Pathol. 2008, 4, 59–65. [Google Scholar] [CrossRef]
- Garcia-Estrada, J.; Gonzalez-Perez, O.; Gonzalez-Castaneda, R.E.; Martinez-Contreras, A.; Luquin, S.; De La Mora, P.G.; Navarro-Ruiz, A. An alpha-lipoic acid-vitamin E mixture reduces post-embolism lipid peroxidation, cerebral infarction, and neurological deficit in rats. Neurosci. Res. 2003, 47, 219–224. [Google Scholar] [CrossRef]
- Mishima, K.; Tanaka, T.; Pu, F.; Egashira, N.; Iwasaki, K.; Hidaka, R.; Matsunaga, K.; Takata, J.; Karube, Y.; Fujiwara, M. Vitamin E isoforms alpha-tocotrienol and gamma-tocopherol prevent cerebral infarction in mice. Neurosci. Lett. 2003, 337, 56–60. [Google Scholar] [CrossRef]
- Miller, E.R., 3rd; Pastor-Barriuso, R.; Dalal, D.; Riemersma, R.A.; Appel, L.J.; Guallar, E. Meta-analysis: High-dosage vitamin E supplementation may increase all-cause mortality. Ann. Intern. Med. 2005, 142, 37–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullegaddi, R.; Powers, H.J.; Gariballa, S. Antioxidant supplementation enhances antioxidant capacity and mitigates oxidative damage following acute ischaemic stroke. Eur. J. Clin. Nutr. 2005, 59, 1367–1373. [Google Scholar] [CrossRef]
- Ducruet, A.F.; Mack, W.J.; Mocco, J.; Hoh, D.J.; Coon, A.L.; D’Ambrosio, A.L.; Winfree, C.J.; Pinsky, D.J.; ConnollyJr, E.S. Preclinical Evaluation of Postischemic Dehydroascorbic Acid Administration in a Large-Animal Stroke Model. Transl. Stroke Res. 2011, 2, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Agus, D.B.; Winfree, C.J.; Kiss, S.; Mack, W.J.; McTaggart, R.A.; Choudhri, T.F.; Kim, L.J.; Mocco, J.; Pinsky, D.J.; et al. Dehydroascorbic acid, a blood-brain barrier transportable form of vitamin C, mediates potent cerebroprotection in experimental stroke. Proc. Natl. Acad. Sci. USA 2001, 98, 11720–11724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, N.; Yanaka, K.; Nagase, S.; Hirayama, A.; Nose, T. The antioxidant EPC-K1 ameliorates brain injury by inhibiting lipid peroxidation in a rat model of transient focal cerebral ischaemia. Acta Neurochir. 2003, 145, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.M.; Pennypacker, K.R. Targeting antioxidant enzyme expression as a therapeutic strategy for ischemic stroke. Neurochem. Int. 2017, 107, 23–32. [Google Scholar] [CrossRef]
- Matsumoto, S.; Murozono, M.; Kanazawa, M.; Nara, T.; Ozawa, T.; Watanabe, Y. Edaravone and cyclosporine A as neuroprotective agents for acute ischemic stroke. Acute Med. Surg. 2018, 5, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.-S.; Jin, H.; Sun, X.; Huang, S.; Zhang, F.-L.; Guo, Z.-N.; Yang, Y. Free Radical Damage in Ischemia-Reperfusion Injury: An Obstacle in Acute Ischemic Stroke after Revascularization Therapy. Oxidative Med. Cell. Longev. 2018, 2018, 1–17. [Google Scholar] [CrossRef]
- Uemura, T.; Watanabe, K.; Ko, K.; Higashi, K.; Kogure, N.; Kitajima, M.; Takayama, H.; Takao, K.; Sugita, Y.; Sakamoto, A.; et al. Protective Effects of Brain Infarction by N -Acetylcysteine Derivatives. Stroke 2018, 49, 1727–1733. [Google Scholar] [CrossRef]
- Gong, Z.; Pan, J.; Shen, Q.; Li, M.; Peng, Y. Mitochondrial dysfunction induces NLRP3 inflammasome activation during cerebral ischemia/reperfusion injury. J. Neuroinflamm. 2018, 15, 1–17. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Gurung, P.; Lukens, J.R.; Kanneganti, T.-D. Mitochondria: Diversity in the regulation of the NLRP3 inflammasome. Trends Mol. Med. 2015, 21, 193–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alishahi, M.; Farzaneh, M.; Ghaedrahmati, F.; Nejabatdoust, A.; Sarkaki, A.; Khoshnam, S.E. NLRP3 inflammasome in ischemic stroke: As possible therapeutic target. Int. J. Stroke 2019, 14, 574–591. [Google Scholar] [CrossRef] [PubMed]
- Lammerding, L.; Slowik, A.; Johann, S.; Beyer, C.; Zendedel, A. Poststroke Inflammasome Expression and Regulation in the Peri-Infarct Area by Gonadal Steroids after Transient Focal Ischemia in the Rat Brain. Neuroendocrinology 2016, 103, 460–475. [Google Scholar] [CrossRef] [PubMed]
- Fann, D.Y.; Lee, S.Y.; Manzanero, S.; Tang, S.C.; Gelderblom, M.; Chunduri, P.; Bernreuther, C.; Glatzel, M.; Cheng, Y.L.; Thundyil, J.; et al. Intravenous immunoglobulin suppresses NLRP1 and NLRP3 inflam-masome-mediated neuronal death in ischemic stroke. Cell Death Dis. 2013, 4, e790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ismael, S.; Zhao, L.; Nasoohi, S.; Ishrat, T. Inhibition of the NLRP3-inflammasome as a potential approach for neuropro-tection after stroke. Sci. Rep. 2018, 8, 5971. [Google Scholar] [CrossRef] [Green Version]
- Su, S.-H.; Wu, Y.-F.; Lin, Q.; Wang, D.-P.; Hai, J. URB597 protects against NLRP3 inflammasome activation by inhibiting autophagy dysfunction in a rat model of chronic cerebral hypoperfusion. J. Neuroinflamm. 2019, 16, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Mo, Z.; Tang, C.; Li, H.; Lei, J.; Zhu, L.; Kou, L.; Li, H.; Luo, S.; Li, C.; Chen, W.; et al. Eicosapentaenoic acid prevents inflammation induced by acute cerebral infarction through inhibition of NLRP3 inflammasome activation. Life Sci. 2020, 242, 117133. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Ning, N.; Zhou, Q.; Khoshnam, S.E.; Farzaneh, M. Mitochondria as a therapeutic target for ischemic stroke. Free. Radic. Biol. Med. 2020, 146, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Di Lisa, F.; Menabò, R.; Canton, M.; Barile, M.; Bernardi, P. Opening of the Mitochondrial Permeability Transition Pore Causes Depletion of Mitochondrial and Cytosolic NAD+and Is a Causative Event in the Death of Myocytes in Postischemic Reperfusion of the Heart. J. Biol. Chem. 2001, 276, 2571–2575. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.; Chen, Y.; Watkins, S.C.; Nathaniel, P.D.; Guo, F.; Kochanek, P.M.; Jenkins, L.W.; Szabo, C.; Clark, R.S. Identifi-cation of poly-ADP-ribosylated mitochondrial proteins after traumatic brain injury. J. Neurochem. 2008, 104, 1700–1711. [Google Scholar] [CrossRef]
