
Causal Links between Hypovitaminosis D and Dysregulation of the T Cell Connection of Immunity Associated with Obesity and Concomitant Pathologies

 ,
, {kind=link}
Abstract
:1. Introduction
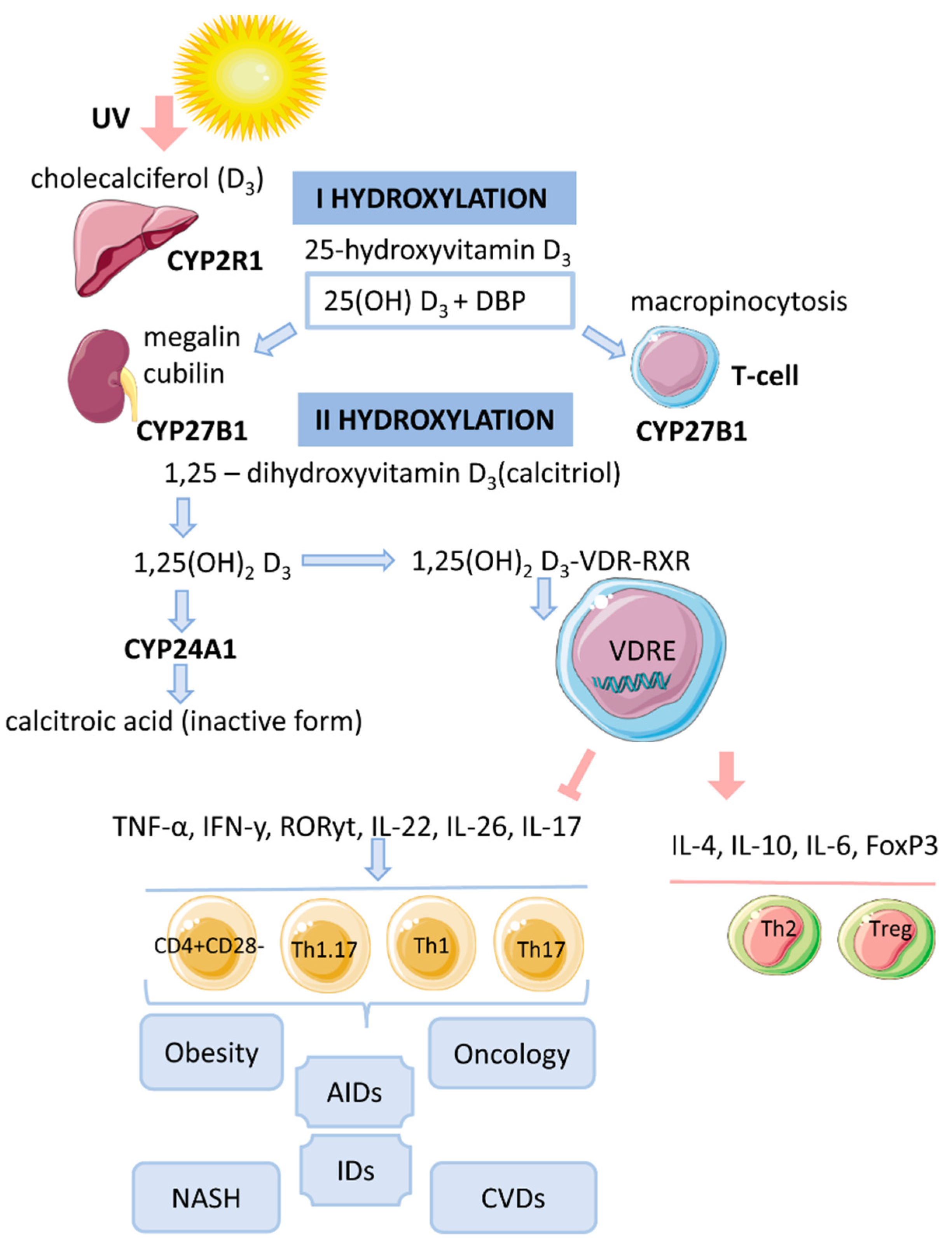
2. Vitamin D Metabolism
3. Cytosolic Complex 1,25(OH)2D3-VDR-RXRF
4. Genomic Mechanism of Action of Vitamin D (T-lymphocytes)
5. Vitamin D and T-lymphocytes
5.1. Th1 and Th2 Cells
5.2. Non-Pathogenic Th17 Cells and Treg
5.3. Th1.17-Cells
5.4. Memory T Cells
6. Hypovitaminosis D in Obesity
- (1)
- (2)
- (3)
- Different ability to activate vitamin D in adipose tissue of lean and obese individuals [58]. In adipocytes, high expression of the enzymes 1α-hydroxylase (mitochondrial CYP27B1) and 25-hydroxylase CYP2J2 was found [55]. However, their expression is lower in obese people compared to lean people [60].
