
Radiosynthesis and Evaluation of Talazoparib and Its Derivatives as PARP-1-Targeting Agents

 , and
, and 
Abstract
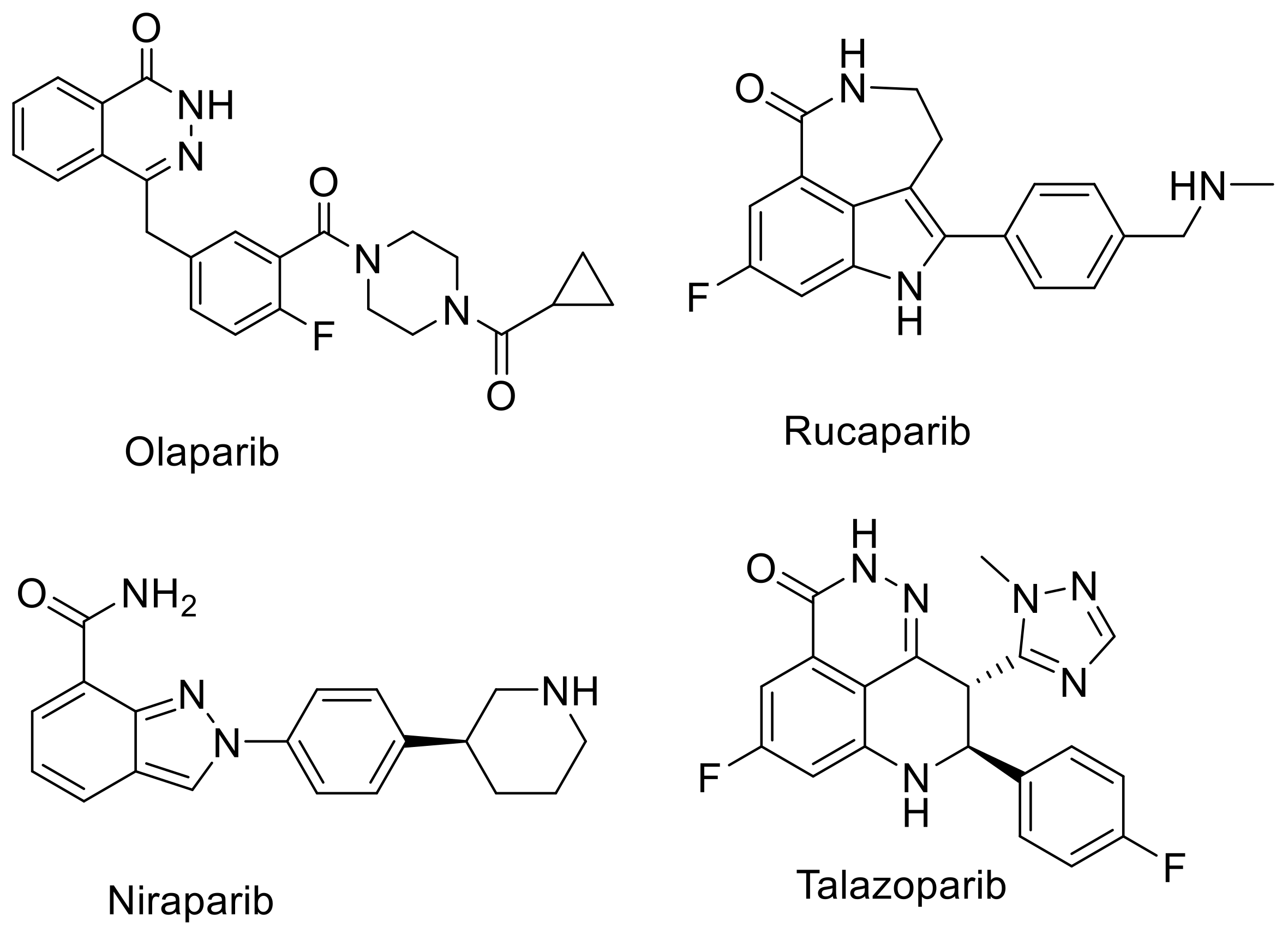
:1. Introduction
2. Method and General Information
2.1. General Information
2.2. Synthesis of Methyl 2-(4-bromophenyl)-7-fluoro-3-(1-methyl-1H-1,2,4-triazol-5-yl)-4-oxo-1,2,3,4-tetrahydroquinoline-5-carboxylate (2b) and Methyl 7-fluoro-2-(4-iodophenyl)-3-(1-methyl-1H-1,2,4-triazol-5-yl)-4-oxo-1,2,3,4-tetrahydroquinoline-5-carboxylate (2c)
2.3. Synthesis of 8-(4-bromophenyl)-5-fluoro-9-(1-methyl-1H-1,2,4-triazol-5-yl)-2,7,8,9-tetrahydro-3H-pyrido[4,3,2-de]phthalazin-3-one (3b) and 8-(4-iodophenyl)-5-fluoro-9-(1-methyl-1H-1,2,4-triazol-5-yl)-2,7,8,9-tetrahydro-3H-pyrido[4,3,2-de]phthalazin-3-one (3c)
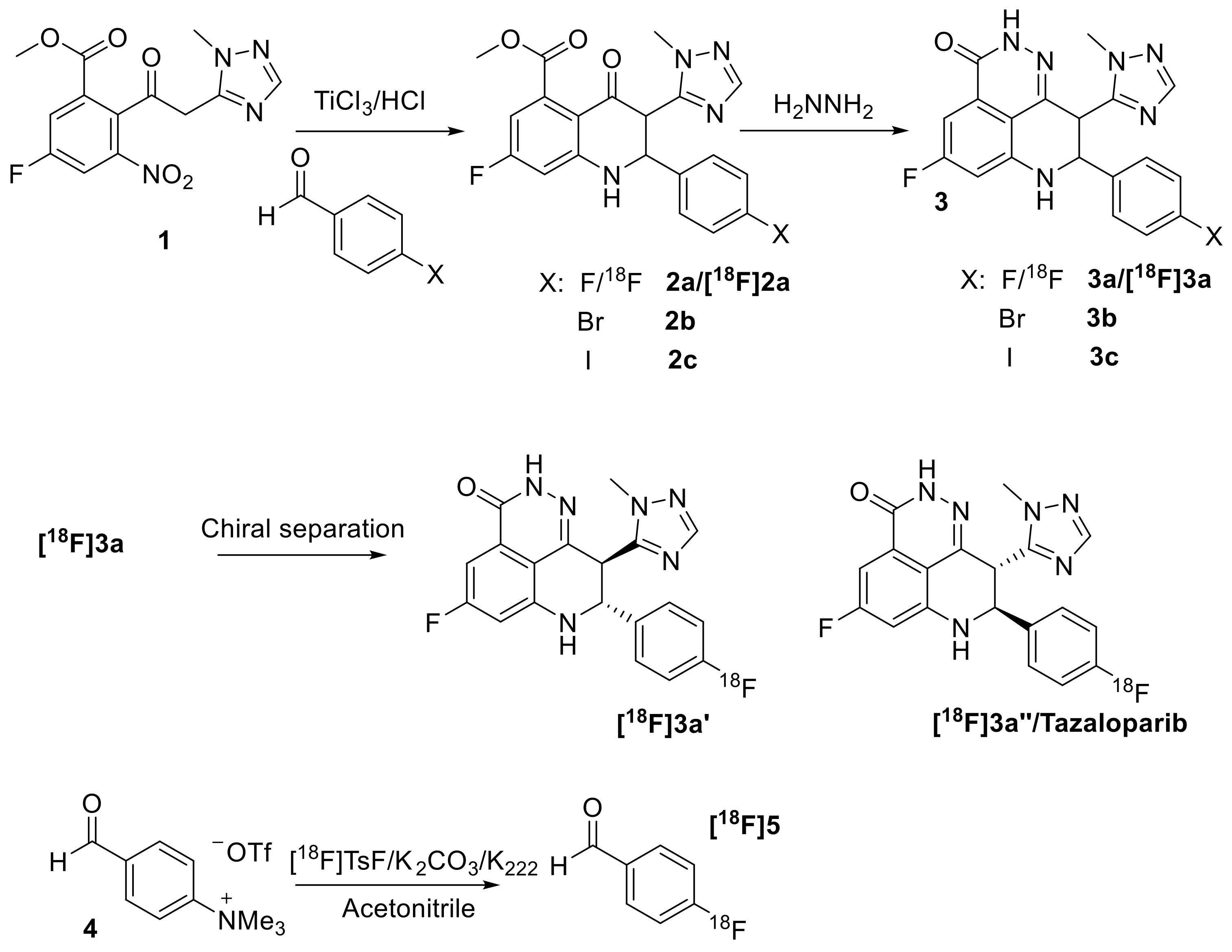
2.4. Synthesis of [18F]Talazoparib (3a″)
2.5. Xenograft Model
2.6. Biodistribution
2.7. Materials and Cell Culture
2.8. Radioligand Cell Uptake Saturation Analyses
2.9. Competitive Assay
3. Results and Discussion
3.1. Organic Synthesis and Radiosynthesis
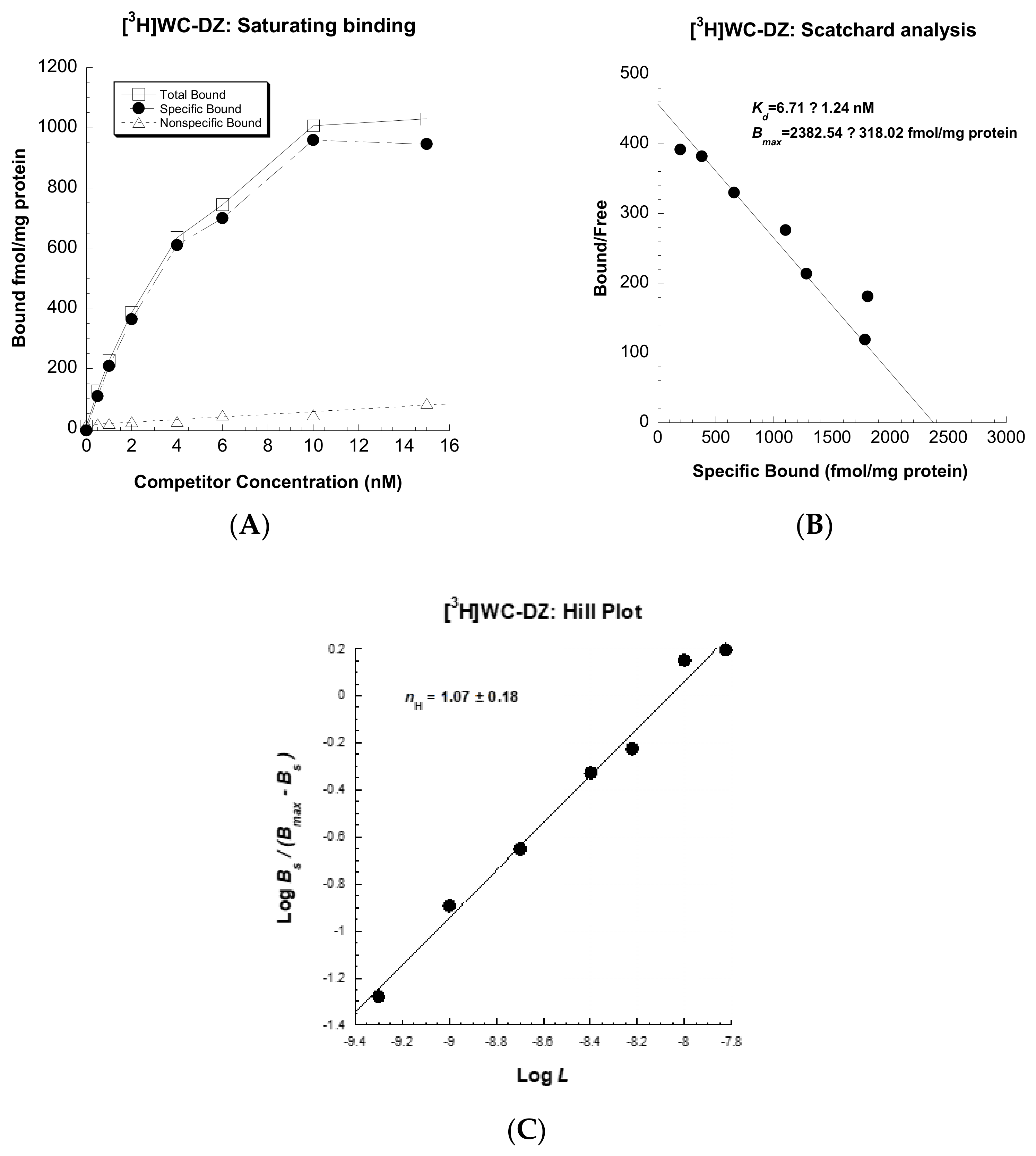
3.2. Synthesis of [3H]WC-DZ
3.3. Saturation Analyses of Radioligand Uptake in U251MG Cells
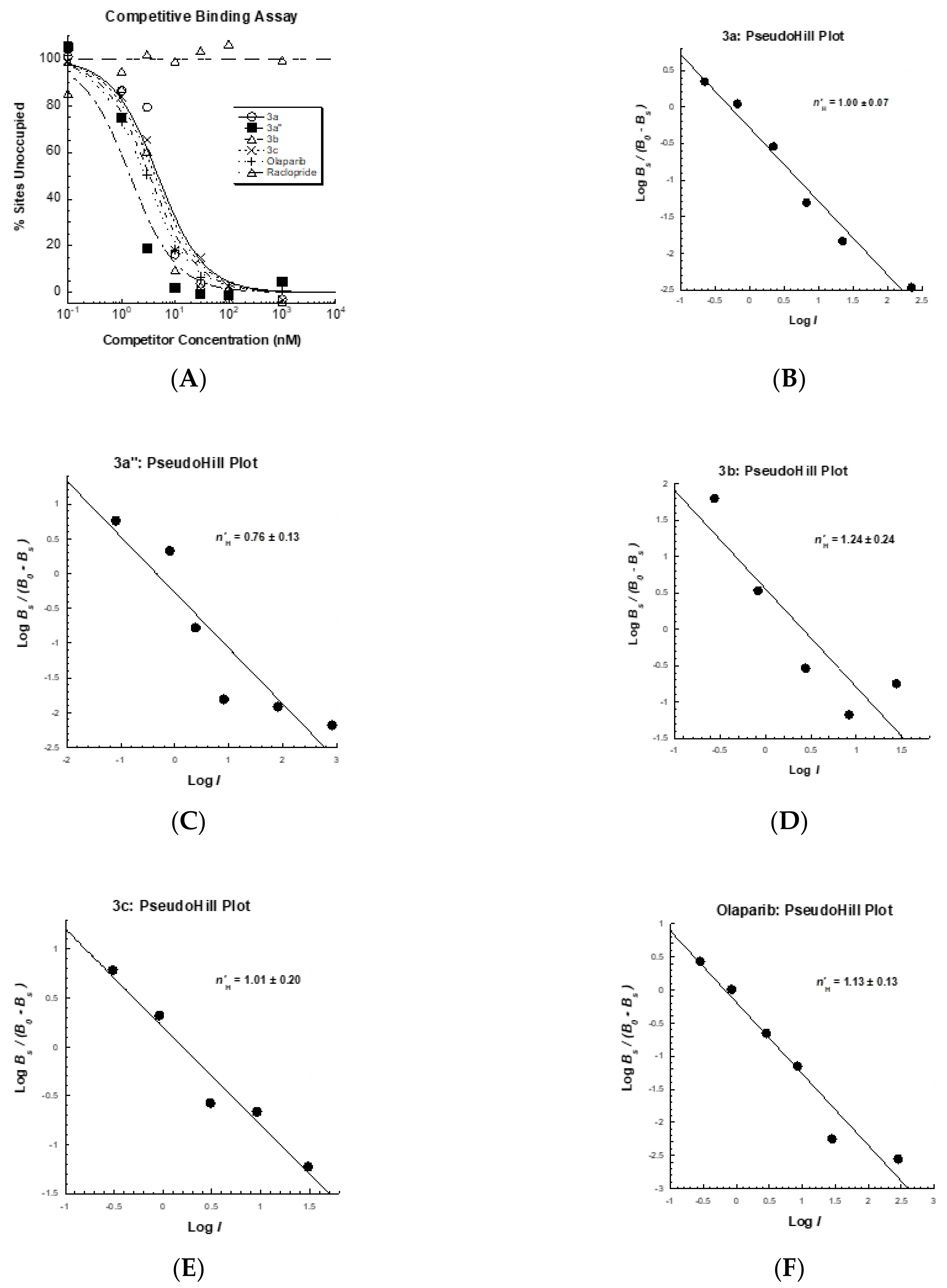
3.4. Competitive Profile of Standard and WC-DZ in U251MG Cells
3.5. Biodistribution Studies of [18F]Talazoparib
