
From Mitochondria to Atherosclerosis: The Inflammation Path

 , ,
, , 
Abstract
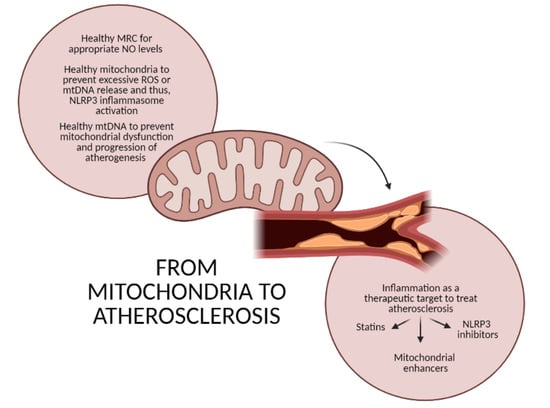
:
{kind=link}
{kind=link}
{kind=link}
1. Mitochondria and Inflammation
2. Atherosclerosis as a Representative Inflammatory Disease
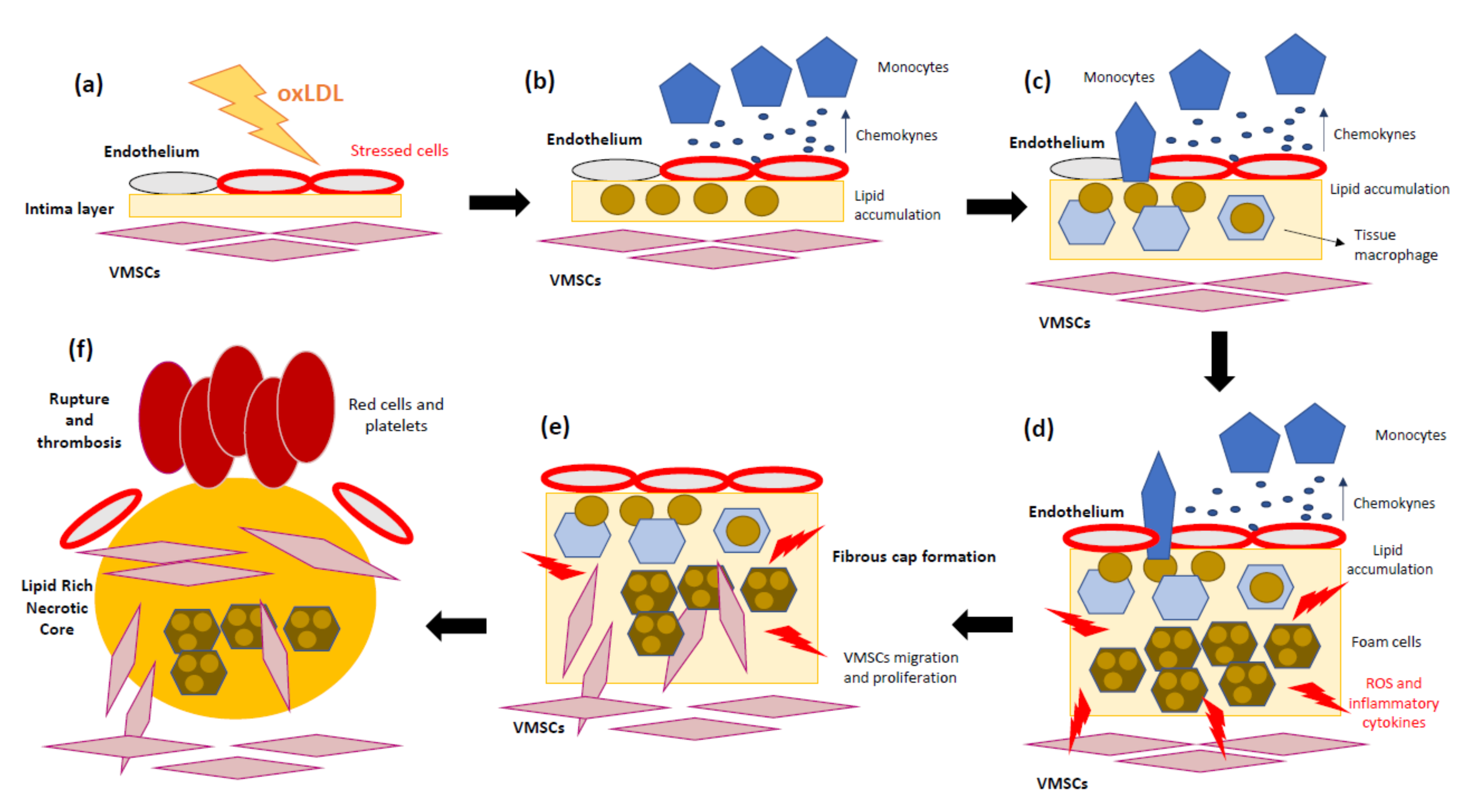
3. The Role of Mitochondria in Atherosclerosis
3.1. Focusing on the Endothelial Origin of Atherosclerosis
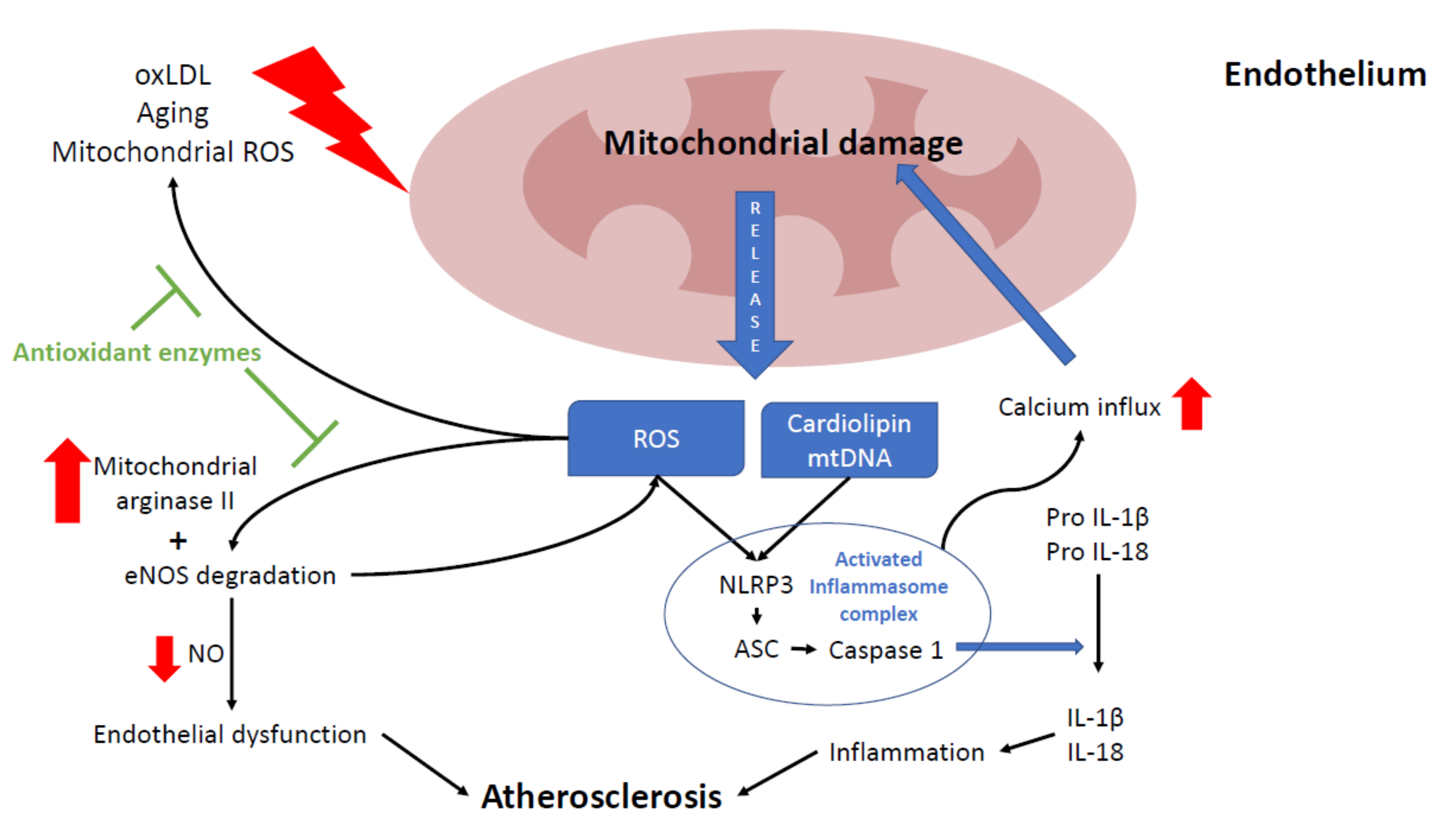
3.2. Mitochondria and NLRP3 Inflammasome
3.3. Mitochondrial Mutations and Atherogenesis
4. Inflammation as a Therapeutic Target
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADP | Adenosine diphosphate |
| AMP | Adenosine monophosphate |
| AMPK | 5′ Adenosine-monophosphate-activated protein kinase |
| ApoE | Apolipoprotein E |
| ASC | Apoptosis-associated speck-like protein containing a caspase recruitment domain |
| ATP | Adenosine triphosphate |
| BRCA | Breast cancer 1 |
| BRCC3 | BRCA1/BRCA2-containing complex subunit 3 |
| CD | Cluster of differentiation |
| cGAS | Cyclic GMP-AMP synthase |
| CoQ | Coenzyme Q10 |
| CpG | Cytosine-guanine sequences |
| CVD | Cardiovascular disease |
| DAG | Diacylglycerol |
| DAMP | Damage-associated molecular pattern |
| DMM | Dimethyl malonate |
| eNOS | Endothelial nitric oxide synthase |
| ECM | Extracellular matrix |
| ECSIT | Evolutionarily conserved signaling intermediate in Toll pathway |
| ER | Endoplasmic reticulum |
| GMP | Guanosine monophosphate |
| GPX | Glutathione peroxidase |
| HDL | High-density lipoprotein |
| HMG-CoA | 3-hydroxy-3-methylglutaryl-coenzyme A |
| ICE | IL converting enzyme |
| IL | Interleukin |
| IRF | Interferon-regulatory factor |
| LC3 | Microtubule-associated protein 1A/1B-light chain 3 |
| LDL-C | Low-density lipoprotein cholesterol |
| LDL-R | Low-density lipoprotein receptor |
| LOX | Lectin-type oxidized LDL receptor |
| LPS | Lipopolysaccharides |
| MAM | Mitochondria-associated ER membrane |
| MAVS | Mitochondrial antiviral signaling protein |
| MCP | Monocyte chemoattractant protein |
| Mfn | Mitofusin |
| MPT | Mitochondrial permeability transition pore |
| MRC | Mitochondrial respiratory chain |
| mtDNA | Mitochondrial deoxyribonucleic acid |
| NAC | N-acetyl-lysine |
| NAD | Nicotinamide adenine dinucleotide |
| NF-κβ | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NLR | Nod-like receptor |
| NLRP3 | NLR family pyrin domain containing 3 |
| NO | Nitric oxide |
| oxLDL | Oxidized low-density lipoprotein |
| OXPHOS | Oxidative phosphorylation |
| PAMP | Pathogen-associated molecule pattern |
| PARP | Poly(ADP-ribose) polymerase |
| PGC | PPAR helper activator |
| PKD | Protein kinase D |
| PPAR | Peroxisome-proliferator-activated receptor |
| PRR | Pattern recognition receptor |
| PCSK9 | Proprotein convertase subtilisin/kexin type 9 |
| PTEN | Phosphatase tensin homologue |
| RIG-I | Retinoic acid-inducible gene I |
| RLR | RIG-like receptor |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| SAMS | Statin-associated muscle symptoms |
| SIRT | Sirtuin |
| SOD | Superoxide dismutase |
| STING | Stimulator of interferon genes |
| TDZ | Thiazolidinedione |
| TLR | Toll-like receptor |
| TRAF6 | TNF-receptor-associated factor 6 |
| VEC | Vascular endothelial cell |
| VSMC | Vascular smooth muscle cell |
| ΔΨm | Mitochondrial membrane potential |
References
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than just a powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, Y.; Rosal, K.; Chung, B.C. Function of CYP11A1 in the mitochondria. Mol. Cell Endocrinol. 2017, 441, 55–61. [Google Scholar] [CrossRef]
- Bravo-Sagua, R.; Parra, V.; Lopez-Crisosto, C.; Diaz, P.; Quest, A.F.; Lavandero, S. Calcium Transport and Signaling in Mitochondria. Compr Physiol 2017, 7, 623–634. [Google Scholar] [CrossRef]
- Paul, B.T.; Manz, D.H.; Torti, F.M.; Torti, S.V. Mitochondria and Iron: Current questions. Expert Rev. Hematol. 2017, 10, 65–79. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.S.; Ho, P.C. Mitochondria: A master regulator in macrophage and T cell immunity. Mitochondrion 2018, 41, 45–50. [Google Scholar] [CrossRef]
- Blajszczak, C.; Bonini, M.G. Mitochondria targeting by environmental stressors: Implications for redox cellular signaling. Toxicology 2017, 391, 84–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, S.Y.; Seol, D.W. The role of mitochondria in apoptosis. BMB Rep. 2008, 41, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolmychkova, K.I.; Zhelankin, A.V.; Karagodin, V.P.; Orekhov, A.N. Mitochondria and inflammation. Patol. Fiziol. Eksp. Ter. 2016, 60, 114–121. [Google Scholar]
- Kuprash, D.V.; Nedospasov, S.A. Molecular and Cellular Mechanisms of Inflammation. Biochemistry (Mosc) 2016, 81, 1237–1239. [Google Scholar] [CrossRef]
- Escamilla-Tilch, M.; Filio-Rodriguez, G.; Garcia-Rocha, R.; Mancilla-Herrera, I.; Mitchison, N.A.; Ruiz-Pacheco, J.A.; Sanchez-Garcia, F.J.; Sandoval-Borrego, D.; Vazquez-Sanchez, E.A. The interplay between pathogen-associated and danger-associated molecular patterns: An inflammatory code in cancer? Immunol. Cell Biol. 2013, 91, 601–610. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Nace, G.; Evankovich, J.; Eid, R.; Tsung, A. Dendritic cells and damage-associated molecular patterns: Endogenous danger signals linking innate and adaptive immunity. J. Innate Immun. 2012, 4, 6–15. [Google Scholar] [CrossRef]
- Elieh Ali Komi, D.; Sharma, L.; Dela Cruz, C.S. Chitin and Its Effects on Inflammatory and Immune Responses. Clin. Rev. Allergy Immunol 2018, 54, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [Green Version]
- Archibald, J.M. Endosymbiosis and Eukaryotic Cell Evolution. Curr. Biol. 2015, 25, R911–R921. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R.; et al. NF-kappaB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [Green Version]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Gao, C. Regulation of MAVS activation through post-translational modifications. Curr. Opin. Immunol. 2018, 50, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498. [Google Scholar] [CrossRef]
- Subramanian, N.; Natarajan, K.; Clatworthy, M.R.; Wang, Z.; Germain, R.N. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 2013, 153, 348–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.; Wang, H.; Hajishengallis, G.N.; Martin, M. TLR-signaling networks: An integration of adaptor molecules, kinases, and cross-talk. J. Dent. Res. 2011, 90, 417–427. [Google Scholar] [CrossRef]
- Banoth, B.; Cassel, S.L. Mitochondria in innate immune signaling. Transl. Res. 2018, 202, 52–68. [Google Scholar] [CrossRef] [PubMed]
