
Pulmonary MicroRNA Changes Alter Angiogenesis in Chronic Obstructive Pulmonary Disease and Lung Cancer


Abstract
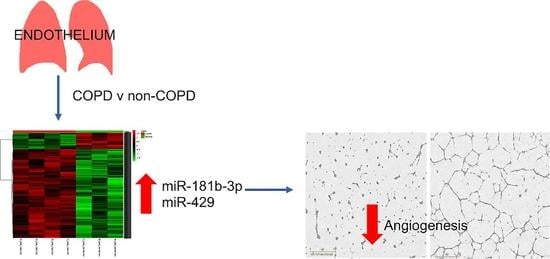
:
1. Introduction
2. Materials and Methods
2.1. Patient Recruitment
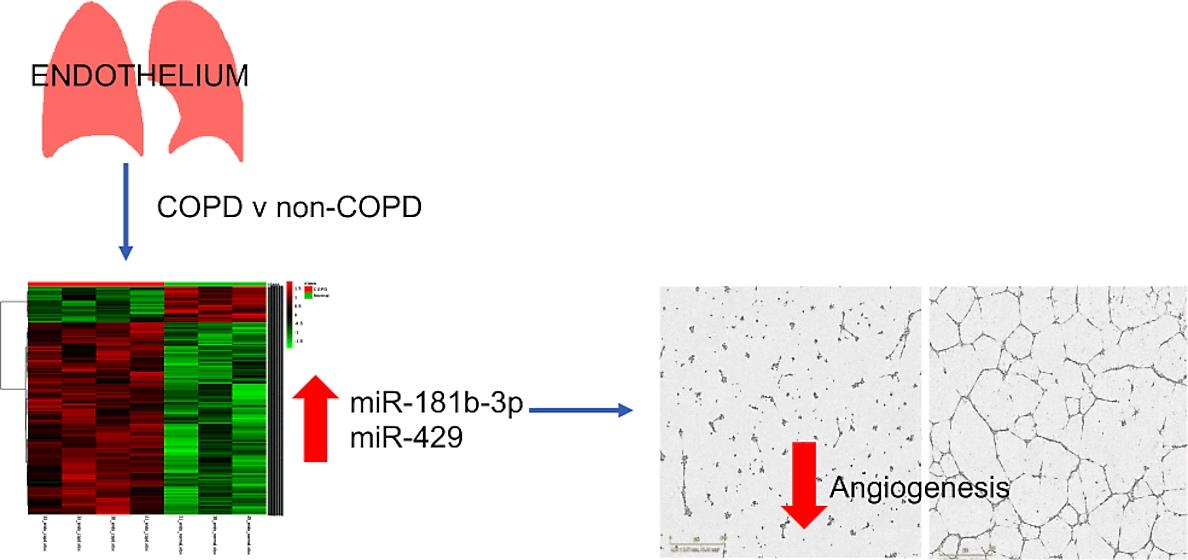
2.2. Tissue Collection and Extraction of Human Pulmonary Endothelial Cells (HPECs)
2.3. Processing of RNA and Conduct of Arrays
2.4. Validation of Array Data
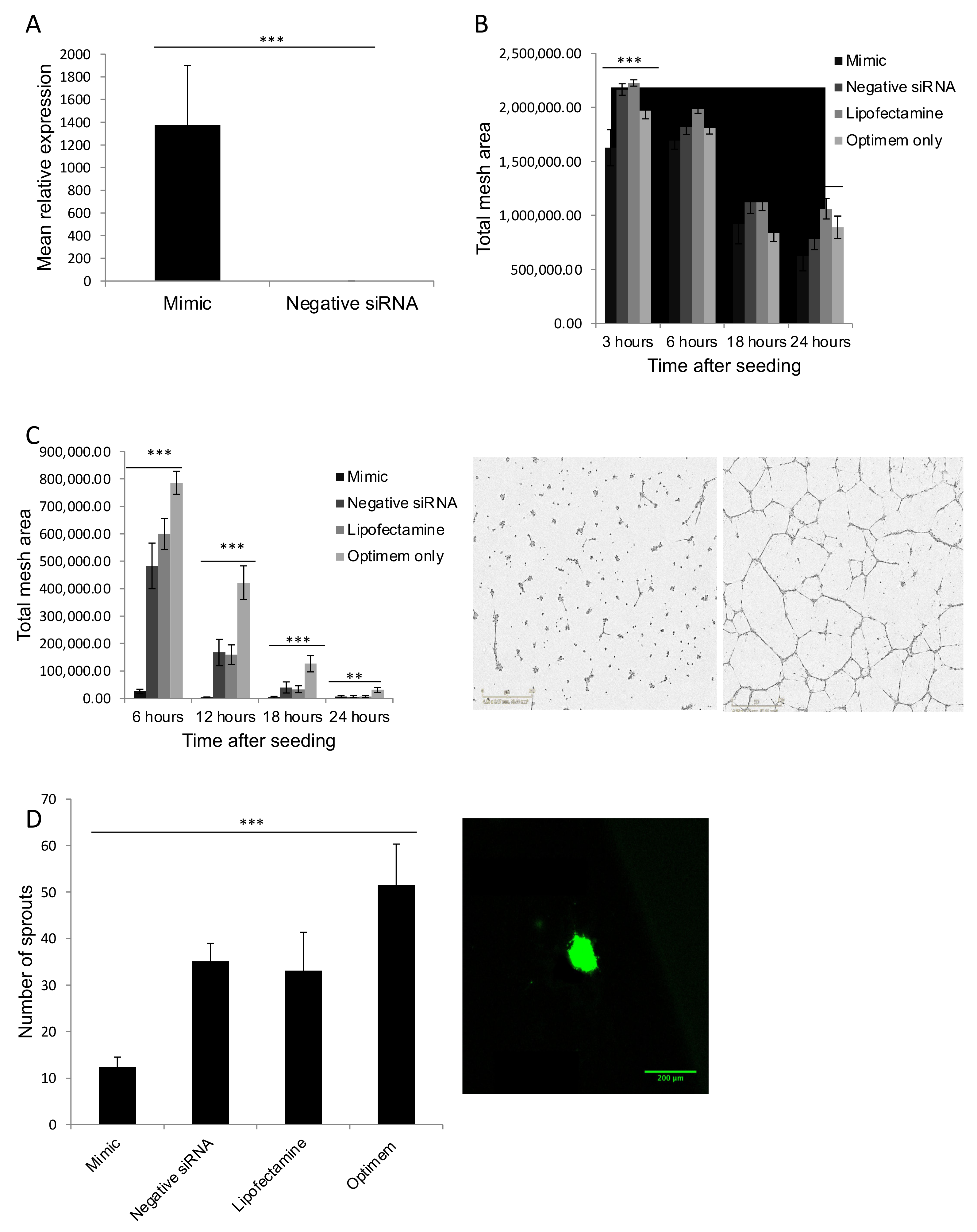
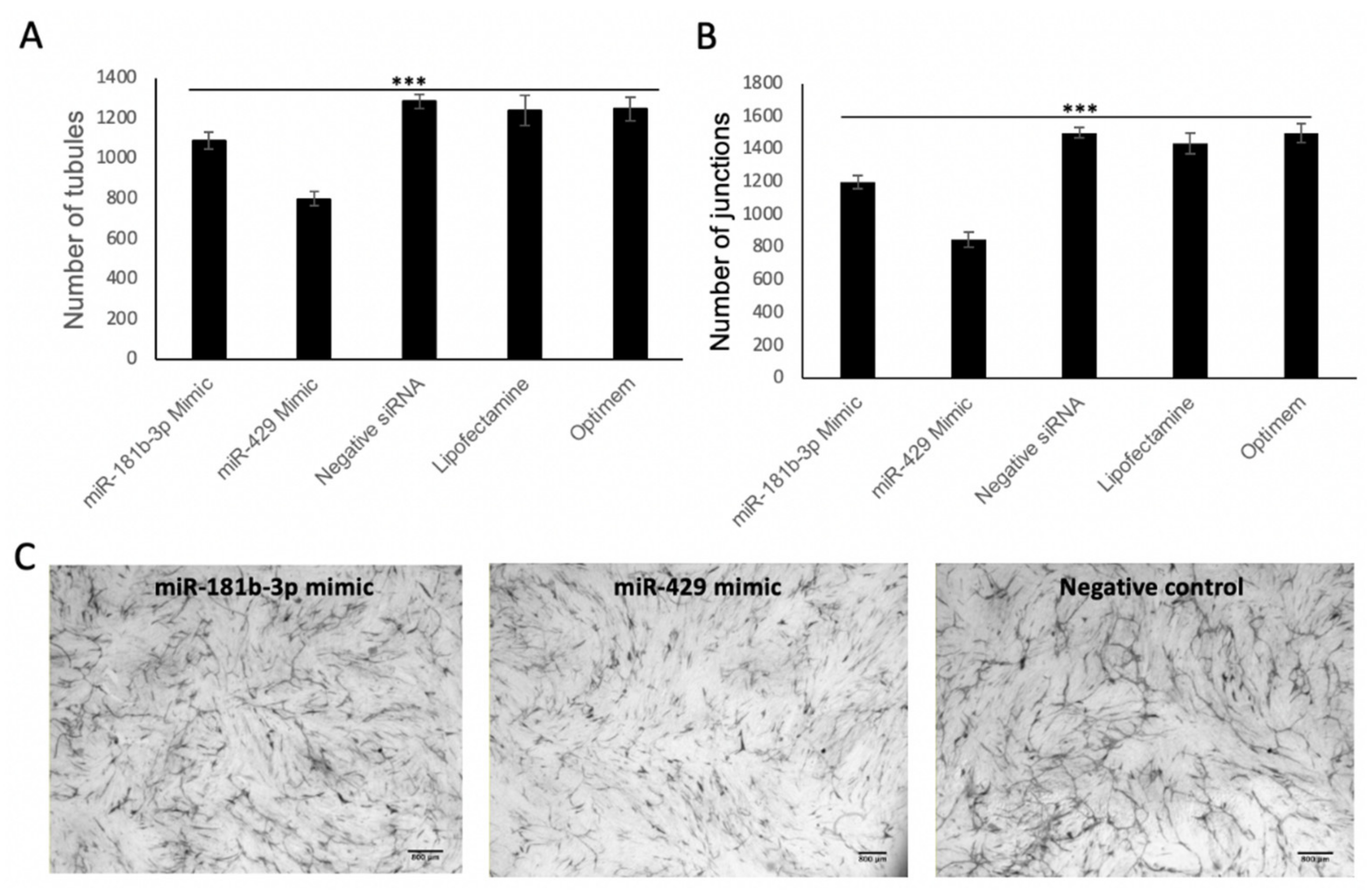
2.5. miR Overexpression
2.6. Cell Growth Assay
2.7. Cell Cycle Analysis
2.8. Scratch Wound Assay
2.9. Matrigel Assay
2.10. Spheroid Assay
2.11. Static Co-Cultures
2.12. Endothelial–Fibroblast Co-Cultures
2.13. Statistical Analysis
3. Results
3.1. Patient Characteristics Were Not Significantly Different between Groups Other than FEV1
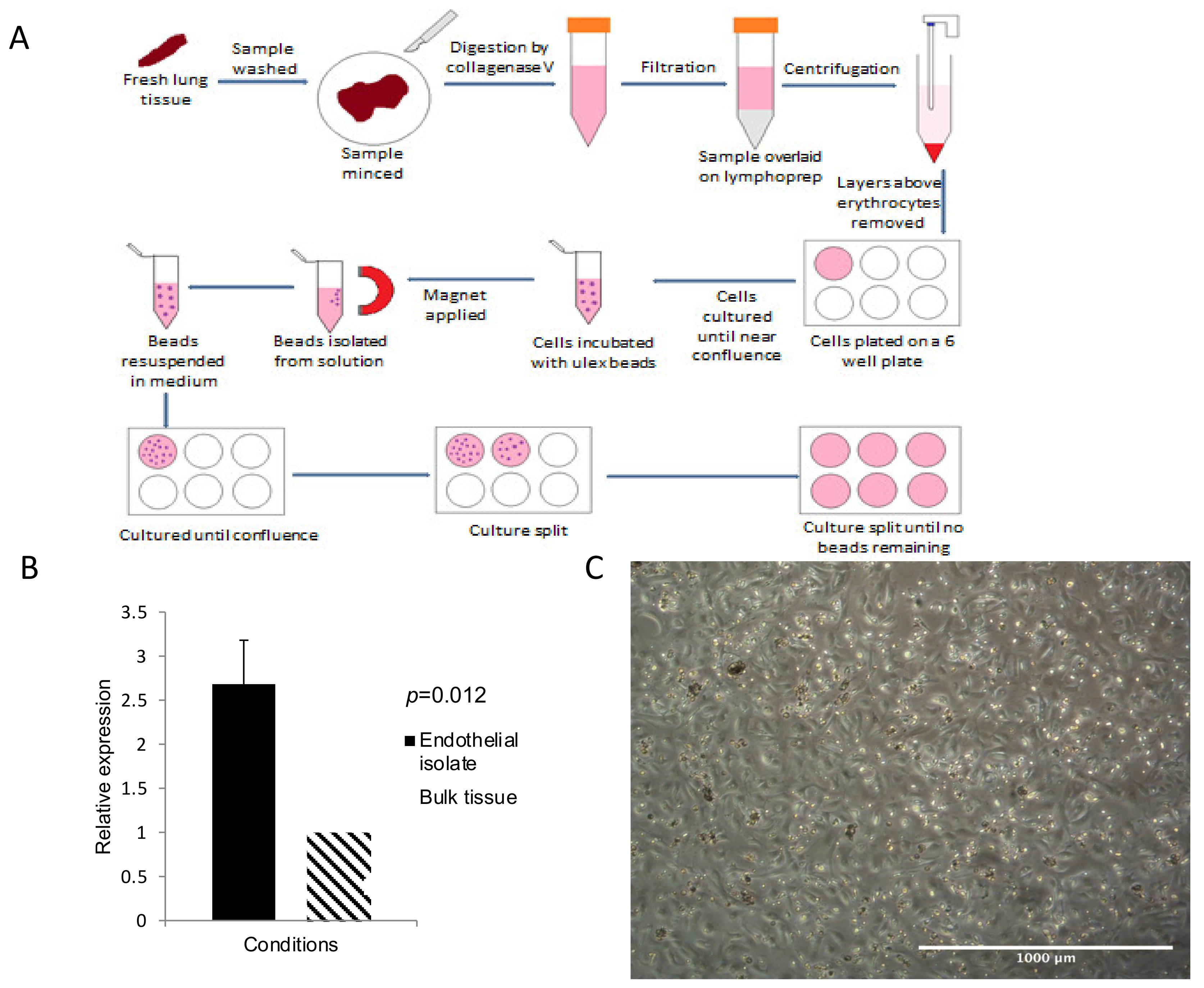
3.2. CD31 Expression Was Significantly Increased in Extracted Pulmonary Endothelial Cells
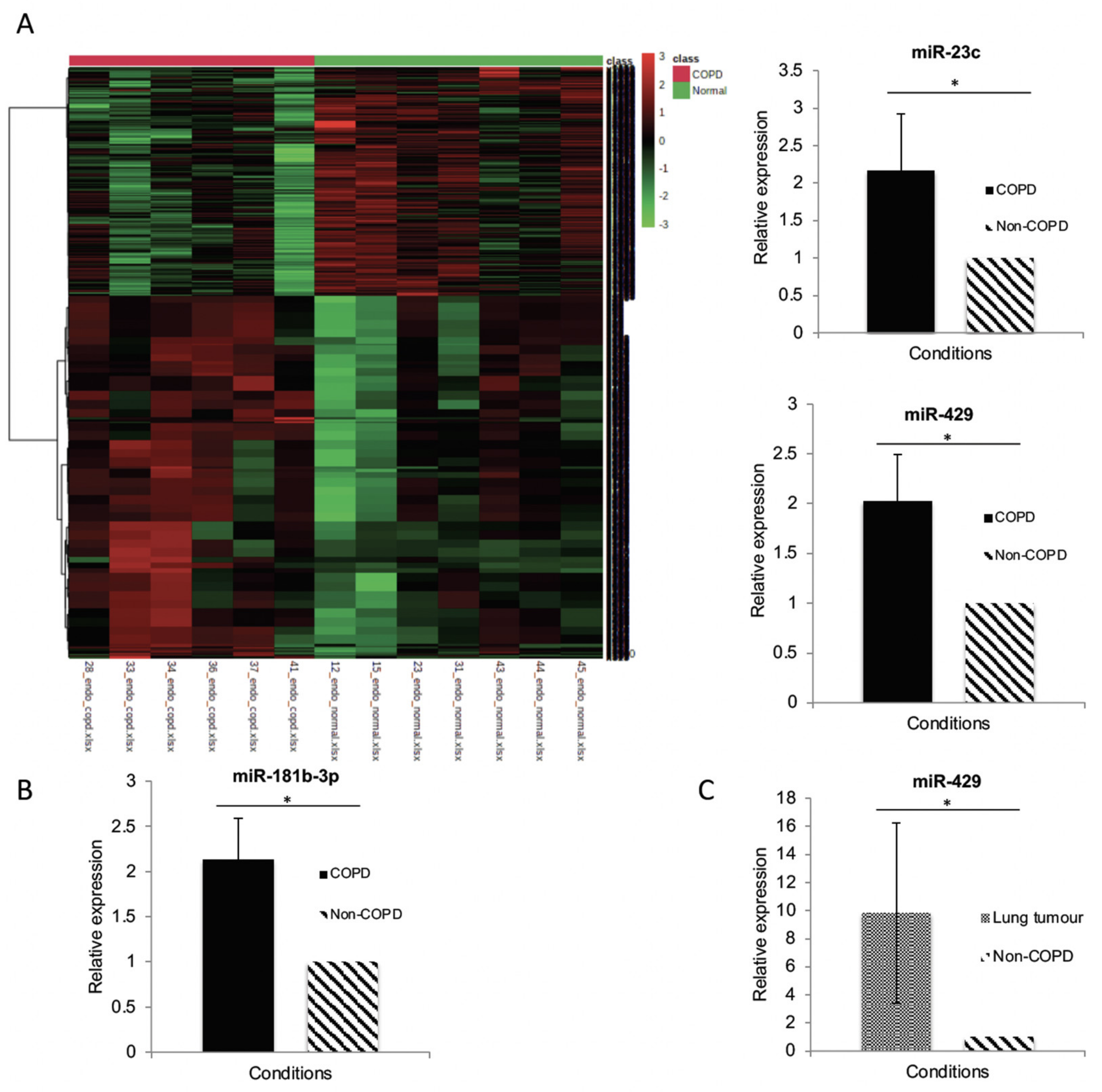
3.3. MiR Expression in HPECs Is Significantly Different in Patients with COPD
3.4. MiR-429 Expression Is Increased in HPECs in COPD and Lung Tumour
3.5. MiR Mimics Successfully Increased miR Expression Levels without Altering Cell Growth or Cell Cycle
3.6. MiR-181b-3p and MiR-429 Significantly Reduce Endothelial Tube Formation and Angiogenesis In Vitro
3.7. MiR-181b-3p and miR-429 Do Not Affect Wound Adhesion
3.8. MiR-181b-3p and miR-429 Do Not Affect Neutrophil Migration
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Solutions Used in This Study
Appendix A.1. HUVEC Media
Appendix A.2. Methocellulose Solution
Appendix A.3. Collagen Mix
References
- GOLD—Global Initiative for Chronic Obstructive Lung Disease. Global Strategy for the Diagnosis, Management and Prevention of Chronic Obstructive Pulmonary Disease. 2021. Available online: https://goldcopd.org/2021-gold-reports/ (accessed on 8 June 2021).
- Kim, W.J.; Silverman, E.K.; Hoffman, E.; Criner, G.J.; Mosenifar, Z.; Sciurba, F.C.; Make, B.J.; Carey, V.; Estepar, R.S.; Diaz, A.; et al. CT metrics of airway disease and emphysema in severe COPD. Chest 2009, 136, 396–404. [Google Scholar] [CrossRef] [Green Version]
- Qaseem, A.; Wilt, T.J.; Weinberger, S.E.; Hanania, N.A.; Criner, G.; van der Molen, T.; Marciniuk, D.D.; Denberg, T.; Schünemann, H.; Wedzicha, W.; et al. Diagnosis and Management of Stable Chronic Obstructive Pulmonary Disease: A Clinical Practice Guideline Update from the American College of Physicians, American College of Chest Physicians, American Thoracic Society, and European Respiratory Society. Ann. Intern. Med. 2011, 155, 179–191. [Google Scholar] [CrossRef]
- WHO. COPD Predicted to be third Leading Cause of DEATH in 2030. Available online: https://www.who.int/respiratory/copd/World_Health_Statistics_2008/en/ (accessed on 8 June 2021).