- Owens, K.; Park, J.H.; Schuh, R.A.; Kristian, T. Mitochondrial Dysfunction and NAD+ Metabolism Alterations in the Pathophysiology of Acute Brain Injury. Transl. Stroke Res. 2013, 4, 618–634. [Google Scholar] [CrossRef]
- Misawa, T.; Takahama, M.; Kozaki, T.; Lee, H.; Zou, J.; Saitoh, T.; Akira, S. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immunol. 2013, 14, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Heid, M.E.; Keyel, P.A.; Kamga, C.; Shiva, S.; Watkins, S.C.; Salter, R.D. Mitochondrial Reactive Oxygen Species Induces NLRP3-Dependent Lysosomal Damage and Inflammasome Activation. J. Immunol. 2013, 191, 5230–5238. [Google Scholar] [CrossRef] [Green Version]
- Formentini, L.; Sanchez-Arago, M.; Sanchez-Cenizo, L.; Cuezva, J.M. The mitochondrial ATPase inhibitory factor 1 trig-gers a ROS-mediated retrograde prosurvival and proliferative response. Mol. Cell 2012, 45, 731–742. [Google Scholar] [CrossRef] [Green Version]
- Fann, D.Y.; Lim, Y.A.; Cheng, Y.L.; Lok, K.Z.; Chunduri, P.; Baik, S.H.; Drummond, G.R.; Dheen, S.T.; Sobey, C.G.; Jo, D.G.; et al. Evidence that NF-kappaB and MAPK Signaling Promotes NLRP Inflammasome Activation in Neurons Following Ischemic Stroke. Mol. Neurobiol. 2018, 55, 1082–1096. [Google Scholar] [CrossRef]
- Liang, X.; Zhang, D.; Liu, W.; Yan, Y.; Zhou, F.; Wu, W.; Yan, Z. Reactive oxygen species trigger NF-kappaB-mediated NLRP3 inflammasome activation induced by zinc oxide nanoparticles in A549 cells. Toxicol. Ind. Health 2017, 33, 737–745. [Google Scholar] [CrossRef]
- Fann, D.Y.-W.; Santro, T.; Manzanero, S.; Widiapradja, A.; Cheng, Y.-L.; Lee, S.-Y.; Chunduri, P.; Jo, D.-G.; Stranahan, A.M.; Mattson, M.P.; et al. Intermittent fasting attenuates inflammasome activity in ischemic stroke. Exp. Neurol. 2014, 257, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, J.; Li, S.; Li, Y.; Wang, X.; Liu, B.; Fu, Q.; Ma, S.-P. Curcumin attenuates glutamate neurotoxicity in the hippocampus by suppression of ER stress-associated TXNIP/NLRP3 inflammasome activation in a manner dependent on AMPK. Toxicol. Appl. Pharmacol. 2015, 286, 53–63. [Google Scholar] [CrossRef]
- Wang, X.; Li, R.; Wang, X.; Fu, Q.; Ma, S.-P. Umbelliferone ameliorates cerebral ischemia–reperfusion injury via upregulating the PPAR gamma expression and suppressing TXNIP/NLRP3 inflammasome. Neurosci. Lett. 2015, 600, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Ishrat, T.; Mohamed, I.N.; Pillai, B.; Soliman, S.; Fouda, A.Y.; Ergul, A.; El-Remessy, A.B.; Fagan, S.C. Thioredox-in-interacting protein: A novel target for neuroprotection in experimental thromboembolic stroke in mice. Mol. Neurobiol. 2015, 51, 766–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, G.; Jiang, N.; Hu, Y.; Zhang, Y.; Wang, G.; Yin, M.; Ma, X.; Zhou, K.; Qi, J.; Yu, B.; et al. Ruscogenin Attenuates Cerebral Ischemia-Induced Blood-Brain Barrier Dysfunction by Suppressing TXNIP/NLRP3 Inflammasome Activation and the MAPK Pathway. Int. J. Mol. Sci. 2016, 17, 1418. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Zuo, D.; Yu, L.; Zhang, L.; Tang, J.; Cui, C.; Bao, L.; Zan, K.; Zhang, Z.; Yang, X.; et al. ROS/TXNIP pathway contributes to thrombin induced NLRP3 inflammasome activation and cell apoptosis in microglia. Biochem. Biophys. Res. Commun. 2017, 485, 499–505. [Google Scholar] [CrossRef]
- McArthur, K.; Whitehead, L.W.; Heddleston, J.M.; Li, L.; Padman, B.S.; Oorschot, V.; Geoghegan, N.D.; Chappaz, S.; Davidson, S.; Chin, H.S.; et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 2018, 359, eaao6047. [Google Scholar] [CrossRef] [Green Version]
- Krysko, D.V.; Agostinis, P.; Krysko, O.; Garg, A.D.; Bachert, C.; Lambrecht, B.N.; Vandenabeele, P. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 2011, 32, 157–164. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Kroemer, G. Mitochondria: Master regulators of danger signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 780–788. [Google Scholar] [CrossRef]
- Grazioli, S.; Pugin, J. Mitochondrial Damage-Associated Molecular Patterns: From Inflammatory Signaling to Human Diseases. Front. Immunol. 2018, 9, 832. [Google Scholar] [CrossRef]
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial Cardiolipin Is Required for Nlrp3 Inflammasome Activation. Immunity 2013, 39, 311–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelfand, E.W. Intravenous Immune Globulin in Autoimmune and Inflammatory Diseases. N. Engl. J. Med. 2012, 367, 2015–2025. [Google Scholar] [CrossRef] [PubMed]
- Guarda, G.; Braun, M.; Staehli, F.; Tardivel, A.; Mattmann, C.; Förster, I.; Farlik, M.; Decker, T.; Du Pasquier, R.A.; Romero, P.; et al. Type I Interferon Inhibits Interleukin-1 Production and Inflammasome Activation. Immunity 2011, 34, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Kuo, P.C.; Scofield, B.A.; Yu, I.C.; Chang, F.L.; Ganea, D.; Yen, J.H. Interferon-beta Modulates Inflammatory Response in Cerebral Ischemia. J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Xiao, G.; Luo, W. Minocycline Suppresses NLRP3 Inflammasome Activation in Experimental Ischemic Stroke. Neuroimmunomodulation 2016, 23, 230–238. [Google Scholar] [CrossRef]