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Do, K.; Chen, A.P. Molecular pathways: Targeting PARP in cancer treatment. Clin. Cancer Res. 2013, 19, 977–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiewer, M.J.; Knudsen, K.E. Transcriptional roles of PARP1 in cancer. Mol. Cancer Res. 2014, 12, 1069–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiber, V.; Amé, J.C.; Dollé, P.; Schultz, I.; Rinaldi, B.; Fraulob, V.; Ménissier-de Murcia, J.; de Murcia, G. Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. J. Biol. Chem. 2002, 277, 23028–23036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferraris, D.V. Evolution of Poly(ADP-ribose) Polymerase-1 (PARP-1) Inhibitors. From Concept to Clinic. J. Med. Chem. 2010, 53, 4561–4584. [Google Scholar] [CrossRef] [PubMed]
- Basu, B.; Sandhu, S.K.; de Bono, J.S. PARP inhibitors: Mechanism of action and their potential role in the prevention and treatment of cancer. Drugs 2012, 72, 1579–1590. [Google Scholar] [CrossRef]
- Carney, B.; Kossatz, S.; Reiner, T. Molecular Imaging of PARP. J. Nucl. Med. 2017, 58, 1025–1030. [Google Scholar] [CrossRef] [Green Version]
- Michel, L.S.; Dyroff, S.; Brooks, F.J.; Spayd, K.J.; Lim, S.; Engle, J.T.; Phillips, S.; Tan, B.; Wang-Gillam, A.; Bognar, C.; et al. PET of Poly (ADP-Ribose) Polymerase Activity in Cancer: Preclinical Assessment and First In-Human Studies. Radiology 2017, 282, 453–463. [Google Scholar] [CrossRef] [Green Version]