7. Molecular Mechanism of Action of Vitamin D on T Cells under Hypoxic Conditions Associated with Obesity
8. Hypovitaminosis D in Obesity and Infectious Diseases
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, R.; Nikolajczyk, B.S. Tissue Immune Cells Fuel Obesity-Associated Inflammation in Adipose Tissue and Beyond. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhi, C.; Huang, J.; Wang, J.; Cao, H.; Bai, Y.; Guo, J.; Su, Z. Connection between Gut Microbiome and the Development of Obesity. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 1987–1998. [Google Scholar] [CrossRef] [PubMed]
- Vallianou, N.; Stratigou, T.; Christodoulatos, G.S.; Dalamaga, M. Understanding the Role of the Gut Microbiome and Microbial Metabolites in Obesity and Obesity-Associated Metabolic Disorders: Current Evidence and Perspectives. Curr. Obes. Rep. 2019, 8, 317–332. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois, C.; Gorwood, J.; Barrail-Tran, A.; Lagathu, C.; Capeau, J.; Desjardins, D.; Le Grand, R.; Damouche, A.; Béréziat, V.; Lambotte, O. Specific Biological Features of Adipose Tissue, and Their Impact on HIV Persistence. Front. Microbiol. 2019, 10, 2837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeffery, L.E.; Henley, P.; Marium, N.; Filer, A.; Sansom, D.M.; Hewison, M.; Raza, K. Decreased Sensitivity to 1,25-Dihydroxyvitamin D3 in T Cells from the Rheumatoid Joint. J. Autoimmun. 2018, 88, 50–60. [Google Scholar] [CrossRef]
- Zhao, R.; He, Q.; Sha, S.; Song, J.; Qin, J.; Liu, P.; Sun, Y.; Sun, L.; Hou, X.; Chen, L. Increased AHR Transcripts Correlate With Pro-Inflammatory T-Helper Lymphocytes Polarization in Both Metabolically Healthy Obesity and Type 2 Diabetic Patients. Front. Immunol. 2020, 11, 1644. [Google Scholar] [CrossRef]
- Van Raemdonck, K.; Umar, S.; Szekanecz, Z.; Zomorrodi, R.K.; Shahrara, S. Impact of Obesity on Autoimmune Arthritis and Its Cardiovascular Complications. Autoimmun. Rev. 2018, 17, 821–835. [Google Scholar] [CrossRef]
- De Oliveira Boldrini, V.; Dos Santos Farias, A.; Degasperi, G.R. Deciphering Targets of Th17 Cells Fate: From Metabolism to Nuclear Receptors. Scand. J. Immunol. 2019, 90, e12793. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Yokote, K.; Nakayama, T. The Obesity-Related Pathology and Th17 Cells. Cell. Mol. Life Sci. CMLS 2017, 74, 1231–1245. [Google Scholar] [CrossRef]
- Chatterjee, S.; Das, S. P2X7 Receptor as a Key Player in Oxidative Stress-Driven Cell Fate in Nonalcoholic Steatohepatitis. Oxid. Med. Cell. Longev. 2015, 2015, e172493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Chen, F.; Wang, J.; Zeng, Z.; Yang, Q.; Shao, S. Th17 and Treg Lymphocytes in Obesity and Type 2 Diabetic Patients. Clin. Immunol. 2018, 197, 77–85. [Google Scholar] [CrossRef]
- Coccurello, R.; Volonté, C. P2X7 Receptor in the Management of Energy Homeostasis: Implications for Obesity, Dyslipidemia, and Insulin Resistance. Front. Endocrinol. 2020, 11, 199. [Google Scholar] [CrossRef] [PubMed]
- Kongsbak, M.; von Essen, M.R.; Levring, T.B.; Schjerling, P.; Woetmann, A.; Ødum, N.; Bonefeld, C.M.; Geisler, C. Vitamin D-Binding Protein Controls T Cell Responses to Vitamin D. BMC Immunol. 2014, 15, 35. [Google Scholar] [CrossRef] [Green Version]
- Bendix, M.; Dige, A.; Deleuran, B.; Dahlerup, J.F.; Jørgensen, S.P.; Bartels, L.E.; Husted, L.B.; Harsløf, T.; Langdahl, B.; Agnholt, J. Flow Cytometry Detection of Vitamin D Receptor Changes during Vitamin D Treatment in Crohn’s Disease. Clin. Exp. Immunol. 2015, 181, 19–28. Available online: https://0-onlinelibrary-wiley-com.brum.beds.ac.uk/doi/full/10.1111/cei.12613 (accessed on 14 September 2021).