- Naderi, Y.; Sabetkasaei, M.; Parvardeh, S.; Zanjani, T.M. Neuroprotective effect of minocycline on cognitive impairments induced by transient cerebral ischemia/reperfusion through its anti-inflammatory and anti-oxidant properties in male rat. Brain Res. Bull. 2017, 131, 207–213. [Google Scholar] [CrossRef]
- Tanaka, M.; Ishihara, Y.; Mizuno, S.; Ishida, A.; Vogel, C.F.; Tsuji, M.; Yamazaki, T.; Itoh, K. Progression of vasogenic edema induced by activated microglia under permanent middle cerebral artery occlusion. Biochem. Biophys. Res. Commun. 2018, 496, 582–587. [Google Scholar] [CrossRef]
- Yew, W.P.; Djukic, N.D.; Jayaseelan, J.S.P.; Walker, F.R.; Roos, K.A.A.; Chataway, T.K.; Muyderman, H.; Sims, N.R. Early treatment with minocycline following stroke in rats improves functional recovery and differentially modifies responses of peri-infarct microglia and astrocytes. J. Neuroinflamm. 2019, 16, 6. [Google Scholar] [CrossRef] [PubMed]
- Amiri-Nikpour, M.R.; Nazarbaghi, S.; Hamdi-Holasou, M.; Rezaei, Y. An open-label evaluator-blinded clinical study of minocycline neuroprotection in ischemic stroke: Gender-dependent effect. Acta Neurol. Scand. 2015, 131, 45–50. [Google Scholar] [CrossRef]
- Srivastava, M.P.; Bhasin, A.; Bhatia, R.; Garg, A.; Gaikwad, S.; Prasad, K.; Singh, M.; Tripathi, M. Efficacy of minocycline in acute ischemic stroke: A single-blinded, placebo-controlled trial. Neurol. India 2012, 60, 23–28. [Google Scholar] [CrossRef]
- Ai, Q.-D.; Chen, C.; Chu, S.; Zhang, Z.; Luo, Y.; Guan, F.-F.; Lin, M.-Y.; Liu, D.; Wang, S.-S.; Chen, N.-H. IMM-H004 therapy for permanent focal ischemic cerebral injury via CKLF1/CCR4-mediated NLRP3 inflammasome activation. Transl. Res. 2019, 212, 36–53. [Google Scholar] [CrossRef]
- Hong, P.; Li, F.-X.; Gu, R.-N.; Fang, Y.-Y.; Lai, L.-Y.; Wang, Y.-W.; Tao, T.; Xu, S.; You, Z.-J.; Zhang, H.-F. Inhibition of NLRP3 Inflammasome Ameliorates Cerebral Ischemia-Reperfusion Injury in Diabetic Mice. Neural Plast. 2018, 2018, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Zhong, D.; Chen, H.; Jin, J.; Liu, Q.; Li, G. NLRP3 inflammasome activates interleukin-23/interleukin-17 axis during ischaemia-reperfusion injury in cerebral ischaemia in mice. Life Sci. 2019, 227, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Shen, T.; Hu, J.; Zhang, L.; Zhang, Y.; Bao, L.; Cui, C.; Jin, G.; Zan, K.; Zhang, Z.; et al. Purinergic 2X7 receptor/NLRP3 pathway triggers neuronal apoptosis after ischemic stroke in the mouse. Exp. Neurol. 2017, 292, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Shichita, T.; Okada, M.; Komine, R.; Noguchi, Y.; Yoshimura, A.; Morita, R. Bruton’s tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat. Commun. 2015, 6, 7360. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, J.; Fang, Y.; Liu, Y.; Chen, T.; Sun, H.; Zhou, X.-F.; Liao, H. Nafamostat mesilate improves function recovery after stroke by inhibiting neuroinflammation in rats. Brain Behav. Immun. 2016, 56, 230–245. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.-Y.; Li, M.; Feng, X.; Wang, J.; Cao, L.; Shen, X.-K.; Chen, J.; Sun, M.; Sheng, R.; Han, F.; et al. Combined NADPH and the NOX inhibitor apocynin provides greater anti-inflammatory and neuroprotective effects in a mouse model of stroke. Free. Radic. Biol. Med. 2017, 104, 333–345. [Google Scholar] [CrossRef]
- Zhou, Y.; Gu, Y.; Liu, J. BRD4 suppression alleviates cerebral ischemia-induced brain injury by blocking glial activation via the inhibition of inflammatory response and pyroptosis. Biochem. Biophys. Res. Commun. 2019, 519, 481–488. [Google Scholar] [CrossRef]
- Chamorro, A.; Hallenbeck, J. The Harms and Benefits of Inflammatory and Immune Responses in Vascular Disease. Stroke 2006, 37, 291–293. [Google Scholar] [CrossRef]
- Chan, D.C. Mitochondrial Dynamics and Its Involvement in Disease. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 235–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta (BBA) Bioenerg. 2011, 1813, 1269–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarpulla, R.C. Nuclear control of respiratory gene expression in mammalian cells. J. Cell. Biochem. 2006, 97, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Schon, E.A.; Manfredi, G. Neuronal degeneration and mitochondrial dysfunction. J. Clin. Investig. 2003, 111, 303–312. [Google Scholar] [CrossRef] [Green Version]
- Austin, S.; St-Pierre, J. PGC1alpha and mitochondrial metabolism--emerging concepts and relevance in ageing and neu-rodegenerative disorders. J. Cell Sci. 2012, 125, 4963–4971. [Google Scholar] [CrossRef] [Green Version]
- Bost, F.; Kaminski, L. The metabolic modulator PGC-1α in cancer. Am. J. Cancer Res. 2019, 9, 198–211. [Google Scholar]
- Chen, S.-D.; Yang, D.-I.; Lin, T.-K.; Shaw, F.-Z.; Liou, C.-W.; Chuang, Y.-C. Roles of Oxidative Stress, Apoptosis, PGC-1α and Mitochondrial Biogenesis in Cerebral Ischemia. Int. J. Mol. Sci. 2011, 12, 7199–7215. [Google Scholar] [CrossRef] [Green Version]
- Yin, W.; Signore, A.P.; Iwai, M.; Cao, G.; Gao, Y.; Chen, J. Rapidly Increased Neuronal Mitochondrial Biogenesis After Hypoxic-Ischemic Brain Injury. Stroke 2008, 39, 3057–3063. [Google Scholar] [CrossRef] [PubMed]
