
The Role of Microbiome and Virome in Idiopathic Pulmonary Fibrosis

 , , and
, , and 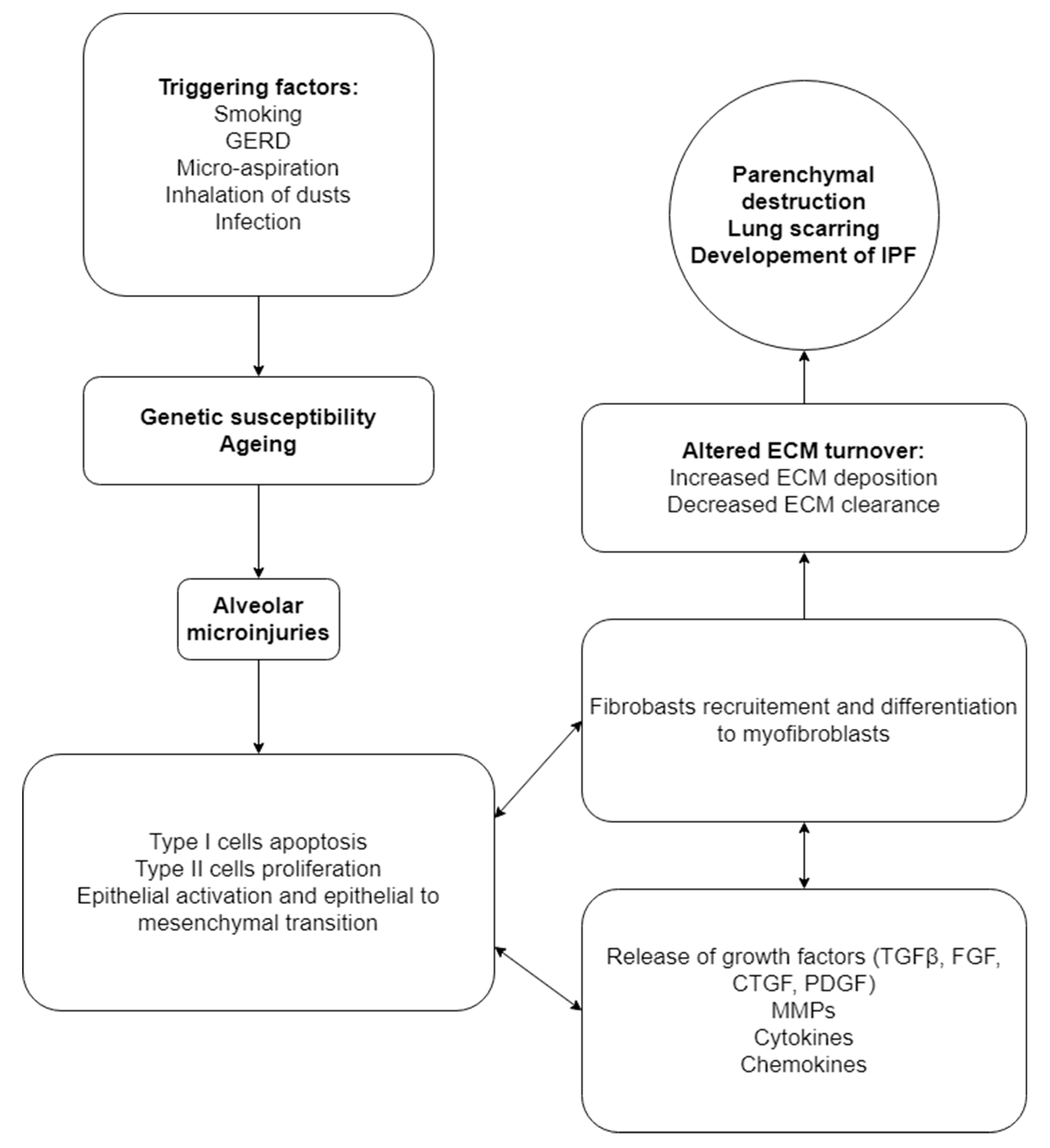{kind=link}
{kind=link}
Abstract
:1. Introduction
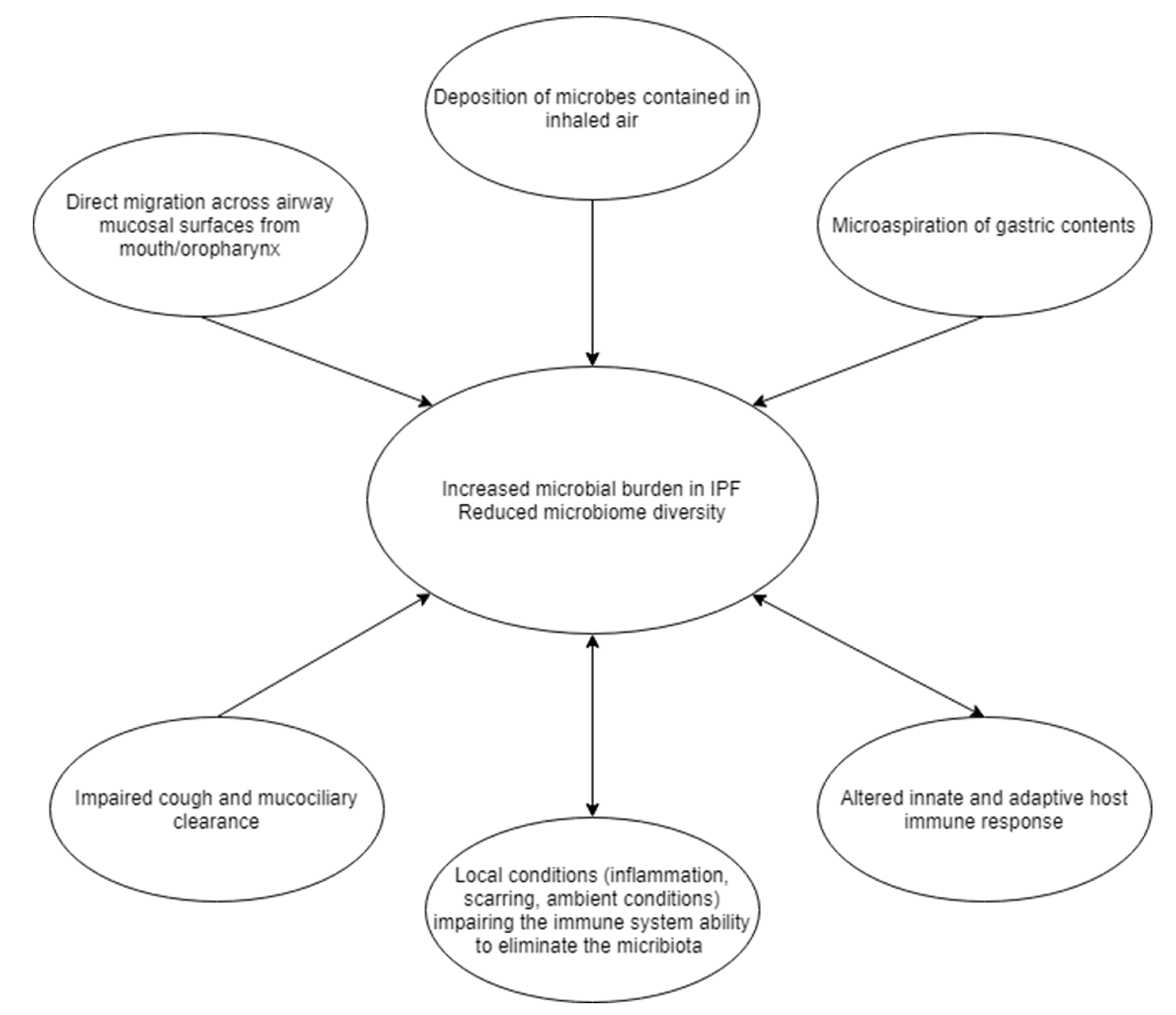
2. Lung Microbiome and Virome in Healthy Subjects
3. Lung Microbiome in Stable IPF
4. Lung Virome in Stable IPF
5. Lung Microbiome in Acute Exacerbation of IPF
6. Lung Virome in Acute Exacerbation of IPF
7. Microbial Involvement and Host Response in IPF Progression
8. Lung Microbiome as a Treatment Target in IPF
9. The “Gut-Lung” Axis in Lung Disease and IPF
10. COVID-19 and IPF
11. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dickson, R.P.; Erb-Downward, J.R.; Martinez, F.J.; Huffnagle, G.B. The Microbiome and the Respiratory Tract. Ann. Rev. Physiol. 2016, 78, 481–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickson, R.P.; Erb-Downward, J.R.; Prescott, H.C.; Martinez, F.J.; Curtis, J.L.; Lama, V.N.; Huffnagle, G.B. Analysis of culture-dependent versus culture-independent techniques for identification of bacteria in clinically obtained bronchoalveolar lavage fluid. J. Clin. Microbiol. 2014, 52, 3605–3613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbas, A.A.; Diamond, J.M.; Chehoud, C.; Chang, B.; Kotzin, J.J.; Young, J.C.; Imai, I.; Haas, A.R.; Cantu, E.; Lederer, D.J.; et al. The Perioperative Lung Transplant Virome: Torque Teno Viruses Are Elevated in Donor Lungs and Show Divergent Dynamics in Primary Graft Dysfunction. Am. J. Transplant. 2017, 17, 1313–1324. [Google Scholar] [CrossRef] [Green Version]
- Wylie, K.M.; Weinstock, G.M.; Storch, G.A. Virome genomics: A tool for defining the human virome. Curr. Opin. Microbiol. 2013, 16, 479–484. [Google Scholar] [CrossRef] [Green Version]
- Young, J.C.; Chehoud, C.; Bittinger, K.; Bailey, A.; Diamond, J.M.; Cantu, E.; Haas, A.R.; Abbas, A.; Frye, L.; Christie, J.D.; et al. Viral Metagenomics Reveal Blooms of Anelloviruses in the Respiratory Tract of Lung Transplant Recipients. Am. J. Transplant. 2015, 15, 200–209. [Google Scholar] [CrossRef] [Green Version]
- Hewitt, R.J.; Molyneaux, P.L. The respiratory microbiome in idiopathic pulmonary fibrosis. Ann. Transl. Med. 2017, 5, 250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef]
- King, T.E., Jr.; Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. Lancet 2011, 378, 1949–1961. [Google Scholar] [CrossRef]
- Tzouvelekis, A.; Ntolios, P.; Karampitsakos, T.; Tzilas, V.; Anevlavis, S.; Bouros, E.; Steiropoulos, P.; Koulouris, N.; Stratakos, G.; Froudarakis, M.; et al. Safety and efficacy of pirfenidone in severe Idiopathic Pulmonary Fibrosis: A real-world observational study. Pulm. Pharmacol. Ther. 2017, 46, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D.; Costabel, U.; Hansell, D.M.; King, T.E., Jr.; Lynch, D.A.; Nicholson, A.G.; Ryerson, C.J.; Ryu, J.H.; Selman, M.; Wells, A.U.; et al. An Official American Thoracic Society/European Respiratory Society Statement: Update of the International Multidisciplinary Classification of the Idiopathic Interstitial Pneumonias. Am. J. Respir. Crit. Care Med. 2013, 188, 733–748. [Google Scholar] [CrossRef]