- WHO. Cancer. 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 8 June 2021).
- Brenner, D.R.; McLaughlin, J.R.; Hung, R.J. Previous lung diseases and lung cancer risk: A systematic review and meta-analysis. PLoS ONE 2011, 6, e17479. [Google Scholar] [CrossRef]
- Liebow, A.A. Pulmonary emphysema with special reference to vascular changes. Am. Rev. Respir. Dis. 1959, 80, 67–93. [Google Scholar] [CrossRef]
- Henson, P.M.; Vandivier, R.W.; Douglas, I.S. Cell death, remodeling, and repair in chronic obstructive pulmonary disease? Proc. Am. Thorac. Soc. 2006, 3, 713–717. [Google Scholar] [CrossRef] [PubMed]
- Polverino, F.; Laucho-Contreras, M.E.; Petersen, H.; Bijol, V.; Sholl, L.M.; Choi, M.E.; Divo, M.; Pinto-Plata, V.; Chetta, A.; Tesfaigzi, Y.; et al. A pilot study linking endothelial injury in lungs and kidneys in chronic obstructive pulmonary disease. American journal of respiratory and critical care medicine. Am. J. Respir. Crit. Care Med. 2017, 195, 1464–1476. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, Y.; Tuder, R.M.; Taraseviciene-Stewart, L.; Le Cras, T.D.; Abman, S.; Hirth, P.K.; Waltenberger, J.; Voelkel, N.F. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J. Clin. Invest. 2000, 106, 1311–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuo, M.; Mizuno, S.; Kraskauskas, D.; Bogaard, H.J.; Natarajan, R.; Cool, C.D.; Zamora, M.; Voelkel, N.F. Hypoxia inducible factor-1α in human emphysema lung tissue. Eur. Respir. J. 2011, 37, 775–783. [Google Scholar] [CrossRef] [Green Version]
- Dinh-Xuan, A.T.; Higenbottam, T.W.; Clelland, C.A.; Pepke-Zaba, J.; Cremona, G.; Butt, A.Y.; Large, S.R.; Wells, F.C.; Wallwork, J. Impairment of endothelium-dependent pulmonary-artery relaxation in chronic obstructive lung disease. N. Engl. J. Med. 1991, 324, 1539–1547. [Google Scholar] [CrossRef]
- Eickhoff, P.; Valipour, A.; Kiss, D.; Schreder, M.; Cekici, L.; Geyer, K.; Kohansal, R.; Burghuber, O.C. Determinants of systemic vascular function in patients with stable chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2008, 178, 1211–1218. [Google Scholar] [CrossRef]
- Barr, R.G.; Mesia-Vela, S.; Austin, J.H.; Basner, R.C.; Keller, B.M.; Reeves, A.P.; Shimbo, D.; Stevenson, L. Impaired flow-mediated dilation is associated with low pulmonary function and emphysema in ex-smokers: The Emphysema and Cancer Action Project (EMCAP) Study. Am. J. Respir. Crit. Care Med. 2007, 176, 1200–1207. [Google Scholar] [CrossRef] [Green Version]
- Piccari, L.; Del Pozo, R.; Blanco, I.; García-Lucio, J.; Torralba, Y.; Tura-Ceide, O.; Moises, J.; Sitges, M.; Peinado, V.I.; Barberà, J.A. Association between systemic and pulmonary vascular dysfunction in COPD. Int. J. Chron. Obstruct Pulmon. Dis. 2020, 15, 2037–2047. [Google Scholar] [CrossRef] [PubMed]