- Taheriniya, S.; Arab, A.; Hadi, A.; Fadel, A.; Askari, G. Vitamin D and Thyroid Disorders: A Systematic Review and Meta-Analysis of Observational Studies. BMC Endocr. Disord. 2021, 21, 171. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.K.; Banerjee, D.; Lal, A.; Jha, V. Vitamin D Deficiency, CD4+CD28null Cells and Accelerated Atherosclerosis in Chronic Kidney Disease. Nephrology 2012, 17, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, M.L.; Pagano, M.T.; Pierdominici, M.; Ortona, E. The Role of Vitamin D in Autoimmune Diseases: Could Sex Make the Difference? Biol. Sex. Differ. 2021, 12, 12. [Google Scholar] [CrossRef]
- McGregor, R.; Chauss, D.; Freiwald, T.; Yan, B.; Wang, L.; Nova-Lamperti, E.; Zhang, Z.; Teague, H.; West, E.E.; Bibby, J.; et al. An Autocrine Vitamin D-Driven Th1 Shutdown Program Can Be Exploited for COVID-19. BioRxiv 2020, preprint. [Google Scholar] [CrossRef]
- Sassi, F.; Tamone, C.; D’Amelio, P. Vitamin D: Nutrient, Hormone, and Immunomodulator. Nutrients 2018, 10, 1656. [Google Scholar] [CrossRef] [Green Version]
- Soto, J.R.; Anthias, C.; Madrigal, A.; Snowden, J.A. Insights Into the Role of Vitamin D as a Biomarker in Stem Cell Transplantation. Front. Immunol. 2020, 11, 966. [Google Scholar] [CrossRef] [PubMed]
- Christakos, S.; Dhawan, P.; Verstuyf, A.; Verlinden, L.; Carmeliet, G. Vitamin D: Metabolism, Molecular Mechanism of Action, and Pleiotropic Effects. Physiol. Rev. 2016, 96, 365–408. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, L. Vitamin D and Microbiota: Two Sides of the Same Coin in the Immunomodulatory Aspects. Int. Immunopharmacol. 2020, 79, 106112. [Google Scholar] [CrossRef] [PubMed]
- Borel, P.; Caillaud, D.; Cano, N.J. Vitamin D Bioavailability: State of the Art. Crit. Rev. Food Sci. Nutr. 2015, 55, 1193–1205. [Google Scholar] [CrossRef] [PubMed]
- Bikle, D.D. Vitamin D Metabolism, Mechanism of Action, and Clinical Applications. Chem. Biol. 2014, 21, 319–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Killick, J.; Hay, J.; Morandi, E.; Vermeren, S.; Kari, S.; Angles, T.; Williams, A.; Damoiseaux, J.; Astier, A.L. Vitamin D/CD46 Crosstalk in Human T Cells in Multiple Sclerosis. Front. Immunol. 2020, 11, 598727. [Google Scholar] [CrossRef] [PubMed]
- Vasiliou, J.E.; Lui, S.; Walker, S.A.; Chohan, V.; Xystrakis, E.; Bush, A.; Hawrylowicz, C.M.; Saglani, S.; Lloyd, C.M. Vitamin D Deficiency Induces Th2 Skewing and Eosinophilia in Neonatal Allergic Airways Disease. Allergy 2014, 69, 1380–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, A.C.; Taylor, C.L.; Yaktine, A.L.; Del Valle, H.B. Institute of Medicine (US) Committee to Review Dietary Reference Intakes for Vitamin D and Calcium Dietary Reference Intakes for Calcium and Vitamin D; The National Academies Collection: Reports funded by National Institutes of Health; National Academies Press (US): Washington, DC, USA, 2011. [Google Scholar]
- Adorini, L.; Penna, G. Control of Autoimmune Diseases by the Vitamin D Endocrine System. Nat. Clin. Pract. Rheumatol. 2008, 4, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Prosser, D.E.; Kaufmann, M. 25-Hydroxyvitamin D-24-Hydroxylase (CYP24A1): Its Important Role in the Degradation of Vitamin D. Arch. Biochem. Biophys. 2012, 523, 9–18. [Google Scholar] [CrossRef]
- DeLuca, H.F. Evolution of Our Understanding of Vitamin D. Nutr. Rev. 2008, 66, S73–S87. [Google Scholar] [CrossRef] [PubMed]
- Pike, J.W.; Meyer, M.B. The Vitamin D Receptor: New Paradigms for the Regulation of Gene Expression by 1,25-Dihydroxyvitamin D(3). Endocrinol. Metab. Clin. North. Am. 2010, 39, 255–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todosenko, N.; Vulf, M.; Yurova, K.; Skuratovskaia, D.; Khaziakhmatova, O.; Gazatova, N.; Melashchenko, O.; Urazova, O.; Litvinova, L. The Pathogenic Subpopulation of Th17 Cells in Obesity. Curr. Pharm. Des. 2021. [Google Scholar] [CrossRef]