- Grebe, A.; Hoss, F.; Latz, E. NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ. Res. 2018, 122, 1722–1740. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, D.; Hu, D.; Zhou, X.; Zhou, Y. The role of mitochondria in NLRP3 inflammasome activation. Mol. Immunol. 2018, 103, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Mallat, Z.; Corbaz, A.; Scoazec, A.; Besnard, S.; Leseche, G.; Chvatchko, Y.; Tedgui, A. Expression of interleukin-18 in human atherosclerotic plaques and relation to plaque instability. Circulation 2001, 104, 1598–1603. [Google Scholar] [CrossRef] [Green Version]
- Thomas, H.; Diamond, J.; Vieco, A.; Chaudhuri, S.; Shinnar, E.; Cromer, S.; Perel, P.; Mensah, G.A.; Narula, J.; Johnson, C.O.; et al. Global Atlas of Cardiovascular Disease 2000-2016: The Path to Prevention and Control. Glob. Heart 2018, 13, 143–163. [Google Scholar] [CrossRef]
- Tietge, U.J. Hyperlipidemia and cardiovascular disease: Inflammation, dyslipidemia, and atherosclerosis. Curr Opin Lipidol 2014, 25, 94–95. [Google Scholar] [CrossRef]
- Fisher, E.A.; Feig, J.E.; Hewing, B.; Hazen, S.L.; Smith, J.D. High-density lipoprotein function, dysfunction, and reverse cholesterol transport. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2813–2820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boren, J.; Chapman, M.J.; Krauss, R.M.; Packard, C.J.; Bentzon, J.F.; Binder, C.J.; Daemen, M.J.; Demer, L.L.; Hegele, R.A.; Nicholls, S.J.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: Pathophysiological, genetic, and therapeutic insights: A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2020. [Google Scholar] [CrossRef]
- Mestas, J.; Ley, K. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc. Med. 2008, 18, 228–232. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; C, G.Z.; Wang, X.H.; Liu, D.H. Progression of atherosclerosis in ApoE-knockout mice fed on a high-fat diet. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 3863–3867. [Google Scholar] [PubMed]
- Emini Veseli, B.; Perrotta, P.; De Meyer, G.R.A.; Roth, L.; Van der Donckt, C.; Martinet, W.; De Meyer, G.R.Y. Animal models of atherosclerosis. Eur. J. Pharmacol 2017, 816, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Ridker, P.M.; Hansson, G.K.; Leducq Transatlantic Network on, A. Inflammation in atherosclerosis: From pathophysiology to practice. J. Am. Coll. Cardiol. 2009, 54, 2129–2138. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M. Residual inflammatory risk: Addressing the obverse side of the atherosclerosis prevention coin. Eur Heart J. 2016, 37, 1720–1722. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Xian, X.; Wang, Z.; Bi, Y.; Chen, Q.; Han, X.; Tang, D.; Chen, R. Research Progress on the Relationship between Atherosclerosis and Inflammation. Biomolecules 2018, 8, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahotupa, M. Oxidized lipoprotein lipids and atherosclerosis. Free Radic. Res. 2017, 51, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L. Metalloproteinases in atherosclerosis. Eur. J. Pharmacol. 2017, 816, 93–106. [Google Scholar] [CrossRef] [Green Version]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of plaque formation and rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef] [PubMed]
- Badimon, L.; Padro, T.; Vilahur, G. Atherosclerosis, platelets and thrombosis in acute ischaemic heart disease. Eur. Heart J. Acute Cardiovasc. Care 2012, 1, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Lu, A.; Wu, H. Structural mechanisms of inflammasome assembly. FEBS J. 2015, 282, 435–444. [Google Scholar] [CrossRef]
- Cohen, G.M. Caspases: The executioners of apoptosis. Biochem. J. 1997, 326 (Pt. 1), 1–16. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Kuida, K.; Tsutsui, H.; Ku, G.; Hsiao, K.; Fleming, M.A.; Hayashi, N.; Higashino, K.; Okamura, H.; Nakanishi, K.; et al. Activation of interferon-gamma inducing factor mediated by interleukin-1beta converting enzyme. Science 1997, 275, 206–209. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, V.A.; Fitzgerald, K.A. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell 2016, 165, 792–800. [Google Scholar] [CrossRef] [Green Version]
- Haneklaus, M.; O’Neill, L.A. NLRP3 at the interface of metabolism and inflammation. Immunol. Rev. 2015, 265, 53–62. [Google Scholar] [CrossRef]
- He, Y.; Hara, H.; Nunez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [Green Version]
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.L. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid. Redox Signal. 2015, 22, 1111–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, M.G.; Weber, M.D.; Watkins, L.R.; Maier, S.F. Stress-induced neuroinflammatory priming: A liability factor in the etiology of psychiatric disorders. Neurobiol. Stress 2016, 4, 62–70. [Google Scholar] [CrossRef] [Green Version]
- Alexander, M.R.; Moehle, C.W.; Johnson, J.L.; Yang, Z.; Lee, J.K.; Jackson, C.L.; Owens, G.K. Genetic inactivation of IL-1 signaling enhances atherosclerotic plaque instability and reduces outward vessel remodeling in advanced atherosclerosis in mice. J. Clin. Invest. 2012, 122, 70–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libby, P. Interleukin-1 Beta as a Target for Atherosclerosis Therapy: Biological Basis of CANTOS and Beyond. J. Am. Coll. Cardiol. 2017, 70, 2278–2289. [Google Scholar] [CrossRef]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nunez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klinkner, A.M.; Waites, C.R.; Kerns, W.D.; Bugelski, P.J. Evidence of foam cell and cholesterol crystal formation in macrophages incubated with oxidized LDL by fluorescence and electron microscopy. J. Histochem. Cytochem. 1995, 43, 1071–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheedy, F.J.; Grebe, A.; Rayner, K.J.; Kalantari, P.; Ramkhelawon, B.; Carpenter, S.B.; Becker, C.E.; Ediriweera, H.N.; Mullick, A.E.; Golenbock, D.T.; et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 2013, 14, 812–820. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westerterp, M.; Fotakis, P.; Ouimet, M.; Bochem, A.E.; Zhang, H.; Molusky, M.M.; Wang, W.; Abramowicz, S.; la Bastide-van Gemert, S.; Wang, N.; et al. Cholesterol Efflux Pathways Suppress Inflammasome Activation, NETosis, and Atherogenesis. Circulation 2018, 138, 898–912. [Google Scholar] [CrossRef] [Green Version]
- Usui, F.; Shirasuna, K.; Kimura, H.; Tatsumi, K.; Kawashima, A.; Karasawa, T.; Hida, S.; Sagara, J.; Taniguchi, S.; Takahashi, M. Critical role of caspase-1 in vascular inflammation and development of atherosclerosis in Western diet-fed apolipoprotein E-deficient mice. Biochem. Biophys. Res. Commun. 2012, 425, 162–168. [Google Scholar] [CrossRef]
- Gage, J.; Hasu, M.; Thabet, M.; Whitman, S.C. Caspase-1 deficiency decreases atherosclerosis in apolipoprotein E-null mice. Can. J. Cardiol 2012, 28, 222–229. [Google Scholar] [CrossRef]
- Zheng, F.; Xing, S.; Gong, Z.; Mu, W.; Xing, Q. Silence of NLRP3 suppresses atherosclerosis and stabilizes plaques in apolipoprotein E-deficient mice. Mediators Inflamm. 2014, 2014, 507208. [Google Scholar] [CrossRef] [Green Version]