- Minet, C.; Vivodtzev, I.; Tamisier, R.; Arbib, F.; Wuyam, B.; Timsit, J.F.; Monneret, D.; Borel, J.C.; Baguet, J.P.; Levy, P.; et al. Reduced six-minute walking distance, high fat-free-mass index and hypercapnia are associated with endothelial dysfunction in COPD. Respir Physiol. Neurobiol. 2012, 183, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Vukic Dugac, A.; Ruzic, A.; Samarzija, M.; Badovinac, S.; Kehler, T.; Jakopovic, M. Persistent endothelial dysfunction turns the frequent exacerbator COPD from respiratory disorder into a progressive pulmonary and systemic vascular disease. Med. Hypotheses 2015, 84, 155–158. [Google Scholar] [CrossRef]
- Takahashi, T.; Kobayashi, S.; Fujino, N.; Suzuki, T.; Ota, C.; He, M.; Yamada, M.; Suzuki, S.; Yanai, M.; Kurosawa, S.; et al. Increased circulating endothelial microparticles in COPD patients: A potential biomarker for COPD exacerbation susceptibility. Thorax 2012, 67, 1067–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nana-Sinkam, S.P.; Hunter, M.G.; Nuovo, G.J.; Schmittgen, T.D.; Gelinas, R.; Galas, D.; Marsh, C.B. Integrating the MicroRNome into the study of lung disease. Am. J. Respir. Crit. Care Med. 2009, 179, 4–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ezzie, M.E.; Crawford, M.; Cho, J.H.; Orellana, R.; Zhang, S.; Gelinas, R.; Batte, K.; Yu, L.; Nuovo, G.; Galas, D.; et al. Gene expression networks in COPD: microRNA and mRNA regulation. Thorax 2012, 67, 122–131. [Google Scholar] [CrossRef] [Green Version]
- Molina-Pinelo, S.; Pastor, M.D.; Suarez, R.; Romero-Romero, B.; Gonzalez De la Pena, M.; Salinas, A.; Garcia-Carbonero, R.; De Miguel, M.J.; Rodriguez-Panadero, F.; Carnero, A.; et al. MicroRNA clusters: Dysregulation in lung adenocarcinoma and COPD. Eur. Respir J. 2014, 43, 1740–1749. [Google Scholar] [CrossRef]
- Serban, K.A.; Rezania, S.; Petrusca, D.N.; Poirier, C.; Cao, D.; Justice, M.J.; Patel, M.; Tsvetkova, I.; Kamocki, K.; Mikosz, A.; et al. Structural and functional characterization of endothelial microparticles released by cigarette smoke. Sci. Rep. 2016, 6, 31596. [Google Scholar] [CrossRef]
- Mizuno, S.; Bogaard, H.J.; Gomez-Arroyo, J.; Alhussaini, A.; Kraskauskas, D.; Cool, C.D.; Voelkel, N.F. MicroRNA-199a-5p is associated with hypoxia-inducible factor-1α expression in lungs from patients with COPD. Chest 2012, 142, 663–672. [Google Scholar] [CrossRef] [Green Version]
- Long, Y.-J.; Liu, X.-P.; Chen, S.-S.; Zong, D.-D.; Chen, Y.; Chen, P. miR-34a is involved in CSE-induced apoptosis of human pulmonary microvascular endothelial cells by targeting Notch-1 receptor protein. Respir. Res. 2018, 19, 21. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, X.; Herbert, J.M.; Lodhia, P.; Bradford, J.; Turner, A.M.; Newby, P.M.; Thickett, D.; Naidu, U.; Blakey, D.; Barry, S.; et al. Identification of novel vascular targets in lung cancer. Br. J. Cancer 2015, 112, 485–494. [Google Scholar] [CrossRef] [Green Version]