- Colin, E.M.; Asmawidjaja, P.S.; van Hamburg, J.P.; Mus, A.M.C.; van Driel, M.; Hazes, J.M.W.; van Leeuwen, J.P.T.M.; Lubberts, E. 1,25-Dihydroxyvitamin D3 Modulates Th17 Polarization and Interleukin-22 Expression by Memory T Cells from Patients with Early Rheumatoid Arthritis. Arthritis Rheum. 2010, 62, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.H.; Chung, Y.; Dong, C. Vitamin D Suppresses Th17 Cytokine Production by Inducing C/EBP Homologous Protein (CHOP) Expression. J. Biol. Chem. 2010, 285, 38751–38755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D.; Luo, F.; Xing, J.-C.; Zhang, F.; Xu, J.-Z.; Zhang, Z.-H. 1,25(OH)2 D3 Inhibited Th17 Cells Differentiation via Regulating the NF-ΚB Activity and Expression of IL-17. Cell Prolif. 2018, 51, e12461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golubovskaya, V.; Wu, L. Different Subsets of T Cells, Memory, Effector Functions, and CAR-T Immunotherapy. Cancers 2016, 8, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavallari, J.F.; Denou, E.; Foley, K.P.; Khan, W.I.; Schertzer, J.D. Different Th17 Immunity in Gut, Liver, and Adipose Tissues during Obesity: The Role of Diet, Genetics, and Microbes. Gut Microbes 2016, 7, 82–89. [Google Scholar] [CrossRef] [Green Version]
- Pandolfi, J.B.; Ferraro, A.A.; Sananez, I.; Gancedo, M.C.; Baz, P.; Billordo, L.A.; Fainboim, L.; Arruvito, L. ATP-Induced Inflammation Drives Tissue-Resident Th17 Cells in Metabolically Unhealthy Obesity. J. Immunol. 2016, 196, 3287–3296. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Yosef, N.; Gaublomme, J.; Wu, C.; Lee, Y.; Clish, C.B.; Kaminski, J.; Xiao, S.; Meyer Zu Horste, G.; Pawlak, M.; et al. CD5L/AIM Regulates Lipid Biosynthesis and Restrains Th17 Cell Pathogenicity. Cell 2015, 163, 1413–1427. [Google Scholar] [CrossRef] [Green Version]
- Castro, G.; Liu, X.; Ngo, K.; De Leon-Tabaldo, A.; Zhao, S.; Luna-Roman, R.; Yu, J.; Cao, T.; Kuhn, R.; Wilkinson, P.; et al. RORγt and RORα Signature Genes in Human Th17 Cells. PLOS ONE 2017, 12, e0181868. [Google Scholar] [CrossRef] [Green Version]
- Di Luccia, B.; Gilfillan, S.; Cella, M.; Colonna, M.; Huang, S.C.-C. ILC3s Integrate Glycolysis and Mitochondrial Production of Reactive Oxygen Species to Fulfill Activation Demands. J. Exp. Med. 2019, 216, 2231–2241. [Google Scholar] [CrossRef] [PubMed]
- Caza, T.; Landas, S. Functional and Phenotypic Plasticity of CD4(+) T Cell Subsets. BioMed Res. Int. 2015, 2015, 521957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, S.; Suto, A.; Iwamoto, T.; Kashiwakuma, D.; Kagami, S.-I.; Suzuki, K.; Takatori, H.; Tamachi, T.; Hirose, K.; Onodera, A.; et al. Sox5 and C-Maf Cooperatively Induce Th17 Cell Differentiation via RORγt Induction as Downstream Targets of Stat3. J. Exp. Med. 2014, 211, 1857–1874. [Google Scholar] [CrossRef] [PubMed]
- Bending, D.; Peña, H.D.L.; Veldhoen, M.; Phillips, J.M.; Uyttenhove, C.; Stockinger, B.; Cooke, A. Highly Purified Th17 Cells from BDC2.5NOD Mice Convert into Th1-like Cells in NOD/SCID Recipient Mice. J. Clin. Investig. 2009, 119, 565–572. [Google Scholar] [CrossRef] [Green Version]
- Mann, E.H.; Ho, T.-R.; Pfeffer, P.E.; Matthews, N.C.; Chevretton, E.; Mudway, I.; Kelly, F.J.; Hawrylowicz, C.M. Vitamin D Counteracts an IL-23-Dependent IL-17A+IFN-Γ+ Response Driven by Urban Particulate Matter. Am. J. Respir. Cell Mol. Biol. 2017, 57, 355–366. [Google Scholar] [CrossRef]
- Liaskou, E.; Jeffery, L.E.; Trivedi, P.J.; Reynolds, G.M.; Suresh, S.; Bruns, T.; Adams, D.H.; Sansom, D.M.; Hirschfield, G.M. Loss of CD28 Expression by Liver-Infiltrating T Cells Contributes to Pathogenesis of Primary Sclerosing Cholangitis. Gastroenterology 2014, 147, 221–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.; Wang, R.; Horng, T. MTOR Is Key to T Cell Transdifferentiation. Cell Metab. 2019, 29, 241–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paintlia, A.S.; Paintlia, M.K.; Hollis, B.W.; Singh, A.K.; Singh, I. Interference with RhoA-ROCK Signaling Mechanism in Autoreactive CD4+ T Cells Enhances the Bioavailability of 1,25-Dihydroxyvitamin D3 in Experimental Autoimmune Encephalomyelitis. Am. J. Pathol. 2012, 181, 993–1006. [Google Scholar] [CrossRef] [Green Version]