- Tzouvelekis, A.; Karampitsakos, T.; Gomatou, G.; Bouros, E.; Tzilas, V.; Manali, E.; Tomos, I.; Trachalaki, A.; Kolilekas, L.; Korbila, I.; et al. Lung cancer in patients with Idiopathic Pulmonary Fibrosis. A retrospective multicenter study in Greece. Pulm. Pharmacol. Ther. 2020, 60, 101880. [Google Scholar] [CrossRef] [PubMed]
- Bouros, D.; Tzouvelekis, A. Idiopathic pulmonary fibrosis: On the move. Lancet Respir. Med. 2014, 2, 17–19. [Google Scholar] [CrossRef]
- Antoniou, K.M.; Tsitoura, E.; Vasarmidi, E.; Symvoulakis, E.K.; Aidinis, V.; Tzilas, V.; Tzouvelekis, A.; Bouros, D. Precision medicine in idiopathic pulmonary fibrosis therapy: From translational research to patient-centered care. Curr. Opin. Pharmacol. 2021, 57, 71–80. [Google Scholar] [CrossRef]
- Molyneaux, P.L.; Maher, T.M. The role of infection in the pathogenesis of idiopathic pulmonary fibrosis. Eur. Respir. Rev. 2013, 22, 376–381. [Google Scholar] [CrossRef] [Green Version]
- Spagnolo, P.; Molyneaux, P.L.; Bernardinello, N.; Cocconcelli, E.; Biondini, D.; Fracasso, F.; Tiné, M.; Saetta, M.; Maher, T.M.; Balestro, E. The Role of the Lung’s Microbiome in the Pathogenesis and Progression of Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2019, 20, 5618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seibold, M.A.; Wise, A.L.; Speer, M.C.; Steele, M.P.; Brown, K.K.; Loyd, J.E.; Fingerlin, T.E.; Zhang, W.; Gudmundsson, G.; Groshong, S.D.; et al. A Common MUC5B Promoter Polymorphism and Pulmonary Fibrosis. N. Engl. J. Med. 2011, 364, 1503–1512. [Google Scholar] [CrossRef] [Green Version]
- Tomos, I.P.; Tzouvelekis, A.; Aidinis, V.; Manali, E.D.; Bouros, E.; Bouros, D.; Papiris, S.A. Extracellular matrix remodeling in idiopathic pulmonary fibrosis. It is the ‘bed’ that counts and not ‘the sleepers’. Expert Rev. Respir. Med. 2017, 11, 299–309. [Google Scholar] [CrossRef]
- Dickson, R.P.; Erb-Downward, J.R.; Huffnagle, G.B. Towards an ecology of the lung: New conceptual models of pulmonary microbiology and pneumonia pathogenesis. Lancet Respir. Med. 2014, 2, 238–246. [Google Scholar] [CrossRef] [Green Version]
- Venkataraman, A.; Bassis, C.M.; Beck, J.M.; Young, V.B.; Curtis, J.L.; Huffnagle, G.B.; Schmidt, T.M. Application of a neutral community model to assess structuring of the human lung microbiome. mBio 2015, 6, e02284-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charlson, E.S.; Bittinger, K.; Haas, A.R.; Fitzgerald, A.S.; Frank, I.; Yadav, A.; Bushman, F.D.; Collman, R.G. Topographical Continuity of Bacterial Populations in the Healthy Human Respiratory Tract. Am. J. Respir. Crit. Care Med. 2011, 184, 957–963. [Google Scholar] [CrossRef] [Green Version]
- Dickson, R.P.; Erb-Downward, J.R.; Freeman, C.M.; McCloskey, L.; Beck, J.M.; Huffnagle, G.B.; Curtis, J.L. Spatial Variation in the Healthy Human Lung Microbiome and the Adapted Island Model of Lung Biogeography. Ann. Am. Thorac. Soc. 2015, 12, 821–830. [Google Scholar] [CrossRef]