- Tusher, V.G.; Tibshirani, R.; Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 2001, 98, 5116–5121. [Google Scholar] [CrossRef] [Green Version]
- Patterson, T.A.; Lobenhofer, E.K.; Fulmer-Smentek, S.B.; Collins, P.J.; Chu, T.M.; Bao, W.; Fang, H.; Kawasaki, E.S.; Hager, J.; Tikhonova, I.R.; et al. Performance comparison of one-color and two-color platforms within the MicroArray Quality Control (MAQC) project. Nat. Biotechnol. 2006, 24, 1140–1150. [Google Scholar] [CrossRef]
- Paraskevopoulou, M.D.; Georgakilas, G.; Kostoulas, N.; Vlachos, I.S.; Vergoulis, T.; Reczko, M.; Filippidis, C.; Dalamagas, T.; Hatzigeorgiou, A.G. DIANA-LncBase: Experimentally verified and computationally predicted microRNA targets on long non-coding RNAs. Nucleic Acids Res. 2013, 41, W169–W173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reczko, M.; Maragkakis, M.; Alexiou, P.; Grosse, I.; Hatzigeorgiou, A.G. Functional microRNA targets in protein coding sequences. Bioinformatics 2012, 28, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Kolesnikov, N.; Hastings, E.; Keays, M.; Melnichuk, O.; Tang, Y.A.; Williams, E.; Dylag, M.; Kurbatova, N.; Brandizi, M.; Burdett, T.; et al. ArrayExpress update—Simplifying data submissions. Nucleic Acids Res. 2015, 43, D1113–D1116. [Google Scholar] [CrossRef]
- Good, R.J.; Hernandez-Lagunas, L.; Allawzi, A.; Maltzahn, J.K.; Vohwinkel, C.U.; Upadhyay, A.K.; Kompella, U.B.; Birukov, K.G.; Carpenter, T.C.; Sucharov, C.C.; et al. MicroRNA dysregulation in lung injury: The role of the miR-26a/EphA2 axis in regulation of endothelial permeability. American journal of physiology. Lung Cell. Mol. Physiol. 2018, 315, L584–L594. [Google Scholar] [CrossRef]
- Korde, A.; Jin, L.; Zhang, J.G.; Ramaswamy, A.; Hu, B.; Kolahian, S.; Guardela, B.J.; Herazo-Maya, J.; Siegfried, J.M.; Stabile, L.; et al. Lung endothelial microRNA-1 regulates tumor growth and angiogenesis. Am. J. Respir Crit. Care Med. 2017, 196, 1443–1455. [Google Scholar] [CrossRef]
- Kaur, S.; Leszczynska, K.; Abraham, S.; Scarcia, M.; Hiltbrunner, S.; Marshall, C.J.; Mavria, G.; Bicknell, R.; Heath, V.L. RhoJ/TCL regulates endothelial motility and tube formation and modulates actomyosin contractility and focal adhesion numbers. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 657–664. [Google Scholar] [CrossRef] [Green Version]