- Stetler, R.A.; Leak, R.K.; Yin, W.; Zhang, L.; Wang, S.; Gao, Y.; Chen, J. Mitochondrial biogenesis contributes to ischemic neuroprotection afforded by LPS pre-conditioning. J. Neurochem. 2012, 123, 125–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.D.; Lin, T.K.; Lin, J.W.; Yang, D.I.; Lee, S.Y.; Shaw, F.Z.; Liou, C.W.; Chuang, Y.C. Activation of calci-um/calmodulin-dependent protein kinase IV and peroxisome proliferator-activated receptor gamma coactivator-1alpha signaling pathway protects against neuronal injury and promotes mitochondrial biogenesis in the hippocampal CA1 sub-field after transient global ischemia. J. Neurosci. Res. 2010, 88, 3144–3154. [Google Scholar]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jäger, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of Reactive Oxygen Species and Neurodegeneration by the PGC-1 Transcriptional Coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef] [Green Version]
- Della-Morte, D.; Dave, K.R.; DeFazio, R.A.; Bao, Y.C.; Raval, A.P.; Perez-Pinzon, M.A. Resveratrol pretreatment protects rat brain from cerebral ischemic damage via a sirtuin 1–uncoupling protein 2 pathway. Neuroscience 2009, 159, 993–1002. [Google Scholar] [CrossRef] [Green Version]
- Santel, A.; Fuller, M.T. Control of mitochondrial morphology by a human mitofusin. J. Cell Sci. 2001, 114, 867–874. [Google Scholar]
- Cipolat, S.; De Brito, O.M.; Zilio, B.D.; Scorrano, L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef] [Green Version]
- Zuchner, S.; Mersiyanova, I.V.; Muglia, M.; Bissar-Tadmouri, N.; Rochelle, J.; Dadali, E.L.; Zappia, M.; Nelis, E.; Patitucci, A.; Senderek, J.; et al. Mutations in the mitochondrial GTPase mi-tofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004, 36, 449–451. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Chen, H.; Fiket, M.; Alexander, C.; Chan, D.C. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J. Cell Biol. 2007, 178, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Duvezin-Caubet, S.; Jagasia, R.; Wagener, J.; Hofmann, S.; Trifunovic, A.; Hansson, A.; Chomyn, A.; Bauer, M.F.; Attardi, G.; Larsson, N.-G.; et al. Proteolytic Processing of OPA1 Links Mitochondrial Dysfunction to Alterations in Mitochondrial Morphology. J. Biol. Chem. 2006, 281, 37972–37979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Y.; Lin, P.; Chen, M.; Zhang, Y.; Chen, J.; Zheng, M.; Liu, J.; Du, H.; Chen, R.; Pan, X.; et al. Restoration of L-OPA1 alleviates acute ischemic stroke injury in rats via inhibiting neuronal apoptosis and preserving mitochondrial function. Redox Biol. 2020, 34, 101503. [Google Scholar] [CrossRef]
- Lee, C.D.; Folsom, A.R.; Blair, S.N. Physical activity and stroke risk: A meta-analysis. Stroke 2003, 34, 2475–2481. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; He, Z.; Zhang, Q.; Wu, Y.; Yang, X.; Niu, W.; Hu, Y.; Jia, J. Exercise Pretreatment Promotes Mitochondrial Dynamic Protein OPA1 Expression after Cerebral Ischemia in Rats. Int. J. Mol. Sci. 2014, 15, 4453–4463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, C.; Rao, W.; Zhang, L.; Gao, F.; Hui, H.; Wang, K.; Dai, S.; Yang, Y.; Luo, P.; Ma, Y.; et al. Mitofusin 2 Exerts a Protective Role in Ischemia Reperfusion Injury Through Increasing Autophagy. Cell. Physiol. Biochem. 2018, 46, 2311–2324. [Google Scholar] [CrossRef] [Green Version]
- Chung, K.W.; Cho, S.Y.; Hwang, S.J.; Kim, K.H.; Yoo, J.H.; Kwon, O.; Kim, S.M.; Sunwoo, I.N.; Zuchner, S.; Choi, B.O. Early-onset stroke associated with a mutation in mitofusin 2. Neurology 2008, 70, 2010–2011. [Google Scholar] [CrossRef]
- Smirnova, E.; Griparic, L.; Shurland, D.-L.; Van Der Bliek, A.M. Dynamin-related Protein Drp1 Is Required for Mitochondrial Division in Mammalian Cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, D.C. Fusion and Fission: Interlinked Processes Critical for Mitochondrial Health. Annu. Rev. Genet. 2012, 46, 265–287. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.; Disatnik, M.H.; Shen, N.; Sobel, R.A.; Mochly-Rosen, D. Aberrant mitochondrial fission in neurons induced by protein kinase C{delta} under oxidative stress conditions in vivo. Mol. Biol. Cell 2011, 22, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic phosphorylation of dynamin-related GTPase Drp1 par-ticipates in mitochondrial fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Jhun, B.S.; Yoon, Y. High-Glucose Stimulation Increases Reactive Oxygen Species Production Through the Calcium and Mitogen-Activated Protein Kinase-Mediated Activation of Mitochondrial Fission. Antioxidants Redox Signal. 2011, 14, 425–437. [Google Scholar] [CrossRef] [Green Version]
- Bustillo-Zabalbeitia, I.; Montessuit, S.; Raemy, E.; Basanez, G.; Terrones, O.; Martinou, J.C. Specific interaction with car-diolipin triggers functional activation of Dynamin-Related Protein 1. PLoS ONE 2014, 9, e102738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, W.; Wang, J.; Van Houten, B. The role of dynamin-related protein 1 in cancer growth: A promising therapeutic target? Expert Opin. Ther. Targets 2013, 17, 997–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lima, A.R.; Santos, L.; Correia, M.; Soares, P.; Sobrinho-Simões, M.; Melo, M.; Máximo, V. Dynamin-Related Protein 1 at the Crossroads of Cancer. Genes 2018, 9, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, D.H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S.A. S-nitrosylation of Drp1 mediates be-ta-amyloid-related mitochondrial fission and neuronal injury. Science 2009, 324, 102–105. [Google Scholar] [CrossRef] [Green Version]