- Morris, A.; Beck, J.M.; Schloss, P.D.; Campbell, T.B.; Crothers, K.; Curtis, J.L.; Flores, S.C.; Fontenot, A.P.; Ghedin, E.; Huang, L.; et al. Comparison of the respiratory microbiome in healthy nonsmokers and smokers. Am. J. Respir. Crit. Care Med. 2013, 187, 1067–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erb-Downward, J.R.; Thompson, D.L.; Han, M.K.; Freeman, C.M.; McCloskey, L.; Schmidt, L.A.; Young, V.B.; Toews, G.B.; Curtis, J.L.; Sundaram, B.; et al. Analysis of the lung microbiome in the “healthy” smoker and in COPD. PLoS ONE 2011, 6, e16384. [Google Scholar] [CrossRef] [Green Version]
- Molyneaux, P.L.; Cox, M.J.; Willis-Owen, S.A.G.; Mallia, P.; Russell, K.E.; Russell, A.-M.; Murphy, E.; Johnston, S.L.; Schwartz, D.A.; Wells, A.U.; et al. The role of bacteria in the pathogenesis and progression of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 906–913. [Google Scholar] [CrossRef] [Green Version]
- Hilty, M.; Burke, C.; Pedro, H.; Cardenas, P.; Bush, A.; Bossley, C.; Davies, J.; Ervine, A.; Poulter, L.; Pachter, L.; et al. Disordered microbial communities in asthmatic airways. PLoS ONE 2010, 5, e8578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Fastrès, A.; Felice, F.; Roels, E.; Moermans, C.; Corhay, J.-L.; Bureau, F.; Louis, R.; Clercx, C.; Guiot, J. The Lung Microbiome in Idiopathic Pulmonary Fibrosis: A Promising Approach for Targeted Therapies. Int. J. Mol. Sci. 2017, 18, 2735. [Google Scholar] [CrossRef] [Green Version]
- Richter, A.G.; Stockley, R.A.; Harper, L.; Thickett, D.R. Pulmonary infection in Wegener granulomatosis and idiopathic pulmonary fibrosis. Thorax 2009, 64, 692–697. [Google Scholar] [CrossRef] [Green Version]
- Friaza, V.; De La Horra, C.; Rodríguez-Domínguez, M.J.; Martín-Juan, J.; Cantón, R.; Calderón, E.J.; Del Campo, R. Metagenomic analysis of bronchoalveolar lavage samples from patients with idiopathic interstitial pneumonia and its antagonic relation with Pneumocystis jirovecii colonization. J. Microbiol. Methods 2010, 82, 98–101. [Google Scholar] [CrossRef]
- Garzoni, C.; Brugger, S.D.; Qi, W.; Wasmer, S.; Cusini, A.; Dumont, P.; Gorgievski-Hrisoho, M.; Mühlemann, K.; Von Garnier, C.; Hilty, M. Microbial communities in the respiratory tract of patients with interstitial lung disease. Thorax 2013, 68, 1150–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, M.K.; Zhou, Y.; Murray, S.; Tayob, N.; Noth, I.; Lama, V.N.; Moore, B.B.; White, E.S.; Flaherty, K.R.; Huffnagle, G.B.; et al. Lung microbiome and disease progression in idiopathic pulmonary fibrosis: An analysis of the COMET study. Lancet Respir. Med. 2014, 2, 548–556. [Google Scholar] [CrossRef] [Green Version]