- Bai, B.; Yang, Y.; Wang, Q.; Li, M.; Tian, C.; Liu, Y.; Aung, L.H.H.; Li, P.F.; Yu, T.; Chu, X.M. NLRP3 inflammasome in endothelial dysfunction. Cell Death Dis 2020, 11, 776. [Google Scholar] [CrossRef] [PubMed]
- Galea, J.; Armstrong, J.; Gadsdon, P.; Holden, H.; Francis, S.E.; Holt, C.M. Interleukin-1 beta in coronary arteries of patients with ischemic heart disease. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 1000–1006. [Google Scholar] [CrossRef]
- Bevilacqua, M.P.; Pober, J.S.; Majeau, G.R.; Cotran, R.S.; Gimbrone, M.A., Jr. Interleukin 1 (IL-1) induces biosynthesis and cell surface expression of procoagulant activity in human vascular endothelial cells. J. Exp. Med. 1984, 160, 618–623. [Google Scholar] [CrossRef] [Green Version]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef]
- Baylis, R.A.; Gomez, D.; Mallat, Z.; Pasterkamp, G.; Owens, G.K. The CANTOS Trial: One Important Step for Clinical Cardiology but a Giant Leap for Vascular Biology. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e174–e177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavillard, L.E.; Marin-Aguilar, F.; Bullon, P.; Cordero, M.D. Cardiovascular diseases, NLRP3 inflammasome, and western dietary patterns. Pharmacol. Res. 2018, 131, 44–50. [Google Scholar] [CrossRef]
- Zheng, F.; Xing, S.; Gong, Z.; Xing, Q. NLRP3 inflammasomes show high expression in aorta of patients with atherosclerosis. Heart Lung Circ. 2013, 22, 746–750. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Xie, W.L.; Kong, W.W.; Chen, D.; Qu, P. Expression of the NLRP3 Inflammasome in Carotid Atherosclerosis. J. Stroke Cerebrovasc. Dis. 2015, 24, 2455–2466. [Google Scholar] [CrossRef]
- Paramel Varghese, G.; Folkersen, L.; Strawbridge, R.J.; Halvorsen, B.; Yndestad, A.; Ranheim, T.; Krohg-Sorensen, K.; Skjelland, M.; Espevik, T.; Aukrust, P.; et al. NLRP3 Inflammasome Expression and Activation in Human Atherosclerosis. J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Afrasyab, A.; Qu, P.; Zhao, Y.; Peng, K.; Wang, H.; Lou, D.; Niu, N.; Yuan, D. Correlation of NLRP3 with severity and prognosis of coronary atherosclerosis in acute coronary syndrome patients. Heart Vessels 2016, 31, 1218–1229. [Google Scholar] [CrossRef] [PubMed]
- Siasos, G.; Tsigkou, V.; Kosmopoulos, M.; Theodosiadis, D.; Simantiris, S.; Tagkou, N.M.; Tsimpiktsioglou, A.; Stampouloglou, P.K.; Oikonomou, E.; Mourouzis, K.; et al. Mitochondria and cardiovascular diseases-from pathophysiology to treatment. Ann. Transl. Med. 2018, 6, 256. [Google Scholar] [CrossRef]
- Graves, S.M.; Xie, Z.; Stout, K.A.; Zampese, E.; Burbulla, L.F.; Shih, J.C.; Kondapalli, J.; Patriarchi, T.; Tian, L.; Brichta, L.; et al. Dopamine metabolism by a monoamine oxidase mitochondrial shuttle activates the electron transport chain. Nat. Neurosci. 2020, 23, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Kundu, T.K.; Velayutham, M.; Zweier, J.L. Aldehyde oxidase functions as a superoxide generating NADH oxidase: An important redox regulated pathway of cellular oxygen radical formation. Biochemistry 2012, 51, 2930–2939. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Jia, Z.; Trush, M.A. Defining ROS in Biology and Medicine. React. Oxyg. Species (Apex) 2016, 1, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Schroder, K. NADPH oxidase-derived reactive oxygen species: Dosis facit venenum. Exp. Physiol. 2019, 104, 447–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Benna, J.; Hurtado-Nedelec, M.; Marzaioli, V.; Marie, J.C.; Gougerot-Pocidalo, M.A.; Dang, P.M. Priming of the neutrophil respiratory burst: Role in host defense and inflammation. Immunol. Rev. 2016, 273, 180–193. [Google Scholar] [CrossRef]
- Brandes, R.P.; Weissmann, N.; Schroder, K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic. Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef] [PubMed]
- Helmcke, I.; Heumuller, S.; Tikkanen, R.; Schroder, K.; Brandes, R.P. Identification of structural elements in Nox1 and Nox4 controlling localization and activity. Antioxid. Redox Signal 2009, 11, 1279–1287. [Google Scholar] [CrossRef] [PubMed]
- Jha, J.C.; Watson, A.M.D.; Mathew, G.; de Vos, L.C.; Jandeleit-Dahm, K. The emerging role of NADPH oxidase NOX5 in vascular disease. Clin. Sci. 2017, 131, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, G.Z.; Rabinovitch, P.S.; Tabas, I. Macrophage mitochondrial oxidative stress promotes atherosclerosis and nuclear factor-kappaB-mediated inflammation in macrophages. Circ. Res. 2014, 114, 421–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, H.L.; Kuo, L.T.; Sung, F.C.; Yeh, C.C. Association between Polymorphisms of Antioxidant Gene (MnSOD, CAT, and GPx1) and Risk of Coronary Artery Disease. Biomed. Res. Int. 2018, 2018, 5086869. [Google Scholar] [CrossRef] [PubMed]
- Kudryavtseva, A.V.; Krasnov, G.S.; Dmitriev, A.A.; Alekseev, B.Y.; Kardymon, O.L.; Sadritdinova, A.F.; Fedorova, M.S.; Pokrovsky, A.V.; Melnikova, N.V.; Kaprin, A.D.; et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 2016, 7, 44879–44905. [Google Scholar] [CrossRef] [Green Version]
- Velayutham, M.; Hemann, C.; Zweier, J.L. Removal of H(2)O(2) and generation of superoxide radical: Role of cytochrome c and NADH. Free Radic. Biol. Med. 2011, 51, 160–170. [Google Scholar] [CrossRef] [Green Version]
- Naik, E.; Dixit, V.M. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J. Exp. Med. 2011, 208, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Yu, E.P.K.; Reinhold, J.; Yu, H.; Starks, L.; Uryga, A.K.; Foote, K.; Finigan, A.; Figg, N.; Pung, Y.F.; Logan, A.; et al. Mitochondrial Respiration Is Reduced in Atherosclerosis, Promoting Necrotic Core Formation and Reducing Relative Fibrous Cap Thickness. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2322–2332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacinto, T.A.; Meireles, G.S.; Dias, A.T.; Aires, R.; Porto, M.L.; Gava, A.L.; Vasquez, E.C.; Pereira, T.M.C.; Campagnaro, B.P.; Meyrelles, S.S. Increased ROS production and DNA damage in monocytes are biomarkers of aging and atherosclerosis. Biol. Res. 2018, 51, 33. [Google Scholar] [CrossRef] [Green Version]
- Vilne, B.; Skogsberg, J.; Foroughi Asl, H.; Talukdar, H.A.; Kessler, T.; Bjorkegren, J.L.M.; Schunkert, H. Network analysis reveals a causal role of mitochondrial gene activity in atherosclerotic lesion formation. Atherosclerosis 2017, 267, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Blanc, J.; Alves-Guerra, M.C.; Esposito, B.; Rousset, S.; Gourdy, P.; Ricquier, D.; Tedgui, A.; Miroux, B.; Mallat, Z. Protective role of uncoupling protein 2 in atherosclerosis. Circulation 2003, 107, 388–390. [Google Scholar] [CrossRef] [Green Version]
- Arsenijevic, D.; Onuma, H.; Pecqueur, C.; Raimbault, S.; Manning, B.S.; Miroux, B.; Couplan, E.; Alves-Guerra, M.C.; Goubern, M.; Surwit, R.; et al. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat. Genet. 2000, 26, 435–439. [Google Scholar] [CrossRef] [PubMed]