- Grohm, J.; Kim, S.-W.; Mamrak, U.; Tobaben, S.; Cassidy-Stone, A.; Nunnari, J.; Plesnila, N.; Culmsee, C. Inhibition of Drp1 provides neuroprotection in vitro and in vivo. Cell Death Differ. 2012, 19, 1446–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.-X.; Cui, M.; Chen, S.-F.; Dong, Q.; Liu, X.-Y. Amelioration of Ischemic Mitochondrial Injury and Bax-Dependent Outer Membrane Permeabilization by Mdivi-1. CNS Neurosci. Ther. 2014, 20, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Bordt, E.A.; Clerc, P.; Roelofs, B.A.; Saladino, A.J.; Tretter, L.; Adam-Vizi, V.; Cherok, E.; Khalil, A.; Yadava, N.; Ge, S.X.; et al. The Putative Drp1 Inhibitor mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species. Dev. Cell 2017, 40, 583–594.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flippo, K.H.; Lin, Z.; Dickey, A.S.; Zhou, X.; Dhanesha, N.A.; Walters, G.C.; Liu, Y.; Merrill, R.A.; Meller, R.; Simon, R.P.; et al. Deletion of a Neuronal Drp1 Activator Protects against Cerebral Ischemia. J. Neurosci. 2020, 40, 3119–3129. [Google Scholar] [CrossRef]
- Duan, C.; Kuang, L.; Xiang, X.; Zhang, J.; Zhu, Y.; Wu, Y.; Yan, Q.; Liu, L.; Li, T. Drp1 regulates mitochondrial dysfunction and dysregulated metabolism in ischemic injury via Clec16a-, BAX-, and GSH- pathways. Cell Death Dis. 2020, 11, 251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Tian, F.; Kurata, T.; Morimoto, N.; Abe, K. Dynamic changes of mitochondrial fusion and fission proteins after transient cerebral ischemia in mice. J. Neurosci. Res. 2012, 90, 1183–1189. [Google Scholar] [CrossRef]
- Owens, K.; Park, J.H.; Gourley, S.; Jones, H.; Kristian, T. Mitochondrial dynamics: Cell-type and hippocampal region specific changes following global cerebral ischemia. J. Bioenerg. Biomembr. 2015, 47, 13–31. [Google Scholar] [CrossRef] [PubMed]
- Kislin, M.; Sword, J.; Fomitcheva, I.V.; Croom, D.; Pryazhnikov, E.; Lihavainen, E.; Toptunov, D.; Rauvala, H.; Ribeiro, A.S.; Khiroug, L.; et al. Reversible Disruption of Neuronal Mitochondria by Ischemic and Traumatic Injury Revealed by Quantitative Two-Photon Imaging in the Neocortex of Anesthetized Mice. J. Neurosci. 2017, 37, 333–348. [Google Scholar] [CrossRef] [Green Version]
- Patergnani, S.; Pinton, P. Mitophagy and Mitochondrial Balance. Toxic. Assess. 2015, 1241, 181–194. [Google Scholar] [CrossRef]
- Marinković, M.; Šprung, M.; Novak, I. Dimerization of mitophagy receptor BNIP3L/NIX is essential for recruitment of autophagic machinery. Autophagy 2020, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nature 2012, 14, 177–185. [Google Scholar] [CrossRef]
- Murakawa, T.; Yamaguchi, O.; Hashimoto, A.; Hikoso, S.; Takeda, T.; Oka, T.; Yasui, H.; Ueda, H.; Akazawa, Y.; Naka-yama, H.; et al. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmenta-tion. Nat. Commun. 2015, 6, 7527. [Google Scholar] [CrossRef] [Green Version]
- Bhujabal, Z.; Birgisdottir, Å.B.; Sjøttem, E.; Brenne, H.B.; Øvervatn, A.; Habisov, S.; Kirkin, V.; Lamark, T.; Johansen, T. FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep. 2017, 18, 947–961. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okatsu, K.; Oka, T.; Iguchi, M.; Imamura, K.; Kosako, H.; Tani, N.; Kimura, M.; Go, E.; Koyano, F.; Funayama, M.; et al. PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat. Commun. 2012, 3, 1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012, 2, 120080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Yan, H.; Yuan, Y.; Gao, J.; Shen, Z.; Cheng, Y.; Shen, Y.; Wang, R.-R.; Wang, X.; Hu, W.-W.; et al. Cerebral ischemia-reperfusion-induced autophagy protects against neuronal injury by mitochondrial clearance. Autophagy 2013, 9, 1321–1333. [Google Scholar] [CrossRef] [Green Version]
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patergnani, S.; Castellazzi, M.; Bonora, M.; Marchi, S.; Casetta, I.; Pugliatti, M.; Giorgi, C.; Granieri, E.; Pinton, P. Autophagy and mitophagy elements are increased in body fluids of multiple sclerosis-affected individuals. J. Neurol. Neurosurg. Psychiatry 2018, 89, 439–441. [Google Scholar] [CrossRef] [PubMed]
- Moskal, N.; Riccio, V.; Bashkurov, M.; Taddese, R.; Datti, A.; Lewis, P.N.; Angus McQuibban, G. ROCK inhibitors up-regulate the neuroprotective Parkin-mediated mitophagy pathway. Nat. Commun. 2020, 11, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardini, J.P.; Lazarou, M.; Dewson, G. Parkin and mitophagy in cancer. Oncogene 2017, 36, 1315–1327. [Google Scholar] [CrossRef]
- Morciano, G.; Patergnani, S.; Bonora, M.; Pedriali, G.; Tarocco, A.; Bouhamida, E.; Marchi, S.; Ancora, G.; Anania, G.; Wieckowski, M.R.; et al. Mitophagy in Cardiovascular Diseases. J. Clin. Med. 2020, 9, 892. [Google Scholar] [CrossRef] [Green Version]
- Guan, R.; Zou, W.; Dai, X.; Yu, X.; Liu, H.; Chen, Q.; Teng, W. Mitophagy, a potential therapeutic target for stroke. J. Biomed. Sci. 2018, 25, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Zhang, T.; Wang, J.; Zhang, Z.; Zhai, Y.; Yang, G.-Y.; Sun, X. Rapamycin attenuates mitochondrial dysfunction via activation of mitophagy in experimental ischemic stroke. Biochem. Biophys. Res. Commun. 2014, 444, 182–188. [Google Scholar] [CrossRef]