- Invernizzi, R.; Wu, B.G.; Barnett, J.; Ghai, P.; Kingston, S.; Hewitt, R.J.; Feary, J.; Li, Y.; Chua, F.; Wu, Z.; et al. The Respiratory Microbiome in Chronic Hypersensitivity Pneumonitis Is Distinct from That of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2021, 203, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Saito, A.; Chiba, H.; Kuronuma, K.; Ikeda, K.; Kobayashi, T.; Ariki, S.; Takahashi, M.; Sasaki, Y.; Takahashi, H. Impaired diversity of the lung microbiome predicts progression of idiopathic pulmonary fibrosis. Respir. Res. 2018, 19, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Valenzi, E.; Yang, H.; Sembrat, J.C.; Yang, L.; Winters, S.; Nettles, R.; Kass, D.J.; Qin, S.; Wang, X.; Myerburg, M.M.; et al. Topographic heterogeneity of lung microbiota in end-stage idiopathic pulmonary fibrosis: The Microbiome in Lung Explants-2 (MiLEs-2) study. Thorax 2021, 76, 239–247. [Google Scholar] [CrossRef]
- Vergnon, J.M.; de Thé, G.; Weynants, P.; Vincent, M.; Mornex, J.F.; Brune, J. Cryptogenic Fibrosing Alveolitis and Epstein-Barr Virus: An Association? Lancet 1984, 324, 768–771. [Google Scholar] [CrossRef]
- Calabrese, F.; Kipar, A.; Lunardi, F.; Balestro, E.; Perissinotto, E.; Rossi, E.; Nannini, N.; Marulli, G.; Stewart, J.P.; Rea, F. Herpes Virus Infection Is Associated with Vascular Remodeling and Pulmonary Hypertension in Idiopathic Pulmonary Fibrosis. PLoS ONE 2013, 8, e55715. [Google Scholar] [CrossRef] [Green Version]
- Kelly, B.G.; Lok, S.S.; Hasleton, P.S.; Egan, J.J.; Stewart, J.P. A Rearranged Form of Epstein–Barr Virus DNA Is Associated with Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2002, 166, 510–513. [Google Scholar] [CrossRef] [PubMed]
- Manika, K.; Alexiou-Daniel, S.; Papakosta, D.; Papa, A.; Kontakiotis, T.; Patakas, D.; Antoniadis, A. Epstein-Barr virus DNA in bronchoalveolar lavage fluid from patients with idiopathic pulmonary fibrosis. Off. J. WASOG 2007, 24, 134–140. [Google Scholar]
- Stewart, J.P.; Egan, J.J.; Ross, A.J.; Kelly, B.G.; Lok, S.S.; Hasleton, P.S.; Woodcock, A.A. The Detection of Epstein-Barr Virus DNA in Lung Tissue from Patients with Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 1999, 159, 1336–1341. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Ohta, K.; Suzuki, N.; Yamaguchi, M.; Hirai, K.; Horiuchi, T.; Watanabe, J.; Miyamoto, T.; Ito, K. Idiopathic Pulmonary Fibrosis and High Prevalence of Serum Antibodies to Hepatitis C Virus. Am. Rev. Respir. Dis. 1992, 146, 266–268. [Google Scholar] [CrossRef] [PubMed]
- Arase, Y.; Suzuki, F.; Suzuki, Y.; Akuta, N.; Kobayashi, M.; Kawamura, Y.; Yatsuji, H.; Sezaki, H.; Hosaka, T.; Hirakawa, M.; et al. Hepatitis C virus enhances incidence of idiopathic pulmonary fibrosis. World J. Gastroenterol. 2008, 14, 5880–5886. [Google Scholar] [CrossRef] [PubMed]
- Irving, W.L.; Day, S.; Johnston, I.D.A. Idiopathic Pulmonary Fibrosis and Hepatitis C Virus Infection. Am. Rev. Respir. Dis. 1993, 148, 1683–1684. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; Strong, M.J.; Zhuang, Y.; Flemington, E.K.; Kaminski, N.; De Andrade, J.A.; Lasky, J.A. Assessment of viral RNA in idiopathic pulmonary fibrosis using RNA-seq. BMC Pulm. Med. 2020, 20, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Collard, H.R.; Ryerson, C.J.; Corte, T.J.; Jenkins, G.; Kondoh, Y.; Lederer, D.J.; Lee, J.S.; Maher, T.M.; Wells, A.U.; Antoniou, K.M.; et al. Acute Exacerbation of Idiopathic Pulmonary Fibrosis. An International Working Group Report. Am. J. Respir. Crit. Care