- El-Kenawi, A.; Ruffell, B. Inflammation, ROS, and Mutagenesis. Cancer Cell 2017, 32, 727–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chistiakov, D.A.; Shkurat, T.P.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. The role of mitochondrial dysfunction in cardiovascular disease: A brief review. Ann. Med. 2018, 50, 121–127. [Google Scholar] [CrossRef]
- An, P.; Wei, L.L.; Zhao, S.; Sverdlov, D.Y.; Vaid, K.A.; Miyamoto, M.; Kuramitsu, K.; Lai, M.; Popov, Y.V. Hepatocyte mitochondria-derived danger signals directly activate hepatic stellate cells and drive progression of liver fibrosis. Nat. Commun. 2020, 11, 2362. [Google Scholar] [CrossRef] [PubMed]
- Qualls, A.E.; Southern, W.M.; Call, J.A. Mitochondria-Cytokine Crosstalk Following Skeletal Muscle Injury and Disuse: A Mini-Review. Am. J. Physiol. Cell Physiol. 2021. [Google Scholar] [CrossRef]
- Chakraborty, K.; Raundhal, M.; Chen, B.B.; Morse, C.; Tyurina, Y.Y.; Khare, A.; Oriss, T.B.; Huff, R.; Lee, J.S.; St Croix, C.M.; et al. The mito-DAMP cardiolipin blocks IL-10 production causing persistent inflammation during bacterial pneumonia. Nat. Commun. 2017, 8, 13944. [Google Scholar] [CrossRef] [Green Version]
- Colon, E.; Strand, M.L.; Carlsson-Skwirut, C.; Wahlgren, A.; Svechnikov, K.V.; Cohen, P.; Soder, O. Anti-apoptotic factor humanin is expressed in the testis and prevents cell-death in leydig cells during the first wave of spermatogenesis. J. Cell Physiol. 2006, 208, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Niikura, T.; Tajima, H.; Kita, Y. Neuronal cell death in Alzheimer’s disease and a neuroprotective factor, humanin. Curr Neuropharmacol. 2006, 4, 139–147. [Google Scholar] [CrossRef]
- Muzumdar, R.H.; Huffman, D.M.; Calvert, J.W.; Jha, S.; Weinberg, Y.; Cui, L.; Nemkal, A.; Atzmon, G.; Klein, L.; Gundewar, S.; et al. Acute humanin therapy attenuates myocardial ischemia and reperfusion injury in mice. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1940–1948. [Google Scholar] [CrossRef]
- Widmer, R.J.; Flammer, A.J.; Herrmann, J.; Rodriguez-Porcel, M.; Wan, J.; Cohen, P.; Lerman, L.O.; Lerman, A. Circulating humanin levels are associated with preserved coronary endothelial function. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H393–H397. [Google Scholar] [CrossRef]
- Zacharias, D.G.; Kim, S.G.; Massat, A.E.; Bachar, A.R.; Oh, Y.K.; Herrmann, J.; Rodriguez-Porcel, M.; Cohen, P.; Lerman, L.O.; Lerman, A. Humanin, a cytoprotective peptide, is expressed in carotid atherosclerotic [corrected] plaques in humans. PLoS ONE 2012, 7, e31065. [Google Scholar] [CrossRef]
- Oh, Y.K.; Bachar, A.R.; Zacharias, D.G.; Kim, S.G.; Wan, J.; Cobb, L.J.; Lerman, L.O.; Cohen, P.; Lerman, A. Humanin preserves endothelial function and prevents atherosclerotic plaque progression in hypercholesterolemic ApoE deficient mice. Atherosclerosis 2011, 219, 65–73. [Google Scholar] [CrossRef] [Green Version]
- Szewczyk, A.; Jarmuszkiewicz, W.; Koziel, A.; Sobieraj, I.; Nobik, W.; Lukasiak, A.; Skup, A.; Bednarczyk, P.; Drabarek, B.; Dymkowska, D.; et al. Mitochondrial mechanisms of endothelial dysfunction. Pharmacol. Rep. 2015, 67, 704–710. [Google Scholar] [CrossRef]
- Kadlec, A.O.; Chabowski, D.S.; Ait-Aissa, K.; Gutterman, D.D. Role of PGC-1alpha in Vascular Regulation: Implications for Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1467–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pircher, A.; Treps, L.; Bodrug, N.; Carmeliet, P. Endothelial cell metabolism: A novel player in atherosclerosis? Basic principles and therapeutic opportunities. Atherosclerosis 2016, 253, 247–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantzarlis, K.; Tsolaki, V.; Zakynthinos, E. Role of Oxidative Stress and Mitochondrial Dysfunction in Sepsis and Potential Therapies. Oxid. Med. Cell Longev. 2017, 2017, 5985209. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.L. eNOS, metabolic syndrome and cardiovascular disease. Trends Endocrinol. Metab. 2009, 20, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Hong, F.F.; Liang, X.Y.; Liu, W.; Lv, S.; He, S.J.; Kuang, H.B.; Yang, S.L. Roles of eNOS in atherosclerosis treatment. Inflamm. Res. 2019, 68, 429–441. [Google Scholar] [CrossRef]
- Vaisman, B.L.; Andrews, K.L.; Khong, S.M.; Wood, K.C.; Moore, X.L.; Fu, Y.; Kepka-Lenhart, D.M.; Morris, S.M., Jr.; Remaley, A.T.; Chin-Dusting, J.P. Selective endothelial overexpression of arginase II induces endothelial dysfunction and hypertension and enhances atherosclerosis in mice. PLoS ONE 2012, 7, e39487. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Zhao, T.; Zhang, W.; Yu, W.; Liu, B.; Wang, Z.; Qiao, W.; Lu, Q.; Wang, A.; Zhang, M. Poly (ADP-Ribose) Polymerase 1 Mediated Arginase II Activation Is Responsible for Oxidized LDL-Induced Endothelial Dysfunction. Front. Pharmacol. 2018, 9, 882. [Google Scholar] [CrossRef] [PubMed]
- Scarpulla, R.C.; Vega, R.B.; Kelly, D.P. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol. Metab. 2012, 23, 459–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kluge, M.A.; Fetterman, J.L.; Vita, J.A. Mitochondria and endothelial function. Circ. Res. 2013, 112, 1171–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Won, J.C.; Park, J.Y.; Kim, Y.M.; Koh, E.H.; Seol, S.; Jeon, B.H.; Han, J.; Kim, J.R.; Park, T.S.; Choi, C.S.; et al. Peroxisome proliferator-activated receptor-gamma coactivator 1-alpha overexpression prevents endothelial apoptosis by increasing ATP/ADP translocase activity. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 290–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coto, E.; Reguero, J.R.; Avanzas, P.; Pascual, I.; Martin, M.; Hevia, S.; Moris, C.; Diaz-Molina, B.; Lambert, J.L.; Alonso, B.; et al. Gene variants in the NF-KB pathway (NFKB1, NFKBIA, NFKBIZ) and risk for early-onset coronary artery disease. Immunol. Lett. 2019, 208, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C.; Ischiropoulos, H.; Radi, R. Peroxynitrite: Biochemistry, pathophysiology and development of therapeutics. Nat. Rev. Drug Discov. 2007, 6, 662–680. [Google Scholar] [CrossRef]
- Yoshioka, J. Thioredoxin Reductase 2 (Txnrd2) Regulates Mitochondrial Integrity in the Progression of Age-Related Heart Failure. J. Am. Heart Assoc. 2015, 4. [Google Scholar] [CrossRef] [Green Version]
- Kameritsch, P.; Singer, M.; Nuernbergk, C.; Rios, N.; Reyes, A.M.; Schmidt, K.; Kirsch, J.; Schneider, H.; Muller, S.; Pogoda, K.; et al. The mitochondrial thioredoxin reductase system (TrxR2) in vascular endothelium controls peroxynitrite levels and tissue integrity. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, W.; Wang, P.; Song, X.; Mao, Z.; Liu, Z. Highly Sensitive Near-Infrared Imaging of Peroxynitrite Fluxes in Inflammation Progress. Anal. Chem. 2021, 93, 3035–3041. [Google Scholar] [CrossRef] [PubMed]
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef] [Green Version]