- Mura, M.; Swain, R.K.; Zhuang, X.; Vorschmitt, H.; Reynolds, G.; Durant, S.; Beesley, J.F.; Herbert, J.M.; Sheldon, H.; Andre, M.; et al. Identification and angiogenic role of the novel tumor endothelial marker CLEC14A. Oncogene 2012, 31, 293–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noy, P.J.; Lodhia, P.; Khan, K.; Zhuang, X.; Ward, D.G.; Verissimo, A.R.; Bacon, A.; Bicknell, R. Blocking CLEC14A-MMRN2 binding inhibits sprouting angiogenesis and tumour growth. Oncogene 2015, 34, 5821–5831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salisbury, V. High Resolution Imaging and Analysis of Endothelial Tubulogenesis and Blood Vessel Formation. In Physical Sciences of Imaging in the Biomedical Sciences School of Chemistry; University of Birmingham: Birmingham, UK, 2017; p. 267. [Google Scholar]
- Munir, H.; Rainger, G.E.; Nash, G.B.; McGettrick, H. Analyzing the effects of stromal cells on the recruitment of leukocytes from flow. J. Vis. Exp. 2015, e52480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, L.M.; McGettrick, H.M.; Nash, G.B. Static and dynamic assays of cell adhesion relevant to the vasculature. Methods Mol. Biol. 2016, 1430, 231–248. [Google Scholar] [CrossRef]
- Liu, K.X.; Chen, G.P.; Lin, P.L.; Huang, J.C.; Lin, X.; Qi, J.C.; Lin, Q.C. Detection and analysis of apoptosis- and autophagy-related miRNAs of mouse vascular endothelial cells in chronic intermittent hypoxia model. Life Sci. 2018, 193, 194–199. [Google Scholar] [CrossRef]
- Sun, X.; Icli, B.; Wara, A.K.; Belkin, N.; He, S.; Kobzik, L.; Hunninghake, G.M.; Vera, M.P.; Blackwell, T.S.; Baron, R.M.; et al. MicroRNA-181b regulates NF-κB–mediated vascular inflammation. J. Clin. Invest. 2012, 122, 1973–1990. [Google Scholar] [CrossRef]
- Khalyfa, A.; Kheirandish-Gozal, L.; Bhattacharjee, R.; Khalyfa, A.A.; Gozal, D. Circulating microRNAs as potential biomarkers of endothelial dysfunction in obese children. Chest 2016, 149, 786–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatwadekar, A.D.; Yan, Y.; Stepps, V.; Hazra, S.; Korah, M.; Bartelmez, S.; Chaqour, B.; Grant, M.B. MiR-92a corrects CD34+ cell dysfunction in diabetes by modulating core circadian genes involved in progenitor differentiation. Diabetes 2015, 64, 4226–4237. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Tian, F.; Wang, J.; Jing, J.; Zhou, S.S.; Chen, Y.D. Endothelial cell autophagy in atherosclerosis is regulated by miR-30-mediated translational control of ATG6. Cell. Physiol. Biochem. 2015, 37, 1421–1430. [Google Scholar] [CrossRef]
- Zhou, Q.; Gallagher, R.; Ufret-Vincenty, R.; Li, X.; Olson, E.N.; Wang, S. Regulation of angiogenesis and choroidal neovascularization by members of microRNA-23∼27∼24 clusters. Proc. Natl. Acad. Sci. USA 2011, 108, 8287–8292. [Google Scholar] [CrossRef] [Green Version]
- Besnier, M.; Gasparino, S.; Vono, R.; Sangalli, E.; Facoetti, A.; Bollati, V.; Cantone, L.; Zaccagnini, G.; Maimone, B.; Fuschi, P.; et al. miR-210 enhances the therapeutic potential of bone-marrow-derived circulating proangiogenic cells in the setting of limb ischemia. Mol. Ther. J. Am. Soc. Gene Ther. 2018, 26, 1694–1705. [Google Scholar] [CrossRef] [Green Version]