- Schöder, H.; França, P.D.D.S.; Nakajima, R.; Burnazi, E.; Roberts, S.; Brand, C.; Grkovski, M.; Mauguen, A.; Dunphy, M.P.; Ghossein, R.A.; et al. Safety and Feasibility of PARP1/2 Imaging with 18F-PARPi in Patients with Head and Neck Cancer. Clin. Cancer Res. 2020, 26, 3110. [Google Scholar] [CrossRef] [Green Version]
- Makvandi, M.; Pantel, A.; Schwartz, L.; Schubert, E.; Xu, K.; Hsieh, C.J.; Hou, C.; Kim, H.; Weng, C.C.; Winters, H.; et al. A PET imaging agent for evaluating PARP-1 expression in ovarian cancer. J. Clin. Investig. 2018, 128, 2116–2126. [Google Scholar] [CrossRef]
- Carney, B.; Kossatz, S.; Lok, B.H.; Schneeberger, V.; Gangangari, K.K.; Pillarsetty, N.V.K.; Weber, W.A.; Rudin, C.M.; Poirier, J.T.; Reiner, T. Target engagement imaging of PARP inhibitors in small-cell lung cancer. Nat. Commun. 2018, 9, 176. [Google Scholar] [CrossRef] [Green Version]
- Wilson, T.C.; Xavier, M.A.; Knight, J.; Verhoog, S.; Torres, J.B.; Mosley, M.; Hopkins, S.L.; Wallington, S.; Allen, P.D.; Kersemans, V.; et al. PET Imaging of PARP Expression Using (18)F-Olaparib. J. Nucl. Med. 2019, 60, 504–510. [Google Scholar] [CrossRef] [Green Version]
- Makvandi, M.; Lee, H.; Puentes, L.N.; Reilly, S.W.; Rathi, K.S.; Weng, C.C.; Chan, H.S.; Hou, C.; Raman, P.; Martinez, D.; et al. Targeting PARP-1 with Alpha-Particles Is Potently Cytotoxic to Human Neuroblastoma in Preclinical Models. Mol. Cancer Ther. 2019, 18, 1195–1204. [Google Scholar] [CrossRef] [Green Version]