- Spach, K.M.; Hayes, C.E. Vitamin D3 Confers Protection from Autoimmune Encephalomyelitis Only in Female Mice. J. Immunol. 2005, 175, 4119–4126. [Google Scholar] [CrossRef] [Green Version]
- Correale, J.; Ysrraelit, M.C.; Gaitán, M.I. Gender Differences in 1,25 Dihydroxyvitamin D3 Immunomodulatory Effects in Multiple Sclerosis Patients and Healthy Subjects. J. Immunol. 2010, 185, 4948–4958. [Google Scholar] [CrossRef] [Green Version]
- Attia, Y.M.; El-Kersh, D.M.; Ammar, R.A.; Adel, A.; Khalil, A.; Walid, H.; Eskander, K.; Hamdy, M.; Reda, N.; Mohsen, N.E.; et al. Inhibition of Aldehyde Dehydrogenase-1 and p-Glycoprotein-Mediated Multidrug Resistance by Curcumin and Vitamin D3 Increases Sensitivity to Paclitaxel in Breast Cancer. Chem. Biol. Interact. 2020, 315, 108865. [Google Scholar] [CrossRef]
- Joshi, S.; Pantalena, L.-C.; Liu, X.K.; Gaffen, S.L.; Liu, H.; Rohowsky-Kochan, C.; Ichiyama, K.; Yoshimura, A.; Steinman, L.; Christakos, S.; et al. 1,25-Dihydroxyvitamin D(3) Ameliorates Th17 Autoimmunity via Transcriptional Modulation of Interleukin-17A. Mol. Cell. Biol. 2011, 31, 3653–3669. [Google Scholar] [CrossRef] [Green Version]
- Cippitelli, M.; Santoni, A. Vitamin D3: A Transcriptional Modulator of the Interferon-Gamma Gene. Eur. J. Immunol. 1998, 28, 3017–3030. [Google Scholar] [CrossRef]
- Abedin, S.A.; Banwell, C.M.; Colston, K.W.; Carlberg, C.; Campbell, M.J. Epigenetic Corruption of VDR Signalling in Malignancy. Anticancer Res. 2006, 26, 2557–2566. [Google Scholar] [PubMed]
- Pereira-Santos, M.; Costa, P.R.F.; Assis, A.M.O.; Santos, C.a.S.T.; Santos, D.B. Obesity and Vitamin D Deficiency: A Systematic Review and Meta-Analysis. Obes. Rev. Off. J. Int. Assoc. Study Obes. 2015, 16, 341–349. [Google Scholar] [CrossRef]
- Pramono, A.; Jocken, J.W.E.; Essers, Y.P.G.; Goossens, G.H.; Blaak, E.E. Vitamin D and Tissue-Specific Insulin Sensitivity in Humans With Overweight/Obesity. J. Clin. Endocrinol. Metab. 2019, 104, 49–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajhashemy, Z.; Shahdadian, F.; Ziaei, R.; Saneei, P. Serum Vitamin D Levels in Relation to Abdominal Obesity: A Systematic Review and Dose-Response Meta-Analysis of Epidemiologic Studies. Obes. Rev. Off. J. Int. Assoc. Study Obes. 2021, 22, e13134. [Google Scholar] [CrossRef]
- Abbas, M.A. Physiological Functions of Vitamin D in Adipose Tissue. J. Steroid Biochem. Mol. Biol. 2017, 165, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Drincic, A.T.; Armas, L.A.G.; Van Diest, E.E.; Heaney, R.P. Volumetric Dilution, Rather than Sequestration Best Explains the Low Vitamin D Status of Obesity. Obesity 2012, 20, 1444–1448. [Google Scholar] [CrossRef]
- Wamberg, L.; Christiansen, T.; Paulsen, S.K.; Fisker, S.; Rask, P.; Rejnmark, L.; Richelsen, B.; Pedersen, S.B. Expression of Vitamin D-Metabolizing Enzymes in Human Adipose Tissue—the Effect of Obesity and Diet-Induced Weight Loss. Int. J. Obes. 2013, 37, 651–657. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Bae, S.; Yoon, Y. Anti-Adipogenic Effects of 1,25-Dihydroxyvitamin D3 Are Mediated by the Maintenance of the Wingless-Type MMTV Integration Site/β-Catenin Pathway. Int. J. Mol. Med. 2012, 30, 1219–1224. [Google Scholar] [CrossRef] [Green Version]
- Ge, X.; Wang, L.; Li, M.; Xu, N.; Yu, F.; Yang, F.; Li, R.; Zhang, F.; Zhao, B.; Du, J. Vitamin D/VDR Signaling Inhibits LPS-Induced IFNγ and IL-1β in Oral Epithelia by Regulating Hypoxia-Inducible Factor-1α Signaling Pathway. Cell Commun. Signal. CCS 2019, 17, 18. [Google Scholar] [CrossRef] [Green Version]