- Gasparello, J.; Lomazzi, M.; Papi, C.; D’Aversa, E.; Sansone, F.; Casnati, A.; Donofrio, G.; Gambari, R.; Finotti, A. Efficient delivery of microRNA and antimiRNA molecules using an argininocalix [4] arene macrocycle. Mol. Ther. Nucleic Acids. 2019, 18, 748–763. [Google Scholar] [CrossRef] [Green Version]
- Hetheridge, C.; Mavria, G.; Mellor, H. Uses of the in vitro endothelial–fibroblast organotypic co-culture assay in angiogenesis research. Biochem. Soc. Trans. 2011, 39, 1597–1600. [Google Scholar] [CrossRef] [PubMed]
- Leidinger, P.; Keller, A.; Borries, A.; Huwer, H.; Rohling, M.; Huebers, J.; Lenhof, H.P.; Meese, E. Specific peripheral miRNA profiles for distinguishing lung cancer from COPD. Lung Cancer 2011, 74, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Spira, A.; Beane, J.; Pinto-Plata, V.; Kadar, A.; Liu, G.; Shah, V.; Celli, B.; Brody, J.S. Gene Expression Profiling of Human Lung Tissue from Smokers with Severe Emphysema. Am. J. Respir. Cell. Mol. Biol. 2004, 31, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; An, N.; Li, J.; Xia, J.; Tian, Y.; Zhao, P.; Liu, X.; Huang, H.; Gao, J.; Zhang, X. miRNA-206 regulates human pulmonary microvascular endothelial cell apoptosis via targeting in chronic obstructive pulmonary disease. J. Cell. Biochem. 2019, 120, 6223–6236. [Google Scholar] [CrossRef] [PubMed]
- Musri, M.M.; Coll-Bonfill, N.; Maron, B.A.; Peinado, V.I.; Wang, R.S.; Altirriba, J.; Blanco, I.; Oldham, W.M.; Tura-Ceide, O.; García-Lucio, J.; et al. MicroRNA dysregulation in pulmonary arteries from chronic obstructive pulmonary disease. Relationships with vascular remodeling. Am. J. Respir Cell. Mol. Biol. 2018, 59, 490–499. [Google Scholar] [CrossRef]
- Paschalaki, K.E.; Zampetaki, A.; Baker, J.R.; Birrell, M.A.; Starke, R.D.; Belvisi, M.G.; Donnelly, L.E.; Mayr, M.; Randi, A.M.; Barnes, P.J. Downregulation of microRNA-126 augments DNA damage response in cigarette smokers and patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2018, 197, 665–668. [Google Scholar] [CrossRef]
- Yoo, J.O.; Kwak, S.Y.; An, H.J.; Bae, I.H.; Park, M.J.; Han, Y.H. miR-181b-3p promotes epithelial–mesenchymal transition in breast cancer cells through Snail stabilization by directly targeting YWHAG. Biochim. Biophys. Acta 2016, 1863, 1601–1611. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.Y.P.; Zhu, S.Y.; Brooks, D.; Bowlby, R.; Durham, J.S.; Ma, Y.; Moore, R.A.; Mungall, A.J.; Jones, S.; Poh, C.F. Tumor microRNA profile and prognostic value for lymph node metastasis in oral squamous cell carcinoma patients. Oncotarget 2020, 11, 2204–2215. [Google Scholar] [CrossRef]
- Cinegaglia, N.C.; Andrade, S.C.; Tokar, T.; Pinheiro, M.; Severino, F.E.; Oliveira, R.A.; Hasimoto, E.N.; Cataneo, D.C.; Cataneo, A.J.; Defaveri, J.; et al. Integrative transcriptome analysis identifies deregulated microRNA-transcription factor networks in lung adenocarcinoma. Oncotarget 2016, 7, 28920–28934. [Google Scholar] [CrossRef] [Green Version]