- Ellison, P.A.; Olson, A.P.; Barnhart, T.E.; Hoffman, S.L.V.; Reilly, S.W.; Makvandi, M.; Bartels, J.L.; Murali, D.; DeJesus, O.T.; Lapi, S.E.; et al. Improved production of 76Br, 77Br and 80mBr via CoSe cyclotron targets and vertical dry distillation. Nucl. Med. Biol. 2020, 80–81, 32–36. [Google Scholar] [CrossRef]
- Pirovano, G.; Jannetti, S.A.; Carter, L.M.; Sadique, A.; Kossatz, S.; Guru, N.; Demétrio De Souza França, P.; Maeda, M.; Zeglis, B.M.; Lewis, J.S.; et al. Targeted Brain Tumor Radiotherapy Using an Auger Emitter. Clin. Cancer Res. 2020, 26, 2871. [Google Scholar] [CrossRef] [Green Version]
- Riad, A.; Gitto, S.B.; Lee, H.; Winters, H.D.; Martorano, P.M.; Hsieh, C.J.; Xu, K.; Omran, D.K.; Powell, D.J., Jr.; Mach, R.H.; et al. PARP Theranostic Auger Emitters Are Cytotoxic in BRCA Mutant Ovarian Cancer and Viable Tumors from Ovarian Cancer Patients Enable Ex-Vivo Screening of Tumor Response. Molecules 2020, 25, 6029. [Google Scholar] [CrossRef]
- Jannetti, S.A.; Carlucci, G.; Carney, B.; Kossatz, S.; Shenker, L.; Carter, L.M.; Salinas, B.; Brand, C.; Sadique, A.; Donabedian, P.L.; et al. PARP-1-Targeted Radiotherapy in Mouse Models of Glioblastoma. J. Nucl. Med. 2018, 59, 1225–1233. [Google Scholar] [CrossRef] [Green Version]
- Jannetti, S.A.; Zeglis, B.M.; Zalutsky, M.R.; Reiner, T. Poly(ADP-Ribose)Polymerase (PARP) Inhibitors and Radiation Therapy. Front. Pharmacol. 2020, 11, 170. [Google Scholar] [CrossRef] [Green Version]
- Ricks, T.K.; Chiu, H.J.; Ison, G.; Kim, G.; McKee, A.E.; Kluetz, P.; Pazdur, R. Successes and Challenges of PARP Inhibitors in Cancer Therapy. Front. Oncol. 2015, 5, 222. [Google Scholar] [CrossRef] [Green Version]