- Rafieian-Kopaei, M.; Setorki, M.; Doudi, M.; Baradaran, A.; Nasri, H. Atherosclerosis: Process, indicators, risk factors and new hopes. Int. J. Prev. Med. 2014, 5, 927–946. [Google Scholar] [PubMed]
- Ip, W.K.; Medzhitov, R. Macrophages monitor tissue osmolarity and induce inflammatory response through NLRP3 and NLRC4 inflammasome activation. Nat. Commun. 2015, 6, 6931. [Google Scholar] [CrossRef]
- Misawa, T.; Takahama, M.; Kozaki, T.; Lee, H.; Zou, J.; Saitoh, T.; Akira, S. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immuno.l 2013, 14, 454–460. [Google Scholar] [CrossRef]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaron, J.R.; Gangaraju, S.; Rao, M.Y.; Kong, X.; Zhang, L.; Su, F.; Tian, Y.; Glenn, H.L.; Meldrum, D.R. K(+) regulates Ca(2+) to drive inflammasome signaling: Dynamic visualization of ion flux in live cells. Cell Death Dis. 2015, 6, e1954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsnelson, M.A.; Rucker, L.G.; Russo, H.M.; Dubyak, G.R. K+ efflux agonists induce NLRP3 inflammasome activation independently of Ca2+ signaling. J. Immunol. 2015, 194, 3937–3952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, A.J.; Reyes, C.N.; Liang, W.; Becker, C.; Shimada, K.; Wheeler, M.L.; Cho, H.C.; Popescu, N.I.; Coggeshall, K.M.; Arditi, M.; et al. Hexokinase Is an Innate Immune Receptor for the Detection of Bacterial Peptidoglycan. Cell 2016, 166, 624–636. [Google Scholar] [CrossRef] [Green Version]
- da-Silva, W.S.; Gomez-Puyou, A.; de Gomez-Puyou, M.T.; Moreno-Sanchez, R.; De Felice, F.G.; de Meis, L.; Oliveira, M.F.; Galina, A. Mitochondrial bound hexokinase activity as a preventive antioxidant defense: Steady-state ADP formation as a regulatory mechanism of membrane potential and reactive oxygen species generation in mitochondria. J. Biol. Chem. 2004, 279, 39846–39855. [Google Scholar] [CrossRef] [Green Version]
- Rasola, A.; Sciacovelli, M.; Pantic, B.; Bernardi, P. Signal transduction to the permeability transition pore. FEBS Lett. 2010, 584, 1989–1996. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Vashisht, A.A.; Tchieu, J.; Wohlschlegel, J.A.; Dreier, L. Voltage-dependent anion channels (VDACs) recruit Parkin to defective mitochondria to promote mitochondrial autophagy. J. Biol. Chem. 2012, 287, 40652–40660. [Google Scholar] [CrossRef] [Green Version]
- Hughes, M.M.; O’Neill, L.A.J. Metabolic regulation of NLRP3. Immunol. Rev. 2018, 281, 88–98. [Google Scholar] [CrossRef]
- Yu, J.; Nagasu, H.; Murakami, T.; Hoang, H.; Broderick, L.; Hoffman, H.M.; Horng, T. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 15514–15519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauernfeind, F.; Bartok, E.; Rieger, A.; Franchi, L.; Nunez, G.; Hornung, V. Cutting edge: Reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J. Immunol. 2011, 187, 613–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Meszaros, G.; He, W.T.; Xu, Y.; de Fatima Magliarelli, H.; Mailly, L.; Mihlan, M.; Liu, Y.; Puig Gamez, M.; Goginashvili, A.; et al. Protein kinase D at the Golgi controls NLRP3 inflammasome activation. J. Exp. Med. 2017, 214, 2671–2693. [Google Scholar] [CrossRef] [PubMed]
- Misawa, T.; Takahama, M.; Saitoh, T. Mitochondria-Endoplasmic Reticulum Contact Sites Mediate Innate Immune Responses. Adv. Exp. Med. Biol 2017, 997, 187–197. [Google Scholar] [CrossRef]
- de Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef]
- Suarez-Rivero, J.M.; Villanueva-Paz, M.; de la Cruz-Ojeda, P.; de la Mata, M.; Cotan, D.; Oropesa-Avila, M.; de Lavera, I.; Alvarez-Cordoba, M.; Luzon-Hidalgo, R.; Sanchez-Alcazar, J.A. Mitochondrial Dynamics in Mitochondrial Diseases. Diseases 2016, 5, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterky, F.H.; Lee, S.; Wibom, R.; Olson, L.; Larsson, N.G. Impaired mitochondrial transport and Parkin-independent degeneration of respiratory chain-deficient dopamine neurons in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 12937–12942. [Google Scholar] [CrossRef] [Green Version]
- Elliott, E.I.; Sutterwala, F.S. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol Rev. 2015, 265, 35–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Won, J.H.; Hwang, I.; Hong, S.; Lee, H.K.; Yu, J.W. Defective mitochondrial fission augments NLRP3 inflammasome activation. Sci. Rep. 2015, 5, 15489. [Google Scholar] [CrossRef] [Green Version]
- Triantafilou, K.; Hughes, T.R.; Triantafilou, M.; Morgan, B.P. The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome activation. J. Cell Sci. 2013, 126, 2903–2913. [Google Scholar] [CrossRef] [Green Version]
- Ding, Z.; Liu, S.; Wang, X.; Dai, Y.; Khaidakov, M.; Deng, X.; Fan, Y.; Xiang, D.; Mehta, J.L. LOX-1, mtDNA damage, and NLRP3 inflammasome activation in macrophages: Implications in atherogenesis. Cardiovasc. Res. 2014, 103, 619–628. [Google Scholar] [CrossRef] [Green Version]
- Freigang, S.; Ampenberger, F.; Weiss, A.; Kanneganti, T.D.; Iwakura, Y.; Hersberger, M.; Kopf, M. Fatty acid-induced mitochondrial uncoupling elicits inflammasome-independent IL-1alpha and sterile vascular inflammation in atherosclerosis. Nat. Immunol. 2013, 14, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Victor, V.M.; Apostolova, N.; Herance, R.; Hernandez-Mijares, A.; Rocha, M. Oxidative stress and mitochondrial dysfunction in atherosclerosis: Mitochondria-targeted antioxidants as potential therapy. Curr. Med. Chem. 2009, 16, 4654–4667. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Sobenin, I.A.; Orekhov, A.N.; Bobryshev, Y.V. Role of endoplasmic reticulum stress in atherosclerosis and diabetic macrovascular complications. Biomed. Res. Int. 2014, 2014, 610140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, X.M.; Li, W.; Brunk, U.T.; Dalen, H.; Chang, Y.H.; Sevanian, A. Lysosomal destabilization during macrophage damage induced by cholesterol oxidation products. Free Radic. Biol. Med. 2000, 28, 208–218. [Google Scholar] [CrossRef]
- de Zoete, M.R.; Palm, N.W.; Zhu, S.; Flavell, R.A. Inflammasomes. Cold Spring Harb. Perspect. Biol. 2014, 6, a016287. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Rivero, J.M.; de la Mata, M.; Pavon, A.D.; Villanueva-Paz, M.; Povea-Cabello, S.; Cotan, D.; Alvarez-Cordoba, M.; Villalon-Garcia, I.; Ybot-Gonzalez, P.; Salas, J.J.; et al. Intracellular cholesterol accumulation and coenzyme Q10 deficiency in Familial Hypercholesterolemia. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3697–3713. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Rivero, J.M.; Pastor-Maldonado, C.J.; de la Mata, M.; Villanueva-Paz, M.; Povea-Cabello, S.; Alvarez-Cordoba, M.; Villalon-Garcia, I.; Suarez-Carrillo, A.; Talaveron-Rey, M.; Munuera, M.; et al. Atherosclerosis and Coenzyme Q10. Int J. Mol. Sci. 2019, 20, 5195. [Google Scholar] [CrossRef] [Green Version]
- Finsterer, J. Atherosclerosis Can Be Mitochondrial: A Review. Cureus 2020, 12, e6987. [Google Scholar] [CrossRef] [Green Version]