Med. 2016, 194, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Collard, H.R.; Moore, B.B.; Flaherty, K.R.; Brown, K.K.; Kaner, R.J.; King, T.E.; Lasky, J.A.; Loyd, J.E.; Noth, I.; Olman, M.A.; et al. Acute Exacerbations of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2007, 176, 636–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molyneaux, P.L.; Cox, M.J.; Wells, A.U.; Kim, H.C.; Ji, W.; Cookson, W.O.C.; Moffatt, M.F.; Kim, D.S.; Maher, T.M. Changes in the respiratory microbiome during acute exacerbations of idiopathic pulmonary fibrosis. Respir. Res. 2017, 18, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Weng, D.; Chen, X.-Q.; Qiu, H.; Zhang, Y.; Li, Q.-H.; Zhao, M.-M.; Wu, Q.; Chen, T.; Hu, Y.; Wang, L.-S.; et al. The Role of Infection in Acute Exacerbation of Idiopathic Pulmonary Fibrosis. Mediat. Inflamm. 2019, 2019, 1–10. [Google Scholar] [CrossRef]
- Knippenberg, S.; Ueberberg, B.; Maus, R.; Bohling, J.; Ding, N.; Tarres, M.T.; Hoymann, H.-G.; Jonigk, D.; Izykowski, N.; Paton, J.C.; et al. Streptococcus pneumoniae triggers progression of pulmonary fibrosis through pneumolysin. Thorax 2015, 70, 636–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Dwyer, D.N.; Ashley, S.L.; Gurczynski, S.J.; Xia, M.; Wilke, C.; Falkowski, N.R.; Norman, K.C.; Arnold, K.B.; Huffnagle, G.B.; Salisbury, M.L.; et al. Lung microbiota contribute to pulmonary inflammation and disease progression in pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1127–1138. [Google Scholar] [CrossRef]
- Vannella, K.M.; Moore, B.B. Viruses as co-factors for the initiation or exacerbation of lung fibrosis. Fibrogenesis Tissue Repair 2008, 1, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wootton, S.C.; Kim, D.S.; Chiu, C.; Kondoh, Y.; Lee, J.S.; Kistler, A.; Ganem, D.; DeRisi, J.; Collard, H.R. Occult Viral Infection in Acute Exacerbation of Idiopathic Pulmonary Fibrosis. Am. J. Resp. Crit. Care Med. 2011, 183, 1698–1702. [Google Scholar] [CrossRef] [PubMed]
- Bando, M.; Ohno, S.; Oshikawa, K.; Takahashi, M.; Okamoto, H.; Sugiyama, Y. Infection of TT virus in patients with idiopathic pulmonary fibrosis. Respir. Med. 2001, 95, 935–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shulgina, L.; Cahn, A.P.; Chilvers, E.R.; Parfrey, H.; Clark, A.B.; Wilson, E.C.F.; Twentyman, O.P.; Davison, A.G.; Curtin, J.J.; Crawford, M.B.; et al. Treating idiopathic pulmonary fibrosis with the addition of co-trimoxazole: A randomised controlled trial. Thorax 2013, 68, 155–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghu, G.; Anstrom, K.J.; King, T.E., Jr.; Lasky, J.A.; Martinez, F.J. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N. Engl. J. Med. 2012, 366, 1968–1977. [Google Scholar]
- Oldham, J.M.; Ma, S.-F.; Martinez, F.J.; Anstrom, K.J.; Raghu, G.; Schwartz, D.A.; Valenzi, E.; Witt, L.; Lee, C.; Vij, R.; et al. TOLLIP, MUC5B, and the Response toN-Acetylcysteine among Individuals with Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2015, 192, 1475–1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noth, I.; Zhang, Y.; Ma, S.-F.; Flores, C.; Barber, M.; Huang, Y.; Broderick, S.M.; Wade, M.S.; Hysi, P.; Scuirba, J.; et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: A genome-wide association study. Lancet Respir. Med. 2013, 1, 309–317. [Google Scholar] [CrossRef] [Green Version]