- Murai, J.; Huang, S.Y.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Chu, D.; Feng, Y.; Shen, Y.; Aoyagi-Scharber, M.; Post, L.E. Discovery and Characterization of (8S,9R)-5-Fluoro-8-(4-fluorophenyl)-9-(1-methyl-1H-1,2,4-triazol-5-yl)-2,7,8,9-tetrahydro-3H-pyrido[4,3,2-de]phthalazin-3-one (BMN 673, Talazoparib), a Novel, Highly Potent, and Orally Efficacious Poly(ADP-ribose) Polymerase-1/2 Inhibitor, as an Anticancer Agent. J. Med. Chem. 2016, 59, 335–357. [Google Scholar] [CrossRef]
- Vaidyanathan, G.; Zalutsky, M.R. Synthesis of N-succinimidyl 4-[18F]fluorobenzoate, an agent for labeling proteins and peptides with 18F. Nat. Protoc. 2006, 1, 1655–1661. [Google Scholar] [CrossRef]
- Chen, H.; Afrin, S.; Guo, Y.; Chu, W.; Benzinger, T.L.S.; Rogers, B.E.; Garbow, J.R.; Perlmutter, J.S.; Zhou, D.; Xu, J. Radiolabeled 6-(2, 3-Dichlorophenyl)-N4-methylpyrimidine-2, 4-diamine (TH287): A Potential Radiotracer for Measuring and Imaging MTH1. Int. J. Mol. Sci. 2020, 21, 8860. [Google Scholar] [CrossRef]
- Scatchard, G. The Attractions of Proteins for Small Molecules and Ions. Ann. N. Y. Acad. Sci. 1949, 51, 660–672. [Google Scholar] [CrossRef]
- Cheng, Y.; Prusoff, W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef]
- Xu, J.; Tu, Z.; Jones, L.A.; Vangveravong, S.; Wheeler, K.T.; Mach, R.H. [3H]N-[4-(3,4-dihydro-6,7-dimethoxyisoquinolin-2(1H)-yl)butyl]-2-methoxy-5-methyl benzamide: A novel sigma-2 receptor probe. Eur. J. Pharmacol. 2005, 525, 8–17. [Google Scholar] [CrossRef]