- Cordero, M.D.; Alcocer-Gomez, E.; Marin-Aguilar, F.; Rybkina, T.; Cotan, D.; Perez-Pulido, A.; Alvarez-Suarez, J.M.; Battino, M.; Sanchez-Alcazar, J.A.; Carrion, A.M.; et al. Mutation in cytochrome b gene of mitochondrial DNA in a family with fibromyalgia is associated with NLRP3-inflammasome activation. J. Med. Genet. 2016, 53, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Riley, J.S.; Tait, S.W. Mitochondrial DNA in inflammation and immunity. EMBO Rep. 2020, 21, e49799. [Google Scholar] [CrossRef]
- Yang, Y.; Karakhanova, S.; Hartwig, W.; D’Haese, J.G.; Philippov, P.P.; Werner, J.; Bazhin, A.V. Mitochondria and Mitochondrial ROS in Cancer: Novel Targets for Anticancer Therapy. J. Cell Physiol. 2016, 231, 2570–2581. [Google Scholar] [CrossRef]
- Stefano, G.B.; Bjenning, C.; Wang, F.; Wang, N.; Kream, R.M. Mitochondrial Heteroplasmy. Adv. Exp. Med. Biol 2017, 982, 577–594. [Google Scholar] [CrossRef]
- Stewart, J.B.; Chinnery, P.F. The dynamics of mitochondrial DNA heteroplasmy: Implications for human health and disease. Nat. Rev. Genet. 2015, 16, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.P.; Cheng, K.; Ning, M.A.; Li, H.H.; Wang, H.C.; Li, F.; Chen, S.Y.; Qu, F.L.; Guo, W.Y. Association between peripheral blood cells mitochondrial DNA content and severity of coronary heart disease. Atherosclerosis 2017, 261, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Ashar, F.N.; Zhang, Y.; Longchamps, R.J.; Lane, J.; Moes, A.; Grove, M.L.; Mychaleckyj, J.C.; Taylor, K.D.; Coresh, J.; Rotter, J.I.; et al. Association of Mitochondrial DNA Copy Number With Cardiovascular Disease. JAMA Cardiol. 2017, 2, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.B.; Freyer, C.; Elson, J.L.; Larsson, N.G. Purifying selection of mtDNA and its implications for understanding evolution and mitochondrial disease. Nat. Rev. Genet. 2008, 9, 657–662. [Google Scholar] [CrossRef]
- Sazonova, M.A.; Sinyov, V.V.; Barinova, V.A.; Ryzhkova, A.I.; Zhelankin, A.V.; Postnov, A.Y.; Sobenin, I.A.; Bobryshev, Y.V.; Orekhov, A.N. Mosaicism of mitochondrial genetic variation in atherosclerotic lesions of the human aorta. Biomed. Res. Int 2015, 2015, 825468. [Google Scholar] [CrossRef] [Green Version]
- Sobenin, I.A.; Sazonova, M.A.; Postnov, A.Y.; Bobryshev, Y.V.; Orekhov, A.N. Changes of mitochondria in atherosclerosis: Possible determinant in the pathogenesis of the disease. Atherosclerosis 2013, 227, 283–288. [Google Scholar] [CrossRef]
- Volobueva, A.; Grechko, A.; Yet, S.F.; Sobenin, I.; Orekhov, A. Changes in Mitochondrial Genome Associated with Predisposition to Atherosclerosis and Related Disease. Biomolecules 2019, 9, 377. [Google Scholar] [CrossRef] [Green Version]
- Sazonova, M.A.; Sinyov, V.V.; Ryzhkova, A.I.; Galitsyna, E.V.; Khasanova, Z.B.; Postnov, A.Y.; Yarygina, E.I.; Orekhov, A.N.; Sobenin, I.A. Role of Mitochondrial Genome Mutations in Pathogenesis of Carotid Atherosclerosis. Oxid. Med. Cell Longev. 2017, 2017, 6934394. [Google Scholar] [CrossRef] [PubMed]
- Sobenin, I.A.; Sazonova, M.A.; Postnov, A.Y.; Salonen, J.T.; Bobryshev, Y.V.; Orekhov, A.N. Association of mitochondrial genetic variation with carotid atherosclerosis. PLoS ONE 2013, 8, e68070. [Google Scholar] [CrossRef] [Green Version]
- Sobenin, I.A.; Zhelankin, A.V.; Khasanova, Z.B.; Sinyov, V.V.; Medvedeva, L.V.; Sagaidak, M.O.; Makeev, V.J.; Kolmychkova, K.I.; Smirnova, A.S.; Sukhorukov, V.N.; et al. Heteroplasmic Variants of Mitochondrial DNA in Atherosclerotic Lesions of Human Aortic Intima. Biomolecules 2019, 9, 455. [Google Scholar] [CrossRef] [Green Version]
- Dominic, E.A.; Ramezani, A.; Anker, S.D.; Verma, M.; Mehta, N.; Rao, M. Mitochondrial cytopathies and cardiovascular disease. Heart 2014, 100, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Beckman, K.B.; Ames, B.N. Oxidative decay of DNA. J. Biol. Chem. 1997, 272, 19633–19636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, S.M.; Jung, Y.K. A Molecular Approach to Mitophagy and Mitochondrial Dynamics. Mol. Cells 2018, 41, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. Autophagy and Mitophagy in Cardiovascular Disease. Circ. Res. 2017, 120, 1812–1824. [Google Scholar] [CrossRef] [PubMed]
- Ko, M.S.; Yun, J.Y.; Baek, I.J.; Jang, J.E.; Hwang, J.J.; Lee, S.E.; Heo, S.H.; Bader, D.A.; Lee, C.H.; Han, J.; et al. Mitophagy deficiency increases NLRP3 to induce brown fat dysfunction in mice. Autophagy 2020, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.P.; Liu, X.J.; Xie, L.; Shen, X.Z.; Wu, J. Impaired mitophagy triggers NLRP3 inflammasome activation during the progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis. Lab. Invest. 2019, 99, 749–763. [Google Scholar] [CrossRef] [PubMed]
- Orekhov, A.N.; Poznyak, A.V.; Sobenin, I.A.; Nikifirov, N.N.; Ivanova, E.A. Mitochondrion as a Selective Target for the Treatment of Atherosclerosis: Role of Mitochondrial DNA Mutations and Defective Mitophagy in the Pathogenesis of Atherosclerosis and Chronic Inflammation. Curr. Neuropharmacol. 2020, 18, 1064–1075. [Google Scholar] [CrossRef] [PubMed]
- Orekhov, A.N.; Nikiforov, N.N.; Ivanova, E.A.; Sobenin, I.A. Possible Role of Mitochondrial DNA Mutations in Chronification of Inflammation: Focus on Atherosclerosis. J. Clin. Med. 2020, 9, 978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oesterle, A.; Laufs, U.; Liao, J.K. Pleiotropic Effects of Statins on the Cardiovascular System. Circ. Res. 2017, 120, 229–243. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Li, M.; Liu, X. Effects of long-term atorvastatin treatment on cardiac aging. Exp. Ther. Med. 2013, 6, 721–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, X.; Ma, Y.; Ruan, Y.; Fu, G.; Wu, S. Long-term atorvastatin improves age-related endothelial dysfunction by ameliorating oxidative stress and normalizing eNOS/iNOS imbalance in rat aorta. Exp. Gerontol. 2014, 52, 9–17. [Google Scholar] [CrossRef]
- Allen, S.C.; Mamotte, C.D.S. Pleiotropic and Adverse Effects of Statins-Do Epigenetics Play a Role? J. Pharmacol. Exp. Ther. 2017, 362, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Babelova, A.; Sedding, D.G.; Brandes, R.P. Anti-atherosclerotic mechanisms of statin therapy. Curr. Opin. Pharmacol. 2013, 13, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Parizadeh, S.M.; Azarpazhooh, M.R.; Moohebati, M.; Nematy, M.; Ghayour-Mobarhan, M.; Tavallaie, S.; Rahsepar, A.A.; Amini, M.; Sahebkar, A.; Mohammadi, M.; et al. Simvastatin therapy reduces prooxidant-antioxidant balance: Results of a placebo-controlled cross-over trial. Lipids 2011, 46, 333–340. [Google Scholar] [CrossRef]
- Sirtori, C.R. The pharmacology of statins. Pharmacol. Res. 2014, 88, 3–11. [Google Scholar] [CrossRef]
- Kong, F.; Ye, B.; Lin, L.; Cai, X.; Huang, W.; Huang, Z. Atorvastatin suppresses NLRP3 inflammasome activation via TLR4/MyD88/NF-kappaB signaling in PMA-stimulated THP-1 monocytes. Biomed. Pharmacother. 2016, 82, 167–172. [Google Scholar] [CrossRef]