- Tian, F.; Shen, Y.; Chen, Z.; Li, R.; Lu, J.; Ge, Q. Aberrant miR-181b-5p and miR-486-5p expression in serum and tissue of non-small cell lung cancer. Gene 2016, 591, 338–343. [Google Scholar] [CrossRef]
- Iliopoulos, D.; Jaeger, S.A.; Hirsch, H.A.; Bulyk, M.L.; Struhl, K. STAT3 Activation of miR-21 and miR-181b-1 via PTEN and CYLD Are Part of the Epigenetic Switch Linking Inflammation to Cancer. Mol. Cell. 2010, 39, 493–506. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.H.; Na, H.S.; Jeong, S.Y.; Jeong, S.H.; Park, H.R.; Chung, J. Comparison of inflammatory microRNA expression in healthy and periodontitis tissues. Biocell 2011, 35, 43–49, PMID: 22128589. [Google Scholar] [CrossRef] [PubMed]
- Usher, A.K.; Stockley, R.A. The link between chronic periodontitis and COPD: A common role for the neutrophil? BMC Med. 2013, 11, 241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Liu, H.; Wang, H.; Sun, Y. Down-regulation of microRNA-181b is a potential prognostic marker of non-small cell lung cancer. Pathol. Res. Pract. 2013, 209, 490–494. [Google Scholar] [CrossRef]
- Lang, Y.; Xu, S.; Ma, J.; Wu, J.; Jin, S.; Cao, S.; Yu, Y. MicroRNA-429 induces tumorigenesis of human non-small cell lung cancer cells and targets multiple tumor suppressor genes. Biochem. Biophys. Res. Commun. 2014, 450, 154–159. [Google Scholar] [CrossRef]
- Halvorsen, A.R.; Bjaanaes, M.; LeBlanc, M.; Holm, A.M.; Bolstad, N.; Rubio, L.; Penalver, J.C.; Cervera, J.; Mojarrieta, J.C.; Lopez-Guerrero, J.A.; et al. A unique set of 6 circulating microRNAs for early detection of non-small cell lung cancer. Oncotarget 2016, 7, 37250–37259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, J.; Tang, J.; Chen, Q.; Tang, D.; Liu, M.; Luo, M.; Wang, Y.; Wang, J.; Zhao, Z.; Tang, C.; et al. miR-429 regulates alveolar macrophage inflammatory cytokine production and is involved in LPS-induced acute lung injury. Biochem. J. 2015, 471, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, X.; Guan, H.; Mizokami, A.; Keller, E.T.; Xu, X.; Liu, X.; Tan, J.; Hu, L.; Lu, Y.; et al. Exosome-derived microRNAs contribute to prostate cancer chemoresistance. Int. J. Oncol. 2016, 49, 838–846. [Google Scholar] [CrossRef] [Green Version]
- Yarrow, J.C.; Perlman, Z.E.; Westwood, N.J.; Mitchison, T.J. A high-throughput cell migration assay using scratch wound healing, a comparison of image-based readout methods. BMC Biotechnol. 2004, 4, 21. [Google Scholar] [CrossRef] [Green Version]