- Skalitzky, D.J.; Marakovits, J.T.; Maegley, K.A.; Ekker, A.; Yu, X.-H.; Hostomsky, Z.; Webber, S.E.; Eastman, B.W.; Almassy, R.; Li, J.; et al. Tricyclic Benzimidazoles as Potent Poly(ADP-ribose) Polymerase-1 Inhibitors. J. Med. Chem. 2003, 46, 210–213. [Google Scholar] [CrossRef]
- Reilly, S.W.; Makvandi, M.; Xu, K.; Mach, R.H. Rapid Cu-Catalyzed [(211)At]Astatination and [(125)I]Iodination of Boronic Esters at Room Temperature. Org. Lett. 2018, 20, 1752–1755. [Google Scholar] [CrossRef]
- Zhou, D.; Xu, J.; Mpoy, C.; Chu, W.; Kim, S.H.; Li, H.; Rogers, B.E.; Katzenellenbogen, J.A. Preliminary evaluation of a novel 18F-labeled PARP-1 ligand for PET imaging of PARP-1 expression in prostate cancer. Nucl. Med. Biol. 2018, 66, 26–31. [Google Scholar] [CrossRef]
- Zmuda, F.; Malviya, G.; Blair, A.; Boyd, M.; Chalmers, A.J.; Sutherland, A.; Pimlott, S.L. Synthesis and Evaluation of a Radioiodinated Tracer with Specificity for Poly(ADP-ribose) Polymerase-1 (PARP-1) in Vivo. J. Med. Chem. 2015, 58, 8683–8693. [Google Scholar] [CrossRef] [Green Version]
- Salinas, B.; Irwin, C.P.; Kossatz, S.; Bolaender, A.; Chiosis, G.; Pillarsetty, N.; Weber, W.A.; Reiner, T. Radioiodinated PARP1 tracers for glioblastoma imaging. EJNMMI Res. 2015, 5, 123. [Google Scholar] [CrossRef] [Green Version]