- Peljto, A.L.; Zhang, Y.; Fingerlin, T.E.; Ma, S.-F.; Garcia, J.G.N.; Richards, T.J.; Silveira, L.J.; Lindell, K.O.; Steele, M.P.; Loyd, J.E.; et al. Association Between the MUC5B Promoter Polymorphism and Survival in Patients with Idiopathic Pulmonary Fibrosis. JAMA 2013, 309, 2232–2239. [Google Scholar] [CrossRef]
- Roy, M.G.; Livraghi-Butrico, A.; Fletcher, A.A.; McElwee, M.M.; Evans, S.E.; Boerner, R.M.; Alexander, S.N.; Bellinghausen, L.K.; Song, A.S.; Petrova, Y.M.; et al. Muc5b is required for airway defence. Nature 2014, 505, 412–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molyneaux, P.L.; Willis-Owen, S.A.G.; Cox, M.J.; James, P.; Cowman, S.; Loebinger, M.; Blanchard, A.; Edwards, L.M.; Stock, C.; Daccord, C.; et al. Host–Microbial Interactions in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2017, 195, 1640–1650. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Ma, S.-F.; Espindola, M.S.; Vij, R.; Oldham, J.M.; Huffnagle, G.B.; Erb-Downward, J.R.; Flaherty, K.R.; Moore, B.B.; White, E.S.; et al. Microbes Are Associated with Host Innate Immune Response in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2017, 196, 208–219. [Google Scholar] [CrossRef] [Green Version]
- Varney, V.; Parnell, H.; Salisbury, D.; Ratnatheepan, S.; Tayar, R. A double blind randomised placebo controlled pilot study of oral co-trimoxazole in advanced fibrotic lung disease. Pulm. Pharmacol. Ther. 2008, 21, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Hammond, M.; Clark, A.B.; Cahn, A.P.; Chilvers, E.R.; Fraser, W.D.; Livermore, D.M.; Maher, T.M.; Parfrey, H.; Swart, A.M.; Stirling, S.; et al. The Efficacy and Mechanism Evaluation of Treating Idiopathic Pulmonary fibrosis with the Addition of Co-trimoxazole (EME-TIPAC): Study protocol for a randomised controlled trial. Trials 2018, 19, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swarnakar, S.; Mishra, A.; Paul, S.; Bhattacharya, P.; Paul, R. An alternative therapy for idiopathic pulmonary fibrosis by doxycycline through matrix metalloproteinase inhibition. Lung India 2011, 28, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Anstrom, K.J.; Noth, I.; Flaherty, K.R.; Edwards, R.H.; Albright, J.; Baucom, A.; Brooks, M.; Clark, A.B.; Clausen, E.S.; Durheim, M.T.; et al. Design and rationale of a multi-center, pragmatic, open-label randomized trial of antimicrobial therapy—The study of clinical efficacy of antimicrobial therapy strategy using pragmatic design in Idiopathic Pulmonary Fibrosis (CleanUP-IPF) clinical trial. Respir. Res. 2020, 21, 68. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, S.; Wang, N.; Tan, H.-Y.; Zhang, Z.; Feng, Y. The Cross-Talk between Gut Microbiota and Lungs in Common Lung Diseases. Front. Microbiol. 2020, 11, 301. [Google Scholar] [CrossRef] [PubMed]