- Apostolopoulou, M.; Corsini, A.; Roden, M. The role of mitochondria in statin-induced myopathy. Eur J. Clin. Invest. 2015, 45, 745–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abd, T.T.; Jacobson, T.A. Statin-induced myopathy: A review and update. Expert Opin. Drug Saf. 2011, 10, 373–387. [Google Scholar] [CrossRef]
- Ayers, J.; Cook, J.; Koenig, R.A.; Sisson, E.M.; Dixon, D.L. Recent Developments in the Role of Coenzyme Q10 for Coronary Heart Disease: A Systematic Review. Curr. Atheroscler. Rep. 2018, 20, 29. [Google Scholar] [CrossRef] [PubMed]
- De Pinieux, G.; Chariot, P.; Ammi-Said, M.; Louarn, F.; Lejonc, J.L.; Astier, A.; Jacotot, B.; Gherardi, R. Lipid-lowering drugs and mitochondrial function: Effects of HMG-CoA reductase inhibitors on serum ubiquinone and blood lactate/pyruvate ratio. Br. J. Clin. Pharmacol. 1996, 42, 333–337. [Google Scholar] [CrossRef] [Green Version]
- Stroes, E.S.; Thompson, P.D.; Corsini, A.; Vladutiu, G.D.; Raal, F.J.; Ray, K.K.; Roden, M.; Stein, E.; Tokgozoglu, L.; Nordestgaard, B.G.; et al. Statin-associated muscle symptoms: Impact on statin therapy-European Atherosclerosis Society Consensus Panel Statement on Assessment, Aetiology and Management. Eur. Heart J. 2015, 36, 1012–1022. [Google Scholar] [CrossRef]
- Laaksonen, R.; Jokelainen, K.; Laakso, J.; Sahi, T.; Harkonen, M.; Tikkanen, M.J.; Himberg, J.J. The effect of simvastatin treatment on natural antioxidants in low-density lipoproteins and high-energy phosphates and ubiquinone in skeletal muscle. Am. J. Cardiol. 1996, 77, 851–854. [Google Scholar] [CrossRef]
- Caso, G.; Kelly, P.; McNurlan, M.A.; Lawson, W.E. Effect of coenzyme q10 on myopathic symptoms in patients treated with statins. Am. J. Cardiol. 2007, 99, 1409–1412. [Google Scholar] [CrossRef]
- Young, J.M.; Florkowski, C.M.; Molyneux, S.L.; McEwan, R.G.; Frampton, C.M.; George, P.M.; Scott, R.S. Effect of coenzyme Q(10) supplementation on simvastatin-induced myalgia. Am. J. Cardiol. 2007, 100, 1400–1403. [Google Scholar] [CrossRef] [PubMed]
- Bookstaver, D.A.; Burkhalter, N.A.; Hatzigeorgiou, C. Effect of coenzyme Q10 supplementation on statin-induced myalgias. Am. J. Cardiol 2012, 110, 526–529. [Google Scholar] [CrossRef]
- Bogsrud, M.P.; Langslet, G.; Ose, L.; Arnesen, K.E.; Sm Stuen, M.C.; Malt, U.F.; Woldseth, B.; Retterstol, K. No effect of combined coenzyme Q10 and selenium supplementation on atorvastatin-induced myopathy. Scand. Cardiovasc J. 2013, 47, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, A.A.; Strunz, C.M.; Takada, J.Y.; Mansur, A.P. Biochemical markers of muscle damage and high serum concentration of creatine kinase in patients on statin therapy. Biomark Med. 2019, 13, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Rosenson, R.S.; Baker, S.K.; Jacobson, T.A.; Kopecky, S.L.; Parker, B.A.; The National Lipid Association’s Muscle Safety Expert, P. An assessment by the Statin Muscle Safety Task Force: 2014 update. J. Clin. Lipidol. 2014, 8, S58–S71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chokchaiwong, S.; Kuo, Y.T.; Lin, S.H.; Hsu, Y.C.; Hsu, S.P.; Liu, Y.T.; Chou, A.J.; Kao, S.H. Coenzyme Q10 serves to couple mitochondrial oxidative phosphorylation and fatty acid beta-oxidation, and attenuates NLRP3 inflammasome activation. Free Radic. Res. 2018, 52, 1445–1455. [Google Scholar] [CrossRef]
- Cordero, M.D.; Alcocer-Gomez, E.; de Miguel, M.; Culic, O.; Carrion, A.M.; Alvarez-Suarez, J.M.; Bullon, P.; Battino, M.; Fernandez-Rodriguez, A.; Sanchez-Alcazar, J.A. Can coenzyme q10 improve clinical and molecular parameters in fibromyalgia? Antioxid. Redox Signal. 2013, 19, 1356–1361. [Google Scholar] [CrossRef]
- Wang, D.; Yan, X.; Xia, M.; Yang, Y.; Li, D.; Li, X.; Song, F.; Ling, W. Coenzyme Q10 promotes macrophage cholesterol efflux by regulation of the activator protein-1/miR-378/ATP-binding cassette transporter G1-signaling pathway. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1860–1870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Liu, H.; Hao, Y.; Xu, L.; Zhang, T.; Liu, Y.; Guo, L.; Zhu, L.; Pei, Z. Coenzyme Q10 protects against hyperlipidemia-induced cardiac damage in apolipoprotein E-deficient mice. Lipids Health Dis. 2018, 17, 279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, L.; Feng, Y.; Chen, G.C.; Qin, L.Q.; Fu, C.L.; Chen, L.H. Effects of coenzyme Q10 supplementation on inflammatory markers: A systematic review and meta-analysis of randomized controlled trials. Pharmacol Res. 2017, 119, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Liu, Y. Efficacy of coenzyme Q10 in patients with cardiac failure: A meta-analysis of clinical trials. BMC Cardiovasc. Disord. 2017, 17, 196. [Google Scholar] [CrossRef] [Green Version]
- Mehrabani, S.; Askari, G.; Miraghajani, M.; Tavakoly, R.; Arab, A. Effect of coenzyme Q10 supplementation on fatigue: A systematic review of interventional studies. Complement. Ther. Med. 2019, 43, 181–187. [Google Scholar] [CrossRef]
- Mercer, J.R.; Yu, E.; Figg, N.; Cheng, K.K.; Prime, T.A.; Griffin, J.L.; Masoodi, M.; Vidal-Puig, A.; Murphy, M.P.; Bennett, M.R. The mitochondria-targeted antioxidant MitoQ decreases features of the metabolic syndrome in ATM+/-/ApoE-/- mice. Free Radic. Biol. Med. 2012, 52, 841–849. [Google Scholar] [CrossRef]
- Docherty, C.K.; Carswell, A.; Friel, E.; Mercer, J.R. Impaired mitochondrial respiration in human carotid plaque atherosclerosis: A potential role for Pink1 in vascular smooth muscle cell energetics. Atherosclerosis 2018, 268, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Bullon, P.; Marin-Aguilar, F.; Roman-Malo, L. AMPK/Mitochondria in Metabolic Diseases. Exp. Suppl. 2016, 107, 129–152. [Google Scholar] [CrossRef]
- Ewart, M.A.; Kennedy, S. AMPK and vasculoprotection. Pharmacol. Ther. 2011, 131, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Vasamsetti, S.B.; Karnewar, S.; Kanugula, A.K.; Thatipalli, A.R.; Kumar, J.M.; Kotamraju, S. Metformin inhibits monocyte-to-macrophage differentiation via AMPK-mediated inhibition of STAT3 activation: Potential role in atherosclerosis. Diabetes 2015, 64, 2028–2041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, B.; Tannahill, G.M.; Murphy, M.P.; O’Neill, L.A. Metformin Inhibits the Production of Reactive Oxygen Species from NADH:Ubiquinone Oxidoreductase to Limit Induction of Interleukin-1beta (IL-1beta) and Boosts Interleukin-10 (IL-10) in Lipopolysaccharide (LPS)-activated Macrophages. J. Biol. Chem. 2015, 290, 20348–20359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, S.; Shan, Q.; Liu, P.; Feng, T.; Zhang, X.; Xiang, P.; Chen, K.; Xie, H.; Song, P.; Zhou, L.; et al. Metformin ameliorates arsenic trioxide hepatotoxicity via inhibiting mitochondrial complex I. Cell Death Dis. 2017, 8, e3159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattori, Y.; Suzuki, K.; Hattori, S.; Kasai, K. Metformin inhibits cytokine-induced nuclear factor kappaB activation via AMP-activated protein kinase activation in vascular endothelial cells. Hypertension 2006, 47, 1183–1188. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.A.; Choi, H.C. Metformin inhibits inflammatory response via AMPK-PTEN pathway in vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 2012, 425, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Szanto, A.; Nagy, L. The many faces of PPARgamma: Anti-inflammatory by any means? Immunobiology 2008, 213, 789–803. [Google Scholar] [CrossRef] [PubMed]