- Karmaus, P.W.F.; Chen, X.; Lim, S.A.; Herrada, A.A.; Nguyen, T.-L.M.; Xu, B.; Dhungana, Y.; Rankin, S.; Chen, W.; Rosencrance, C.; et al. Metabolic Heterogeneity Underlies Reciprocal Fates of TH17 Cell Stemness and Plasticity. Nature 2019, 565, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Kurebayashi, Y.; Nagai, S.; Ikejiri, A.; Ohtani, M.; Ichiyama, K.; Baba, Y.; Yamada, T.; Egami, S.; Hoshii, T.; Hirao, A.; et al. PI3K-Akt-MTORC1-S6K1/2 Axis Controls Th17 Differentiation by Regulating Gfi1 Expression and Nuclear Translocation of RORγ. Cell Rep. 2012, 1, 360–373. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.-P.; Ma, Y.-P.; Zhuo, L.; Zhang, Z.; Zou, G.-M.; Yang, Y.; Gao, H.-M.; Li, W.-G. 1,25-Dihydroxyvitamin D 3 Inhibits the Proliferation of Rat Mesangial Cells Induced by High Glucose via DDIT4. Oncotarget 2017, 9, 418–427. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Luo, F.; Li, J.; Wan, J.; Zhang, L.; Li, H.; Chen, A.; Chen, J.; Cai, T.; He, X.; et al. DNA Damage-Inducible Transcript 4 Is an Innate Guardian for Human Squamous Cell Carcinoma and an Molecular Vector for Anti-Carcinoma Effect of 1,25(OH)2D3. Exp. Dermatol. 2019, 28, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Lisse, T.S.; Liu, T.; Irmler, M.; Beckers, J.; Chen, H.; Adams, J.S.; Hewison, M. Gene Targeting by the Vitamin D Response Element Binding Protein Reveals a Role for Vitamin D in Osteoblast MTOR Signaling. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2011, 25, 937–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Liu, F.; Qu, H.; Wang, H.; Xiao, X.; Deng, H. 1,25(OH)2D3 Protects β Cell against High Glucose-Induced Apoptosis through MTOR Suppressing. Mol. Cell. Endocrinol. 2015, 414, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Molitoris, J.K.; McColl, K.S.; Swerdlow, S.; Matsuyama, M.; Lam, M.; Finkel, T.H.; Matsuyama, S.; Distelhorst, C.W. Glucocorticoid Elevation of Dexamethasone-Induced Gene 2 (Dig2/RTP801/REDD1) Protein Mediates Autophagy in Lymphocytes. J. Biol. Chem. 2011, 286, 30181–30189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolff, N.C.; McKay, R.M.; Brugarolas, J. REDD1/DDIT4-Independent MTORC1 Inhibition and Apoptosis by Glucocorticoids in Thymocytes. Mol. Cancer Res. 2014, 12, 867–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reuschel, E.L.; Wang, J.; Shivers, D.K.; Muthumani, K.; Weiner, D.B.; Ma, Z.; Finkel, T.H. REDD1 Is Essential for Optimal T Cell Proliferation and Survival. PLOS ONE 2015, 10, e0136323. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Liu, G.; Li, D.; Wei, C.; Hao, J. DDIT4 and Associated LncDDIT4 Modulate Th17 Differentiation through the DDIT4/TSC/MTOR Pathway. J. Immunol. 2018, 200, 1618–1626. [Google Scholar] [CrossRef] [PubMed]
- Savio, L.E.B.; de Andrade Mello, P.; da Silva, C.G.; Coutinho-Silva, R. The P2X7 Receptor in Inflammatory Diseases: Angel or Demon? Front. Pharmacol. 2018, 9, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Bo, C.; Kang, Y.; Li, H. What Else Can CD39 Tell Us? Front. Immunol. 2017, 8, 727. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Guan, X.; Chen, D.; Ma, W. The Th17/Treg Cell Balance: A Gut Microbiota-Modulated Story. Microorganisms 2019, 7, 583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Yao, Y.; Sumi, Y.; Li, A.; To, U.K.; Elkhal, A.; Inoue, Y.; Woehrle, T.; Zhang, Q.; Hauser, C.; et al. Purinergic Signaling: A Fundamental Mechanism in Neutrophil Activation. Sci. Signal. 2010, 3, ra45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, M.-H.; Maresz, K.; Lee, P.-S.; Wu, J.-C.; Ho, C.-T.; Popko, J.; Mehta, D.S.; Stohs, S.J.; Badmaev, V. Inhibition of TNF-α, IL-1α, and IL-1β by Pretreatment of Human Monocyte-Derived Macrophages with Menaquinone-7 and Cell Activation with TLR Agonists In Vitro. J. Med. Food 2016, 19, 663–669. [Google Scholar] [CrossRef]
- Bai, A.; Moss, A.; Kokkotou, E.; Usheva, A.; Sun, X.; Cheifetz, A.; Zheng, Y.; Longhi, M.S.; Gao, W.; Wu, Y.; et al. CD39 and CD161 Modulate Th17 Responses in Crohn’s Disease. J. Immunol. 2014, 193, 3366–3377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miossec, P.; Kolls, J.K. Targeting IL-17 and TH17 Cells in Chronic Inflammation. Nat. Rev. Drug Discov. 2012, 11, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.M.; Lonergan, R.; Costelloe, L.; Kinsella, K.; Moran, B.; O’Farrelly, C.; Tubridy, N.; Mills, K.H.G. CD39+Foxp3+ Regulatory T Cells Suppress Pathogenic Th17 Cells and Are Impaired in Multiple Sclerosis. J. Immunol. 2009, 183, 7602–7610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanzer, A.M.; Chambers, E.S.; Ryanna, K.; Richards, D.F.; Black, C.; Timms, P.M.; Martineau, A.R.; Griffiths, C.J.; Corrigan, C.J.; Hawrylowicz, C.M. Enhanced Production of IL-17A in Patients with Severe Asthma Is Inhibited by 1α,25-Dihydroxyvitamin D3 in a Glucocorticoid-Independent Fashion. J. Allergy Clin. Immunol. 2013, 132, 297–304.e3. [Google Scholar] [CrossRef] [PubMed]