- Sze, M.A.; Dimitriu, P.A.; Hayashi, S.; Elliott, W.M.; McDonough, J.E.; Gosselink, J.V.; Cooper, J.; Sin, D.D.; Mohn, W.W.; Hogg, J.C. The Lung Tissue Microbiome in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2012, 185, 1073–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsland, B.J.; Trompette, A.; Gollwitzer, E.S. The Gut-Lung Axis in Respiratory Disease. Ann. Am. Thorac. Soc. 2015, 12, S150–S156. [Google Scholar]
- Hillman, E.T.; Lu, H.; Yao, T.; Nakatsu, C.H. Microbial Ecology along the Gastrointestinal Tract. Microbes Environ. 2017, 32, 300–313. [Google Scholar] [CrossRef] [Green Version]
- McAleer, J.P.; Kolls, J.K. Contributions of the intestinal microbiome in lung immunity. Eur. J. Immunol. 2018, 48, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Keely, S.J.; Talley, N.J.; Hansbro, P.M. Pulmonary-intestinal cross-talk in mucosal inflammatory disease. Mucosal Immunol. 2011, 5, 7–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budden, K.F.; Gellatly, S.L.; Wood, D.L.A.; Cooper, M.A.; Morrison, M.; Hugenholtz, P.; Hansbro, P.M. Emerging pathogenic links between microbiota and the gut–lung axis. Nat. Rev. Microbiol. 2017, 15, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Kim, W.-U. Microbiota in T-cell homeostasis and inflammatory diseases. Exp. Mol. Med. 2017, 49, e340. [Google Scholar] [CrossRef] [PubMed]
- O’Dwyer, D.N.; Dickson, R.P.; Moore, B.B. The Lung Microbiome, Immunity, and the Pathogenesis of Chronic Lung Disease. J. Immunol. 2016, 196, 4839–4847. [Google Scholar] [CrossRef] [Green Version]
- Ohnmacht, C. Microbiota, regulatory T cell subsets, and allergic disorders. Allergo J. Int. 2016, 25, 114–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scales, B.S.; Dickson, R.P.; Huffnagle, G.B. A tale of two sites: How inflammation can reshape the microbiomes of the gut and lungs. J. Leukoc. Biol. 2016, 100, 943–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.M.; Lee, D.H.; Ahn, B.K.; Hwang, J.J.; Yoon, H.; Shin, C.M.; Park, Y.S.; Kim, Y.S.P.A.N. Protective Effect of Proton Pump Inhibitor for Survival in Patients with Gastroesophageal Reflux Disease and Idiopathic Pulmonary Fibrosis. J. Neurogastroenterol. Motil. 2016, 22, 444–451. [Google Scholar] [CrossRef] [Green Version]
- Raghu, G.; Freudenberger, T.D.; Yang, S.; Curtis, J.R.; Spada, C.; Hayes, J.; Sillery, J.K.; Pope, C.E.; Pellegrini, C.A. High prevalence of abnormal acid gastro-oesophageal reflux in idiopathic pulmonary fibrosis. Eur. Respir. J. 2006, 27, 136–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fagundes, C.T.; Amaral, F.A.; Vieira, A.T.; Soares, A.C.; Pinho, V.; Nicoli, J.R.; Vieira, L.Q.; Teixeira, M.M.; Souza, D.G. Transient TLR Activation Restores Inflammatory Response and Ability to Control Pulmonary Bacterial Infection in Germfree Mice. J. Immunol. 2011, 188, 1411–1420. [Google Scholar] [CrossRef] [Green Version]
- King, S.; Glanville, J.; Sanders, M.E.; Fitzgerald, A.; Varley, D. Effectiveness of probiotics on the duration of illness in healthy children and adults who develop common acute respiratory infectious conditions: A systematic review and meta-analysis. Br. J. Nutr. 2014, 112, 41–54. [Google Scholar] [CrossRef]