- LeBrasseur, N.K.; Kelly, M.; Tsao, T.S.; Farmer, S.R.; Saha, A.K.; Ruderman, N.B.; Tomas, E. Thiazolidinediones can rapidly activate AMP-activated protein kinase in mammalian tissues. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E175–E181. [Google Scholar] [CrossRef] [Green Version]
- Coletta, D.K.; Sriwijitkamol, A.; Wajcberg, E.; Tantiwong, P.; Li, M.; Prentki, M.; Madiraju, M.; Jenkinson, C.P.; Cersosimo, E.; Musi, N.; et al. Pioglitazone stimulates AMP-activated protein kinase signalling and increases the expression of genes involved in adiponectin signalling, mitochondrial function and fat oxidation in human skeletal muscle in vivo: A randomised trial. Diabetologia 2009, 52, 723–732. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Chen, T.; Wu, Y.; Yang, L.; Wang, L.; Fan, X.; Zhang, W.; Feng, J.; Yu, H.; Yang, Y.; et al. Pioglitazone stabilizes atherosclerotic plaque by regulating the Th17/Treg balance in AMPK-dependent mechanisms. Cardiovasc. Diabetol. 2017, 16, 140. [Google Scholar] [CrossRef]
- Ou, H.T.; Chang, K.C.; Li, C.Y.; Wu, J.S. Risks of cardiovascular diseases associated with dipeptidyl peptidase-4 inhibitors and other antidiabetic drugs in patients with type 2 diabetes: A nation-wide longitudinal study. Cardiovasc. Diabetol. 2016, 15, 41. [Google Scholar] [CrossRef] [Green Version]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Dabritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167, 457–470 e413. [Google Scholar] [CrossRef] [Green Version]
- Michishita, E.; Park, J.Y.; Burneskis, J.M.; Barrett, J.C.; Horikawa, I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol. Biol. Cell 2005, 16, 4623–4635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burns, J.; Yokota, T.; Ashihara, H.; Lean, M.E.; Crozier, A. Plant foods and herbal sources of resveratrol. J. Agric. Food Chem. 2002, 50, 3337–3340. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.R.; Li, S.; Lin, C.C. Effect of resveratrol and pterostilbene on aging and longevity. Biofactors 2018, 44, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Riccioni, G.; Gammone, M.A.; Tettamanti, G.; Bergante, S.; Pluchinotta, F.R.; D’Orazio, N. Resveratrol and anti-atherogenic effects. Int. J. Food Sci. Nutr. 2015, 66, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Coll, R.C.; Robertson, A.A.; Chae, J.J.; Higgins, S.C.; Munoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [Green Version]
- van Hout, G.P.; Bosch, L.; Ellenbroek, G.H.; de Haan, J.J.; van Solinge, W.W.; Cooper, M.A.; Arslan, F.; de Jager, S.C.; Robertson, A.A.; Pasterkamp, G.; et al. The selective NLRP3-inflammasome inhibitor MCC950 reduces infarct size and preserves cardiac function in a pig model of myocardial infarction. Eur. Heart J. 2017, 38, 828–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Heijden, T.; Kritikou, E.; Venema, W.; van Duijn, J.; van Santbrink, P.J.; Slutter, B.; Foks, A.C.; Bot, I.; Kuiper, J. NLRP3 Inflammasome Inhibition by MCC950 Reduces Atherosclerotic Lesion Development in Apolipoprotein E-Deficient Mice-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1457–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; He, H.; Chen, Y.; Huang, W.; Cheng, J.; Ye, J.; Wang, A.; Tao, J.; Wang, C.; Liu, Q.; et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J. Exp. Med. 2017, 214, 3219–3238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Li, C.; Yin, H.; Zhang, X.; Li, Y. NLRP3 Inflammasome: A Potential Alternative Therapy Target for Atherosclerosis. Evid Based Complement. Alternat. Med. 2020, 2020, 1561342. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Du, H.; Liu, X.; Fu, X.; Li, X.; Cao, Q. Artemisinin alleviates atherosclerotic lesion by reducing macrophage inflammation via regulation of AMPK/NF-kappaB/NLRP3 inflammasomes pathway. J. Drug Target. 2020, 28, 70–79. [Google Scholar] [CrossRef]
- Yao, Y.; Mao, J.; Xu, S.; Zhao, L.; Long, L.; Chen, L.; Li, D.; Lu, S. Rosmarinic acid inhibits nicotine-induced C-reactive protein generation by inhibiting NLRP3 inflammasome activation in smooth muscle cells. J. Cell Physiol. 2019, 234, 1758–1767. [Google Scholar] [CrossRef]
- Kong, F.; Ye, B.; Cao, J.; Cai, X.; Lin, L.; Huang, S.; Huang, W.; Huang, Z. Curcumin Represses NLRP3 Inflammasome Activation via TLR4/MyD88/NF-kappaB and P2X7R Signaling in PMA-Induced Macrophages. Front. Pharmacol. 2016, 7, 369. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Huang, K.; Lin, X.; Chen, Q.; Lin, S.; Feng, X.; Zhen, C.; Huang, M.; Wang, S. Berberine Attenuates NLRP3 Inflammasome Activation in Macrophages to Reduce the Secretion of Interleukin-1beta. Ann. Clin. Lab. Sci 2017, 47, 720–728. [Google Scholar]
- Ricci, C.; Ruscica, M.; Camera, M.; Rossetti, L.; Macchi, C.; Colciago, A.; Zanotti, I.; Lupo, M.G.; Adorni, M.P.; Cicero, A.F.G.; et al. PCSK9 induces a pro-inflammatory response in macrophages. Sci. Rep. 2018, 8, 2267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenson, R.S.; Hegele, R.A.; Fazio, S.; Cannon, C.P. The Evolving Future of PCSK9 Inhibitors. J. Am. Coll. Cardiol. 2018, 72, 314–329. [Google Scholar] [CrossRef] [PubMed]
- Ruscica, M.; Tokgozoglu, L.; Corsini, A.; Sirtori, C.R. PCSK9 inhibition and inflammation: A narrative review. Atherosclerosis 2019, 288, 146–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suárez-Rivero, J.M.; Pastor-Maldonado, C.J.; Povea-Cabello, S.; Álvarez-Córdoba, M.; Villalón-García, I.; Talaverón-Rey, M.; Suárez-Carrillo, A.; Munuera-Cabeza, M.; Sánchez-Alcázar, J.A. From Mitochondria to Atherosclerosis: The Inflammation Path. Biomedicines 2021, 9, 258. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9030258
Suárez-Rivero JM, Pastor-Maldonado CJ, Povea-Cabello S, Álvarez-Córdoba M, Villalón-García I, Talaverón-Rey M, Suárez-Carrillo A, Munuera-Cabeza M, Sánchez-Alcázar JA. From Mitochondria to Atherosclerosis: The Inflammation Path. Biomedicines. 2021; 9(3):258. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9030258
Chicago/Turabian StyleSuárez-Rivero, Juan M., Carmen J. Pastor-Maldonado, Suleva Povea-Cabello, Mónica Álvarez-Córdoba, Irene Villalón-García, Marta Talaverón-Rey, Alejandra Suárez-Carrillo, Manuel Munuera-Cabeza, and José A. Sánchez-Alcázar. 2021. "From Mitochondria to Atherosclerosis: The Inflammation Path" Biomedicines 9, no. 3: 258. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9030258