- Cao, S.; Shrestha, S.; Li, J.; Yu, X.; Chen, J.; Yan, F.; Ying, G.; Gu, C.; Wang, L.; Chen, G. Melatonin-mediated mitophagy protects against early brain injury after subarachnoid hemorrhage through inhibition of NLRP3 inflammasome activation. Sci. Rep. 2017, 7, 2417. [Google Scholar] [CrossRef] [Green Version]
- Di, Y.; He, Y.-L.; Zhao, T.; Huang, X.; Wu, K.-W.; Liu, S.-H.; Zhao, Y.-Q.; Fan, M.; Wu, L.-Y.; Zhu, L. Methylene Blue Reduces Acute Cerebral Ischemic Injury via the Induction of Mitophagy. Mol. Med. 2015, 21, 420–429. [Google Scholar] [CrossRef]
- He, Q.; Li, Z.; Meng, C.; Wu, J.; Zhao, Y.; Zhao, J. Parkin-Dependent Mitophagy is Required for the Inhibition of ATF4 on NLRP3 Inflammasome Activation in Cerebral Ischemia-Reperfusion Injury in Rats. Cells 2019, 8, 897. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Ma, X.; Yu, W.; Lou, Z.; Mu, D.; Wang, Y.; Shen, B.; Qi, S. Reperfusion Promotes Mitochondrial Dysfunction following Focal Cerebral Ischemia in Rats. PLoS ONE 2012, 7, e46498. [Google Scholar] [CrossRef]
- Yuan, Y.; Zheng, Y.; Zhang, X.; Chen, Y.; Wu, X.; Wu, J.; Shen, Z.; Jiang, L.; Wang, L.; Yang, Y.; et al. BNIP3L/NIX-mediated mitophagy protects against ischemic brain injury independent of PARK2. Autophagy 2017, 13, 1754–1766. [Google Scholar] [CrossRef]
- Shi, R.-Y.; Zhu, S.-H.; Li, V.; Gibson, S.B.; Xu, X.-S.; Kong, J. BNIP3 Interacting with LC3 Triggers Excessive Mitophagy in Delayed Neuronal Death in Stroke. CNS Neurosci. Ther. 2014, 20, 1045–1055. [Google Scholar] [CrossRef]
- Yu, S.; Zheng, S.; Leng, J.; Wang, S.; Zhao, T.; Liu, J. Inhibition of mitochondrial calcium uniporter protects neurocytes from ischemia/reperfusion injury via the inhibition of excessive mitophagy. Neurosci. Lett. 2016, 628, 24–29. [Google Scholar] [CrossRef]
- Baek, S.H.; Noh, A.R.; Kim, K.A.; Akram, M.; Shin, Y.J.; Kim, E.S.; Yu, S.W.; Majid, A.; Bae, O.N. Modulation of mito-chondrial function and autophagy mediates carnosine neuroprotection against ischemic brain damage. Stroke 2014, 45, 2438–2443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rustom, A.; Saffrich, R.; Markovic, I.; Walther, P.; Gerdes, H.-H. Nanotubular Highways for Intercellular Organelle Transport. Science 2004, 303, 1007–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, K.; Shi, X.; Zhang, X.; Dang, S.; Ma, X.; Liu, F.; Xu, M.; Lv, Z.; Han, D.; Fang, X.; et al. Long-distance intercellular connectivity between cardiomyocytes and cardiofibroblasts mediated by membrane nanotubes. Cardiovasc. Res. 2011, 92, 39–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Önfelt, B.; Nedvetzki, S.; Benninger, R.K.P.; Purbhoo, M.A.; Sowinski, S.; Hume, A.N.; Seabra, M.C.; Neil, M.A.A.; French, P.M.W.; Davis, D.M. Structurally Distinct Membrane Nanotubes between Human Macrophages Support Long-Distance Vesicular Traffic or Surfing of Bacteria. J. Immunol. 2006, 177, 8476–8483. [Google Scholar] [CrossRef] [Green Version]
- Koyanagi, M.; Brandes, R.P.; Haendeler, J.; Zeiher, A.M.; Dimmeler, S. Cell-to-cell connection of endothelial progenitor cells with cardiac myocytes by nanotubes: A novel mechanism for cell fate changes? Circ. Res. 2005, 96, 1039–1041. [Google Scholar] [CrossRef] [Green Version]
- Mathivanan, S.; Ji, H.; Simpson, R.J. Exosomes: Extracellular organelles important in intercellular communication. J. Proteom. 2010, 73, 1907–1920. [Google Scholar] [CrossRef]
- Arion, D.; Fletcher, R.S.; Borkow, G.; Camarasa, M.J.; Balzarini, J.; Dmitrienko, G.I.; Parniak, M.A. Differences in the in-hibition of human immunodeficiency virus type 1 reverse transcriptase DNA polymerase activity by analogs of nevirapine and [2′,5′-bis-O-(tert-butyldimethylsilyl)-3′-spiro-5″-(4″-amino-1″, 2″-oxathiole-2″,2″-dioxide] (TSAO). Mol. Pharmacol. 1996, 50, 1057–1064. [Google Scholar] [PubMed]
- Sinclair, K.A.; Yerkovich, S.T.; Hopkins, P.M.; Chambers, D.C. Characterization of intercellular communication and mi-tochondrial donation by mesenchymal stromal cells derived from the human lung. Stem Cell Res. Ther. 2016, 7, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plotnikov, E.Y.; Khryapenkova, T.G.; Vasileva, A.K.; Marey, M.V.; Galkina, S.I.; Isaev, N.K.; Sheval, E.V.; Polyakov, V.Y.; Sukhikh, G.T.; Zorov, D.B. Cell-to-cell cross-talk between mesenchymal stem cells and cardiomyocytes in co-culture. J. Cell. Mol. Med. 2008, 12, 1622–1631. [Google Scholar] [CrossRef] [PubMed]
- Paliwal, S.; Chaudhuri, R.; Agrawal, A.; Mohanty, S. Regenerative abilities of mesenchymal stem cells through mito-chondrial transfer. J. Biomed. Sci. 2018, 25, 31. [Google Scholar] [CrossRef]
- Davis, C.-H.O.; Kim, K.-Y.; Bushong, E.A.; Mills, E.A.; Boassa, D.; Shih, T.; Kinebuchi, M.; Phan, S.; Zhou, Y.; Bihlmeyer, N.A.; et al. Transcellular degradation of axonal mitochondria. Proc. Natl. Acad. Sci. USA 2014, 111, 9633–9638. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from as-trocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.N.; Das, S.R.; Emin, M.T.; Wei, M.; Sun, L.; Westphalen, K.; Rowlands, D.J.; Quadri, S.K.; Bhattacharya, S.; Bhattacharya, J. Mitochondrial transfer from bone-marrow–derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 2012, 18, 759–765. [Google Scholar] [CrossRef] [Green Version]
- Moschoi, R.; Imbert, V.; Nebout, M.; Chiche, J.; Mary, D.; Prebet, T.; Saland, E.; Castellano, R.; Pouyet, L.; Collette, Y.; et al. Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood 2016, 128, 253–264. [Google Scholar] [CrossRef] [Green Version]