- Makvandi, M.; Xu, K.; Lieberman, B.P.; Anderson, R.C.; Effron, S.S.; Winters, H.D.; Zeng, C.; McDonald, E.S.; Pryma, D.A.; Greenberg, R.A.; et al. A Radiotracer Strategy to Quantify PARP-1 Expression In Vivo Provides a Biomarker That Can Enable Patient Selection for PARP Inhibitor Therapy. Cancer Res. 2016, 76, 4516–4524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laird, J.; Lok, B.H.; Carney, B.; Kossatz, S.; de Stanchina, E.; Reiner, T.; Poirier, J.T.; Rudin, C.M. Positron-Emission Tomographic Imaging of a Fluorine 18-Radiolabeled Poly(ADP-Ribose) Polymerase 1 Inhibitor Monitors the Therapeutic Efficacy of Talazoparib in SCLC Patient-Derived Xenografts. J. Thorac. Oncol. 2019, 14, 1743–1752. [Google Scholar] [CrossRef] [PubMed]
- Elmeliegy, M.; Láng, I.; Smolyarchuk, E.A.; Chung, C.H.; Plotka, A.; Shi, H.; Wang, D. Evaluation of the effect of P-glycoprotein inhibition and induction on talazoparib disposition in patients with advanced solid tumours. Br. J. Clin. Pharmacol. 2020, 86, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Kizilbash, S.H.; Gupta, S.K.; Chang, K.; Kawashima, R.; Parrish, K.E.; Carlson, B.L.; Bakken, K.K.; Mladek, A.C.; Schroeder, M.A.; Decker, P.A.; et al. Restricted Delivery of Talazoparib Across the Blood-Brain Barrier Limits the Sensitizing Effects of PARP Inhibition on Temozolomide Therapy in Glioblastoma. Mol. Cancer Ther. 2017, 16, 2735–2746. [Google Scholar] [CrossRef] [Green Version]