- Mercola, J.; Grant, W.B.; Wagner, C.L. Evidence Regarding Vitamin D and Risk of COVID-19 and Its Severity. Nutrients 2020, 12, 3361. [Google Scholar] [CrossRef] [PubMed]
- Bold, J.; Harris, M.; Fellows, L.; Chouchane, M. Nutrition, the Digestive System and Immunity in COVID-19 Infection. Gastroenterol. Hepatol. Bed Bench 2020, 13, 331–340. [Google Scholar] [PubMed]
- Di Renzo, L.; Gualtieri, P.; Pivari, F.; Soldati, L.; Attinà, A.; Leggeri, C.; Cinelli, G.; Tarsitano, M.G.; Caparello, G.; Carrano, E.; et al. COVID-19: Is There a Role for Immunonutrition in Obese Patient? J. Transl. Med. 2020, 18, 415. [Google Scholar] [CrossRef] [PubMed]
- Merzon, E.; Tworowski, D.; Gorohovski, A.; Vinker, S.; Golan Cohen, A.; Green, I.; Frenkel-Morgenstern, M. Low Plasma 25(OH) Vitamin D Level Is Associated with Increased Risk of COVID-19 Infection: An Israeli Population-Based Study. FEBS J. 2020, 287, 3693–3702. [Google Scholar] [CrossRef] [PubMed]
- Baktash, V.; Hosack, T.; Patel, N.; Shah, S.; Kandiah, P.; Van den Abbeele, K.; Mandal, A.K.J.; Missouris, C.G. Vitamin D Status and Outcomes for Hospitalised Older Patients with COVID-19. Postgrad. Med. J. 2021, 97, 442–447. [Google Scholar] [CrossRef]
- Ekiz, T.; Pazarlı, A.C. Relationship between COVID-19 and Obesity. Diabetes Metab. Syndr. 2020, 14, 761–763. [Google Scholar] [CrossRef]
- Di Filippo, L.; Allora, A.; Doga, M.; Formenti, A.M.; Locatelli, M.; Rovere Querini, P.; Frara, S.; Giustina, A. Vitamin D Levels Associate with Blood Glucose and BMI in COVID-19 Patients Predicting Disease Severity. J. Clin. Endocrinol. Metab. 2021, dgab599. [Google Scholar] [CrossRef] [PubMed]
- Dumas, A.; Bernard, L.; Poquet, Y.; Lugo-Villarino, G.; Neyrolles, O. The Role of the Lung Microbiota and the Gut–Lung Axis in Respiratory Infectious Diseases. Cell. Microbiol. 2018, 20, e12966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mülsch, A.; Schray-Utz, B.; Mordvintcev, P.I.; Hauschildt, S.; Busse, R. Diethyldithiocarbamate Inhibits Induction of Macrophage NO Synthase. FEBS Lett. 1993, 321, 215–218. [Google Scholar] [CrossRef] [Green Version]
- Toor, S.M.; Saleh, R.; Sasidharan Nair, V.; Taha, R.Z.; Elkord, E. T-Cell Responses and Therapies against SARS-CoV-2 Infection. Immunology 2021, 162, 30–43. [Google Scholar] [CrossRef]
- Fehr, A.R.; Perlman, S. Coronaviruses: An Overview of Their Replication and Pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Di Renzo, L.; Merra, G.; Esposito, E.; De Lorenzo, A. Are Probiotics Effective Adjuvant Therapeutic Choice in Patients with COVID-19? Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 4062–4063. [Google Scholar] [CrossRef]
- Perlot, T.; Penninger, J.M. ACE2 - from the Renin-Angiotensin System to Gut Microbiota and Malnutrition. Microbes Infect. 2013, 15, 866–873. [Google Scholar] [CrossRef] [PubMed]
- Ruenjaiman, V.; Hirankarn, N.; Palaga, T. Innate Immunity in COVID-19: Drivers of Pathogenesis and Potential Therapeutic Targets. Asian Pac. J. Allergy Immunol. 2021, 39, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Bombardini, T.; Picano, E. Angiotensin-Converting Enzyme 2 as the Molecular Bridge between Epidemiologic and Clinical Features of COVID-19. Can. J. Cardiol. 2020, 36, 784.e1–784.e2. [Google Scholar] [CrossRef] [PubMed]
- Grant, W.B.; Lahore, H.; McDonnell, S.L.; Baggerly, C.A.; French, C.B.; Aliano, J.L.; Bhattoa, H.P. Evidence That Vitamin D Supplementation Could Reduce Risk of Influenza and COVID-19 Infections and Deaths. Nutrients 2020, 12, 988. [Google Scholar] [CrossRef] [Green Version]
- Malek Mahdavi, A. A Brief Review of Interplay between Vitamin D and Angiotensin-Converting Enzyme 2: Implications for a Potential Treatment for COVID-19. Rev. Med. Virol. 2020, 30, e2119. [Google Scholar] [CrossRef]