- Aksoy, P.; White, T.A.; Thompson, M.; Chini, E.N. Regulation of intracellular levels of NAD: A novel role for CD38. Biochem. Biophys. Res. Commun. 2006, 345, 1386–1392. [Google Scholar] [CrossRef]
- Voloboueva, L.A.; Suh, S.W.; Swanson, R.A.; Giffard, R.G. Inhibition of mitochondrial function in astrocytes: Implications for neuroprotection. J. Neurochem. 2007, 102, 1383–1394. [Google Scholar] [CrossRef]
- Bruzzone, S.; Verderio, C.; Schenk, U.; Fedele, E.; Zocchi, E.; Matteoli, M.; De Flora, A. Glutamate-mediated overexpression of CD38 in astrocytes cultured with neurones. J. Neurochem. 2004, 89, 264–272. [Google Scholar] [CrossRef]
- Huang, P.-J.; Kuo, C.-C.; Lee, H.-C.; Shen, C.-I.; Cheng, F.-C.; Wu, S.-F.; Chang, J.-C.; Pan, H.-C.; Lin, S.-Z.; Liu, C.-S.; et al. Transferring Xenogenic Mitochondria Provides Neural Protection against Ischemic Stress in Ischemic Rat Brains. Cell Transplant. 2016, 25, 913–927. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Ma, Z.; Yan, C.; Pu, K.; Wu, M.; Bai, J.; Li, Y.; Wang, Q. Muscle-derived autologous mitochondrial transplan-tation: A novel strategy for treating cerebral ischemic injury. Behav. Brain Res. 2019, 356, 322–331. [Google Scholar] [CrossRef]
- Barry, F.P.; Murphy, J.M.; English, K.; Mahon, B.P. Immunogenicity of Adult Mesenchymal Stem Cells: Lessons from the Fetal Allograft. Stem Cells Dev. 2005, 14, 252–265. [Google Scholar] [CrossRef]
- Dezawa, M.; Kanno, H.; Hoshino, M.; Cho, H.; Matsumoto, N.; Itokazu, Y.; Tajima, N.; Yamada, H.; Sawada, H.; Ishikawa, H.; et al. Specific induction of neuronal cells from bone marrow stromal cells and application for autologous transplantation. J. Clin. Investig. 2004, 113, 1701–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vezzani, B.; Pierantozzi, E.; Sorrentino, V. Mesenchymal stem cells: From the perivascular environment to clinical appli-cations. Histol. Histopathol. 2018, 33, 1235–1246. [Google Scholar] [PubMed]
- Chen, J.; Li, Y.; Wang, L.; Zhang, Z.; Lu, D.; Lu, M.; Chopp, M. Therapeutic Benefit of Intravenous Administration of Bone Marrow Stromal Cells After Cerebral Ischemia in Rats. Stroke 2001, 32, 1005–1011. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Zhang, Z.G.; Li, Y.; Wang, L.; Xu, Y.X.; Gautam, S.C.; Lu, M.; Zhu, Z.; Chopp, M. Intravenous Administration of Human Bone Marrow Stromal Cells Induces Angiogenesis in the Ischemic Boundary Zone After Stroke in Rats. Circ. Res. 2003, 92, 692–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurozumi, K.; Nakamura, K.; Tamiya, T.; Kawano, Y.; Ishii, K.; Kobune, M.; Hirai, S.; Uchida, H.; Sasaki, K.; Ito, Y.; et al. Mesenchymal stem cells that produce neurotrophic factors reduce is-chemic damage in the rat middle cerebral artery occlusion model. Mol. Ther. 2005, 11, 96–104. [Google Scholar] [CrossRef]
- Zhao, M.Z.; Nonoguchi, N.; Ikeda, N.; Watanabe, T.; Furutama, D.; Miyazawa, D.; Funakoshi, H.; Kajimoto, Y.; Nakamura, T.; Dezawa, M.; et al. Novel therapeutic strategy for stroke in rats by bone marrow stromal cells and ex vivo HGF gene transfer with HSV-1 vector. J. Cereb. Blood Flow Metab. 2006, 26, 1176–1188. [Google Scholar] [CrossRef] [Green Version]
- Iihoshi, S.; Honmou, O.; Houkin, K.; Hashi, K.; Kocsis, J.D. A therapeutic window for intravenous administration of au-tologous bone marrow after cerebral ischemia in adult rats. Brain Res. 2004, 1007, 1–9. [Google Scholar] [CrossRef]
- Shimada, I.S.; Spees, J.L. Stem and progenitor cells for neurological repair: Minor issues, major hurdles, and exciting opportunities for paracrine-based therapeutics. J. Cell. Biochem. 2011, 112, 374–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mombo, B.N.; Gerbal-Chaloin, S.; Bokus, A.; Daujat-Chavanieu, M.; Jorgensen, C.; Hugnot, J.-P.; Vignais, M.-L. MitoCeption: Transferring Isolated Human MSC Mitochondria to Glioblastoma Stem Cells. J. Vis. Exp. 2017, e55245. [Google Scholar] [CrossRef]
- Boukelmoune, N.; Chiu, G.S.; Kavelaars, A.; Heijnen, C.J. Mitochondrial transfer from mesenchymal stem cells to neural stem cells protects against the neurotoxic effects of cisplatin. Acta Neuropathol. Commun. 2018, 6, 1–13. [Google Scholar] [CrossRef]
- Liu, K.; Guo, L.; Zhou, Z.; Pan, M.; Yan, C. Mesenchymal stem cells transfer mitochondria into cerebral microvasculature and promote recovery from ischemic stroke. Microvasc. Res. 2019, 123, 74–80. [Google Scholar] [CrossRef]
- Liu, K.; Ji, K.; Guo, L.; Wu, W.; Lu, H.; Shan, P.; Yan, C. Mesenchymal stem cells rescue injured endothelial cells in an in vitro ischemia–reperfusion model via tunneling nanotube like structure-mediated mitochondrial transfer. Microvasc. Res. 2014, 92, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Spees, J.L.; Olson, S.D.; Whitney, M.J.; Prockop, D.J. Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. USA 2006, 103, 1283–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babenko, V.A.; Silachev, D.N.; Zorova, L.D.; Pevzner, I.B.; Khutornenko, A.A.; Plotnikov, E.Y.; Sukhikh, G.T.; Zorov, D.B. Improving the Post-Stroke Therapeutic Potency of Mesenchymal Multipotent Stromal Cells by Cocultivation With Cortical Neurons: The Role of Crosstalk Between Cells. Stem Cells Transl. Med. 2015, 4, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Babenko, V.A.; Silachev, D.N.; Popkov, V.A.; Zorova, L.D.; Pevzner, I.B.; Plotnikov, E.Y.; Sukhikh, G.T.; Zorov, D.B. Miro1 Enhances Mitochondria Transfer from Multipotent Mesenchymal Stem Cells (MMSC) to Neural Cells and Improves the Efficacy of Cell Recovery. Molecules 2018, 23, 687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, N.; Lambie, S.C.; Huynh, C.Q.; Sanford, B.; Patel, M.; Herson, P.S.; Ormond, D.R. Mitochondrial transfer from mesenchymal stem cells improves neuronal metabolism after oxidant injury in vitro: The role of Miro1. Br. J. Pharmacol. 2020, 271678X20928147. [Google Scholar] [CrossRef]