- Zandarashvili, L.; Langelier, M.F.; Velagapudi, U.K.; Hancock, M.A.; Steffen, J.D.; Billur, R.; Hannan, Z.M.; Wicks, A.J.; Krastev, D.B.; Pettitt, S.J.; et al. Structural basis for allosteric PARP-1 retention on DNA breaks. Science 2020, 368. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3a″ a | 3a | 3b | 3c | Olaparib | Raclopride | |
|---|---|---|---|---|---|---|
| Ki (nM) b | 0.65 ± 0.07 | 2.37 ± 0.56 | 1.92 ± 0.41 | 1.73 ± 0.43 | 1.87 ± 0.10 | >20,000 |
| n′H | 0.76 ± 0.13 | 1.00 ± 0.07 | 1.24 ± 0.24 | 1.01 ± 0.20 | 1.13 ± 0.13 | None |
| cLogP c | 2.58 | 2.58 | 3.25 | 3.78 | / | / |
| MW | 380.36 | 380.36 | 441.26 | 488.26 | / | / |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, D.; Chen, H.; Mpoy, C.; Afrin, S.; Rogers, B.E.; Garbow, J.R.; Katzenellenbogen, J.A.; Xu, J. Radiosynthesis and Evaluation of Talazoparib and Its Derivatives as PARP-1-Targeting Agents. Biomedicines 2021, 9, 565. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9050565
Zhou D, Chen H, Mpoy C, Afrin S, Rogers BE, Garbow JR, Katzenellenbogen JA, Xu J. Radiosynthesis and Evaluation of Talazoparib and Its Derivatives as PARP-1-Targeting Agents. Biomedicines. 2021; 9(5):565. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9050565
Chicago/Turabian StyleZhou, Dong, Huaping Chen, Cedric Mpoy, Sadia Afrin, Buck E. Rogers, Joel R. Garbow, John A. Katzenellenbogen, and Jinbin Xu. 2021. "Radiosynthesis and Evaluation of Talazoparib and Its Derivatives as PARP-1-Targeting Agents" Biomedicines 9, no. 5: 565. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9050565