- Shi, Z.M.; Wang, X.F.; Qian, X.; Tao, T.; Wang, L.; Chen, Q.D.; Wang, X.R.; Cao, L.; Wang, Y.Y.; Zhang, J.X.; et al. MiRNA-181b suppresses IGF-1R and functions as a tumor suppressor gene in gliomas. RNA 2013, 19, 552–560. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Ge, S.; Jia, R.; Zhou, Y.; Song, X.; Zhang, H.; Fan, X. Hypoxia-induced miR-181b enhances angiogenesis of retinoblastoma cells by targeting PDCD10 and GATA6. Oncol. Rep. 2015, 33, 2789–2796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartoszewska, S.; Kochan, K.; Piotrowski, A.; Kamysz, W.; Ochocka, R.J.; Collawn, J.F.; Bartoszewski, R. The hypoxia-inducible miR-429 regulates hypoxia-induciblefactor-1a expression in human endothelial cells through a negative feedback loop. FASEB J. 2015, 29, 1467–1479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng-Blichfeldt, J.P.; Alcada, J.; Montero, M.A.; Dean, C.H.; Griesenbach, U.; Griffiths, M.J.; Hind, M. Deficient retinoid-driven angiogenesis may contribute to failure of adult human lung regeneration in emphysema. Thorax 2017, 72, 510–521. [Google Scholar] [CrossRef]
- Kaza, A.K.; Kron, I.L.; Kern, J.A.; Long, S.M.; Fiser, S.M.; Nguyen, R.P.; Tribble, C.G.; Laubach, V.E. Retinoic acid enhances lung growth after pneumonectomy. Ann. Thorac. Surg. 2001, 71, 1645–1650. [Google Scholar] [CrossRef]
- Green, C.E.; Turner, A.M. The role of the endothelium in asthma and chronic obstructive pulmonary disease (COPD). Respir. Res. 2017, 18, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | All n = 13 | Non-COPD n = 7 | COPD n = 6 | p Value |
|---|---|---|---|---|
| Male patients | 5 (38.5%) | 3 (42.9%) | 2 (33.3%) | 1.000 |
| Age | 67.0 (14.0) | 63.0 (18.0) | 69.0 (11.0) | 0.366 |
| BMI | 21.8 (7.1) | 27.2 (13.9) | 21.6 (5.8) | 0.731 |
| Pack years | 26.5 (40.5) | 0 (31) | 26.0 (61.3) | 0.073 |
| Current smoker | 2 (15.4%) | 1 (14.3%) | 1 (16.7%) | 0.086 |
| FEV1pp | 85.1 (36.0) | 96.8 (20.4) | 64.0 (40.4) | 0.008 |
| No cancer * | 2 (15.4%) | 0 | 2 (33.3%) | 0.874 |
| 0 (in situ) | 0 | 0 | 0 | |
| IA | 3 (23.1%) | 2 (28.6%) | 1 (16.7%) | |
| IB | 3 (23.1%) | 2 (28.6%) | 1 (16.7%) | |
| IIA | 0 | 0 | 0 | |
| IIB | 2 (15.4%) | 1 (14.3%) | 1 (16.7%) | |
| IIIA | 2 (15.4%) | 1 (14.3%) | 1 (16.7%) | |
| Lung metastases | 1 (7.7%) | 1 (14.3%) | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Green, C.E.; Clarke, J.; Bicknell, R.; Turner, A.M. Pulmonary MicroRNA Changes Alter Angiogenesis in Chronic Obstructive Pulmonary Disease and Lung Cancer. Biomedicines 2021, 9, 830. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9070830
Green CE, Clarke J, Bicknell R, Turner AM. Pulmonary MicroRNA Changes Alter Angiogenesis in Chronic Obstructive Pulmonary Disease and Lung Cancer. Biomedicines. 2021; 9(7):830. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9070830
Chicago/Turabian StyleGreen, Clara E., Joseph Clarke, Roy Bicknell, and Alice M. Turner. 2021. "Pulmonary MicroRNA Changes Alter Angiogenesis in Chronic Obstructive Pulmonary Disease and Lung Cancer" Biomedicines 9, no. 7: 830. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9070830