- Aygun, H. Vitamin D Can Prevent COVID-19 Infection-Induced Multiple Organ Damage. Naunyn. Schmiedebergs Arch. Pharmacol. 2020, 393, 1157–1160. [Google Scholar] [CrossRef]
- Kumar, D.; Gupta, P.; Banerjee, D. Letter: Does Vitamin D Have a Potential Role against COVID-19? Aliment. Pharmacol. Ther. 2020, 52, 409–411. [Google Scholar] [CrossRef]
- Hanff, T.C.; Harhay, M.O.; Brown, T.S.; Cohen, J.B.; Mohareb, A.M. Is There an Association between COVID-19 Mortality and the Renin-Angiotensin System? A Call for Epidemiologic Investigations. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2020, 71, 870–874. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, M.; Saito, J.; Zhao, H.; Sakamoto, A.; Hirota, K.; Ma, D. Inflammation Triggered by SARS-CoV-2 and ACE2 Augment Drives Multiple Organ Failure of Severe COVID-19: Molecular Mechanisms and Implications. Inflammation 2021, 44, 13–34. [Google Scholar] [CrossRef]
- Hojyo, S.; Uchida, M.; Tanaka, K.; Hasebe, R.; Tanaka, Y.; Murakami, M.; Hirano, T. How COVID-19 Induces Cytokine Storm with High Mortality. Inflamm. Regen. 2020, 40, 37. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tecson, K.M.; McCullough, P.A. Endothelial Dysfunction Contributes to COVID-19-Associated Vascular Inflammation and Coagulopathy. Rev. Cardiovasc. Med. 2020, 21, 315–319. [Google Scholar] [CrossRef]
- Jun Zhang, P.A.M.; Jun Zhang, P.A.M. Vitamin D deficiency in association with endothelial dysfunction: Implications for patients with COVID-19. Rev. Cardiovasc. Med. 2020, 21, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Kanikarla-Marie, P.; Jain, S.K. 1,25(OH)2D3 Inhibits Oxidative Stress and Monocyte Adhesion by Mediating the Upregulation of GCLC and GSH in Endothelial Cells Treated with Acetoacetate (Ketosis). J. Steroid Biochem. Mol. Biol. 2016, 159, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Zemb, P.; Bergman, P.; Camargo, C.A.; Cavalier, E.; Cormier, C.; Courbebaisse, M.; Hollis, B.; Joulia, F.; Minisola, S.; Pilz, S.; et al. Vitamin D Deficiency and the COVID-19 Pandemic. J. Glob. Antimicrob. Resist. 2020, 22, 133–134. [Google Scholar] [CrossRef]
- Jolliffe, D.A.; Camargo, C.A.; Sluyter, J.D.; Aglipay, M.; Aloia, J.F.; Ganmaa, D.; Bergman, P.; Bischoff-Ferrari, H.A.; Borzutzky, A.; Damsgaard, C.T.; et al. Vitamin D Supplementation to Prevent Acute Respiratory Infections: A Systematic Review and Meta-Analysis of Aggregate Data from Randomised Controlled Trials. Lancet Diabetes Endocrinol. 2021, 9, 276–292. [Google Scholar] [CrossRef]
- Skrobot, A.; Demkow, U.; Wachowska, M. Immunomodulatory Role of Vitamin D: A Review. In Current Trends in Immunity and Respiratory Infections; Advances in Experimental Medicine and Biology; Pokorski, M., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 13–23. ISBN 978-3-030-01635-7. [Google Scholar]
- Cantorna, M.T.; Snyder, L.; Lin, Y.-D.; Yang, L. Vitamin D and 1,25(OH)2D Regulation of T Cells. Nutrients 2015, 7, 3011–3021. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Todosenko, N.; Vulf, M.; Yurova, K.; Khaziakhmatova, O.; Mikhailova, L.; Litvinova, L. Causal Links between Hypovitaminosis D and Dysregulation of the T Cell Connection of Immunity Associated with Obesity and Concomitant Pathologies. Biomedicines 2021, 9, 1750. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9121750
Todosenko N, Vulf M, Yurova K, Khaziakhmatova O, Mikhailova L, Litvinova L. Causal Links between Hypovitaminosis D and Dysregulation of the T Cell Connection of Immunity Associated with Obesity and Concomitant Pathologies. Biomedicines. 2021; 9(12):1750. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9121750
Chicago/Turabian StyleTodosenko, Natalia, Maria Vulf, Kristina Yurova, Olga Khaziakhmatova, Larisa Mikhailova, and Larisa Litvinova. 2021. "Causal Links between Hypovitaminosis D and Dysregulation of the T Cell Connection of Immunity Associated with Obesity and Concomitant Pathologies" Biomedicines 9, no. 12: 1750. https://0-doi-org.brum.beds.ac.uk/10.3390/biomedicines9121750