- Wei, W.; Wu, D.; Duan, Y.; Elkin, K.B.; Chandra, A.; Guan, L.; Peng, C.; He, X.; Wu, C.; Ji, X.; et al. Neuroprotection by mesenchymal stem cell (MSC) administration is enhanced by local cooling infusion (LCI) in ischemia. Brain Res. 2019, 1724, 146406. [Google Scholar] [CrossRef]
- Hayakawa, K.; Chan, S.J.; Mandeville, E.T.; Park, J.H.; Bruzzese, M.; Montaner, J.; Arai, K.; Rosell, A.; Lo, E.H. Protective Effects of Endothelial Progenitor Cell-Derived Extracellular Mitochondria in Brain Endothelium. Stem Cells 2018, 36, 1404–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneko, Y.; Tajiri, N.; Shinozuka, K.; Glover, L.E.; Weinbren, N.L.; Cortes, L.; Borlongan, C.V. Cell Therapy for Stroke: Emphasis on Optimizing Safety and Efficacy Profile of Endothelial Progenitor Cells. Curr. Pharm. Des. 2012, 18, 3731–3734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asahara, T.; Murohara, T.; Sullivan, A.; Silver, M.; Van Der Zee, R.; Li, T.; Witzenbichler, B.; Schatteman, G.; Isner, J.M. Isolation of putative progenitor endothelial cells for angiogenesis. Science 1997, 275, 964–966. [Google Scholar] [CrossRef]
- Toyama, K.; Honmou, O.; Harada, K.; Suzuki, J.; Houkin, K.; Hamada, H.; Kocsis, J.D. Therapeutic benefits of angiogenetic gene-modified human mesenchymal stem cells after cerebral ischemia. Exp. Neurol. 2009, 216, 47–55. [Google Scholar] [CrossRef]
- Wang, L.-L.; Chen, D.; Lee, J.; Gu, X.; Alaaeddine, G.; Li, J.; Wei, L.; Yu, S.P. Mobilization of Endogenous Bone Marrow Derived Endothelial Progenitor Cells and Therapeutic Potential of Parathyroid Hormone after Ischemic Stroke in Mice. PLoS ONE 2014, 9, e87284. [Google Scholar] [CrossRef]
- Martí-Fàbregas, J.; Delgado-Mederos, R.; Crespo, J.; Peña, E.; Marín, R.; Jiménez-Xarrié, E.; Fernández-Arcos, A.; Pérez-Pérez, J.; Martínez-Domeño, A.; Camps-Renom, P.; et al. Circulating Endothelial Progenitor Cells and the Risk of Vascular Events after Ischemic Stroke. PLoS ONE 2015, 10, e0124895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, S.; Luo, C.; Cao, B.; Hu, H.; Wang, S.; Yue, H.; Chen, L.; Zhou, Z. Endothelial Progenitor Cells for Ischemic Stroke: Update on Basic Research and Application. Stem Cells Int. 2017, 2017, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Yip, H.-K.; Chang, L.-T.; Chang, W.-N.; Lu, C.-H.; Liou, C.-W.; Lan, M.-Y.; Liu, J.S.; Youssef, A.A.; Chang, H.-W. Level and Value of Circulating Endothelial Progenitor Cells in Patients After Acute Ischemic Stroke. Stroke 2008, 39, 69–74. [Google Scholar] [CrossRef]
- Ghani, U.; Shuaib, A.; Salam, A.; Nasir, A.; Shuaib, U.; Jeerakathil, T.; Sher, F.; O’Rourke, F.; Nasser, A.M.; Schwindt, B.; et al. Endothelial Progenitor Cells During Cerebrovascular Disease. Stroke 2005, 36, 151–153. [Google Scholar] [CrossRef] [Green Version]
- Deb, P.; Sharma, S.; Hassan, K. Pathophysiologic mechanisms of acute ischemic stroke: An overview with emphasis on therapeutic significance beyond thrombolysis. Pathophysiology 2010, 17, 197–218. [Google Scholar] [CrossRef] [PubMed]
- Kaesmacher, J.; Kaesmacher, M.; Maegerlein, C.; Zimmer, C.; Gersing, A.S.; Wunderlich, S.; Friedrich, B.; Boeckh-Behrens, T.; Kleine, J.F. Hemorrhagic Transformations after Thrombectomy: Risk Factors and Clinical Relevance. Cerebrovasc. Dis. 2017, 43, 294–304. [Google Scholar] [CrossRef]
- Heyck, M.; Bonsack, B.; Zhang, H.; Sadanandan, N.; Cozene, B.; Kingsbury, C.; Lee, J.-Y.; Borlongan, C.V. The brain and eye: Treating cerebral and retinal ischemia through mitochondrial transfer. Exp. Biol. Med. 2019, 244, 1485–1492. [Google Scholar] [CrossRef] [PubMed]
- Röther, J. Neuroprotection Does Not Work! Stroke 2008, 39, 523–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carinci, M.; Vezzani, B.; Patergnani, S.; Ludewig, P.; Lessmann, K.; Magnus, T.; Casetta, I.; Pugliatti, M.; Pinton, P.; Giorgi, C. Different Roles of Mitochondria in Cell Death and Inflammation: Focusing on Mitochondrial Quality Control in Ischemic Stroke and Reperfusion. Biomedicines 2021, 9, 169. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9020169
Carinci M, Vezzani B, Patergnani S, Ludewig P, Lessmann K, Magnus T, Casetta I, Pugliatti M, Pinton P, Giorgi C. Different Roles of Mitochondria in Cell Death and Inflammation: Focusing on Mitochondrial Quality Control in Ischemic Stroke and Reperfusion. Biomedicines. 2021; 9(2):169. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9020169
Chicago/Turabian StyleCarinci, Marianna, Bianca Vezzani, Simone Patergnani, Peter Ludewig, Katrin Lessmann, Tim Magnus, Ilaria Casetta, Maura Pugliatti, Paolo Pinton, and Carlotta Giorgi. 2021. "Different Roles of Mitochondria in Cell Death and Inflammation: Focusing on Mitochondrial Quality Control in Ischemic Stroke and Reperfusion" Biomedicines 9, no. 2: 169. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9020169