- Luoto, R.; Ruuskanen, O.; Waris, M.; Kalliomäki, M.; Salminen, S.; Isolauri, E. Prebiotic and probiotic supplementation prevents rhinovirus infections in preterm infants: A randomized, placebo-controlled trial. J. Allergy Clin. Immunol. 2014, 133, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Schuijt, T.J.; Lankelma, J.M.; Scicluna, B.P.; Melo, F.D.S.E.; Roelofs, J.J.T.H.; De Boer, J.D.; Hoogendijk, A.J.; De Beer, R.; De Vos, A.; Belzer, C.; et al. The gut microbiota plays a protective role in the host defence against pneumococcal pneumonia. Gut 2015, 65, 575–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Burnham, E.L.; Janssen, W.J.; Riches, D.W.H.; Moss, M.; Downey, G.P. The fibroproliferative response in acute respiratory distress syndrome: Mechanisms and clinical significance. Eur. Respir. J. 2014, 43, 276–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasarmidi, E.; Tsitoura, E.; Spandidos, D.A.; Tzanakis, N.; Antoniou, K.M. Pulmonary fibrosis in the aftermath of the Covid-19 era (Review). Exp. Ther. Med. 2020, 20, 2557–2560. [Google Scholar] [CrossRef]
- Myall, K.J.; Mukherjee, B.; Castanheira, A.M.; Lam, J.L.; Benedetti, G.; Mak, S.M.; Preston, R.; Thillai, M.; Dewar, A.; Molyneaux, P.L.; et al. Persistent Post-COVID-19 Inflammatory Interstitial Lung Disease: An Observational Study of Corticosteroid Treatment. Ann. Am. Thorac. Soc. 2021. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Barnett, J.; Brill, S.E.; Brown, J.S.; Denneny, E.K.; Hare, S.S.; Heightman, M.; Hillman, T.E.; Jacob, J.; Jarvis, H.C.; et al. ‘Long-COVID’: A cross-sectional study of persisting symptoms, biomarker and imaging abnormalities following hospitalisation for COVID-19. Thorax 2021, 76, 396–398. [Google Scholar] [CrossRef]
- Papiris, S.A.; Bouros, D.; Markopoulou, K.; Kolilekas, L.; Papaioannou, A.I.; Tzilas, V.; Tzouvelekis, A.; Fouka, E.; Papakosta, D.; Daniil, Z.; et al. Early COVID-19 lockdown in Greece and idiopathic pulmonary fibrosis: A beneficial “impact” beyond any expectation. Eur. Respir. J. 2021, 57, 2003111. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ntolios, P.; Tzilas, V.; Bouros, E.; Avdoula, E.; Karakasiliotis, I.; Bouros, D.; Steiropoulos, P. The Role of Microbiome and Virome in Idiopathic Pulmonary Fibrosis. Biomedicines 2021, 9, 442. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9040442
Ntolios P, Tzilas V, Bouros E, Avdoula E, Karakasiliotis I, Bouros D, Steiropoulos P. The Role of Microbiome and Virome in Idiopathic Pulmonary Fibrosis. Biomedicines. 2021; 9(4):442. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9040442
Chicago/Turabian StyleNtolios, Paschalis, Vassilios Tzilas, Evangelos Bouros, Eleni Avdoula, Ioannis Karakasiliotis, Demosthenes Bouros, and Paschalis Steiropoulos. 2021. "The Role of Microbiome and Virome in Idiopathic Pulmonary Fibrosis" Biomedicines 9, no. 4: 442. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9040442