Potentiometric Biosensing of Ascorbic Acid, Uric Acid, and Cysteine in Microliter Volumes Using Miniaturized Nanoporous Gold Electrodes


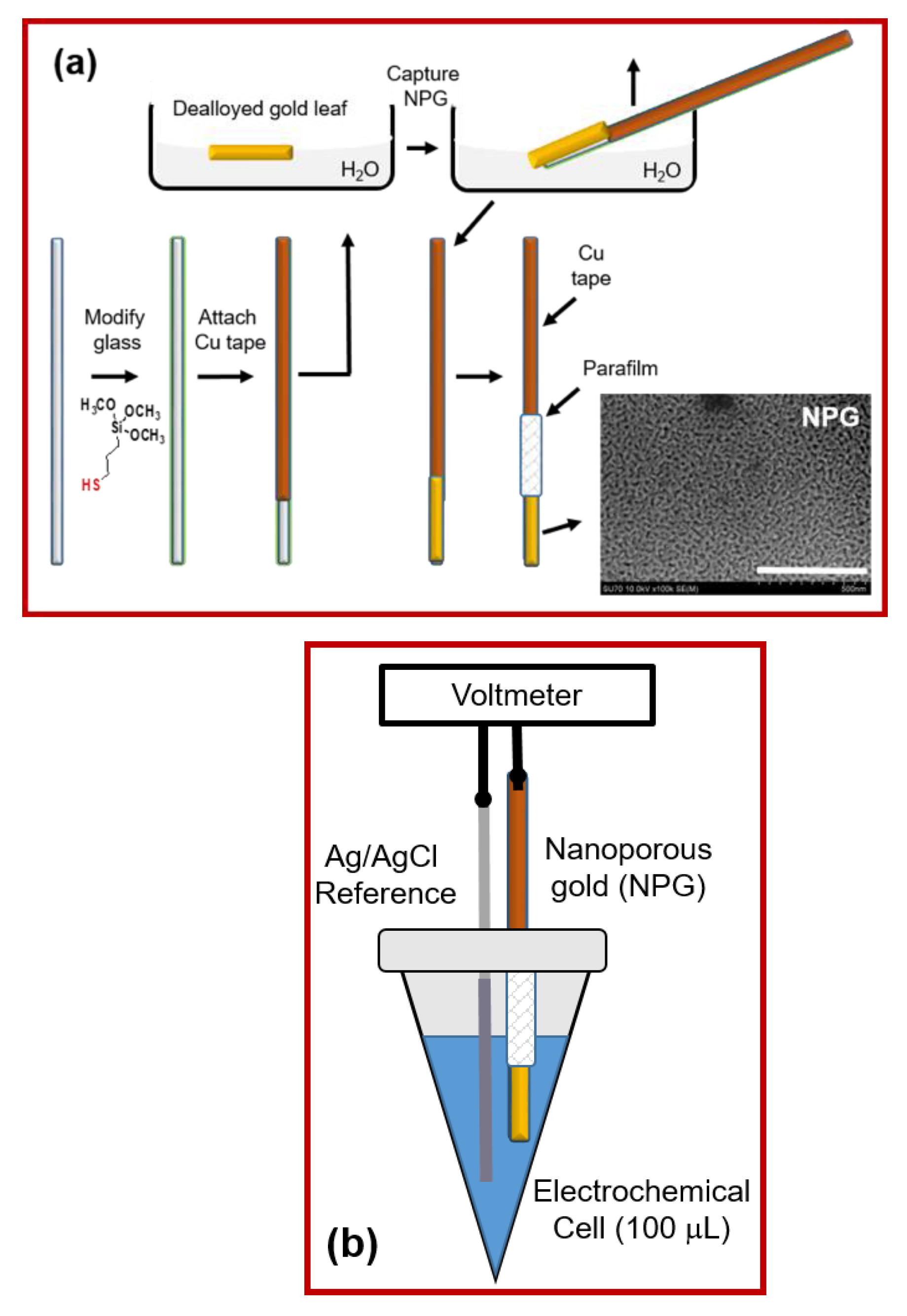{kind=link}
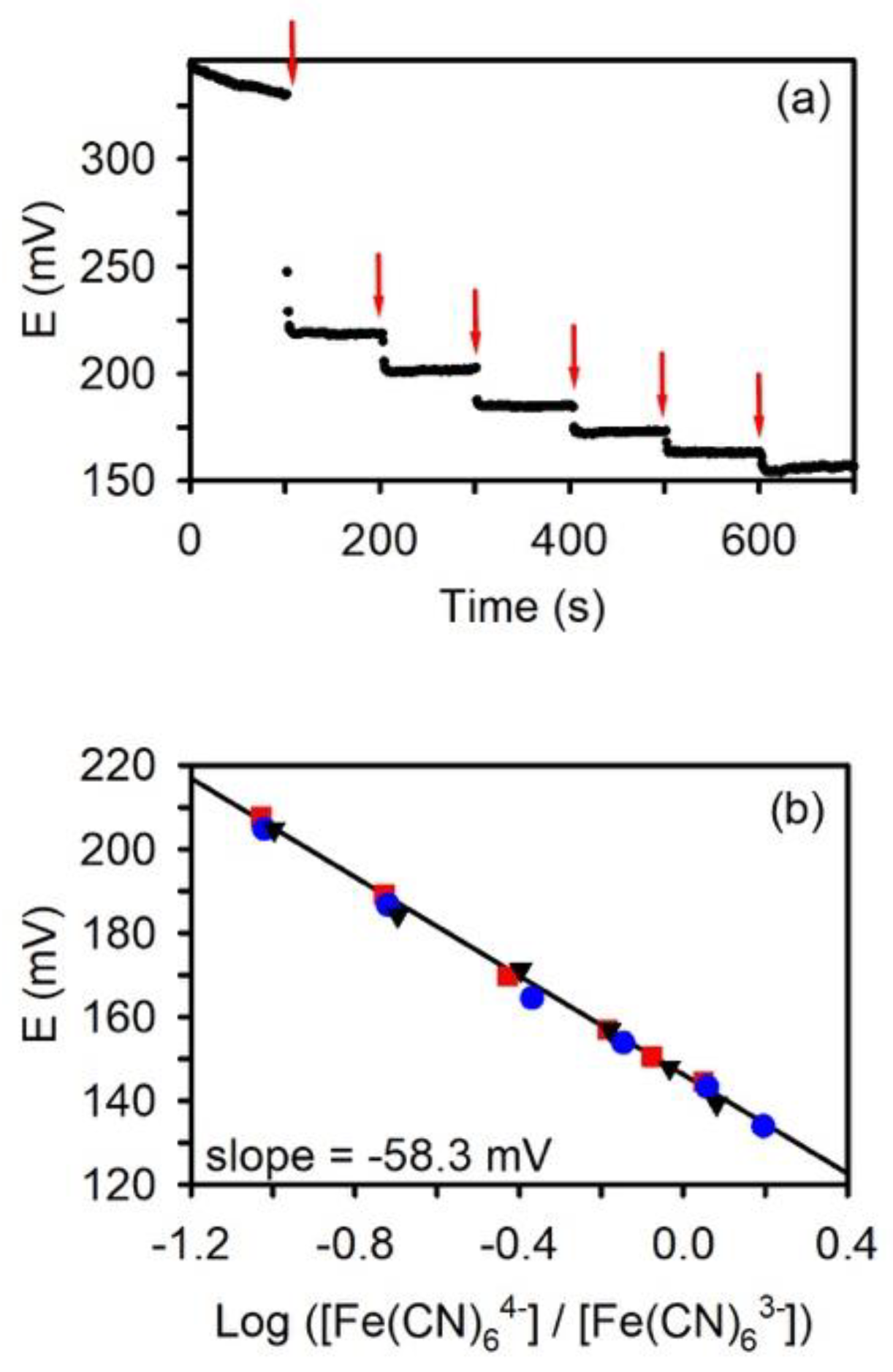{kind=link}
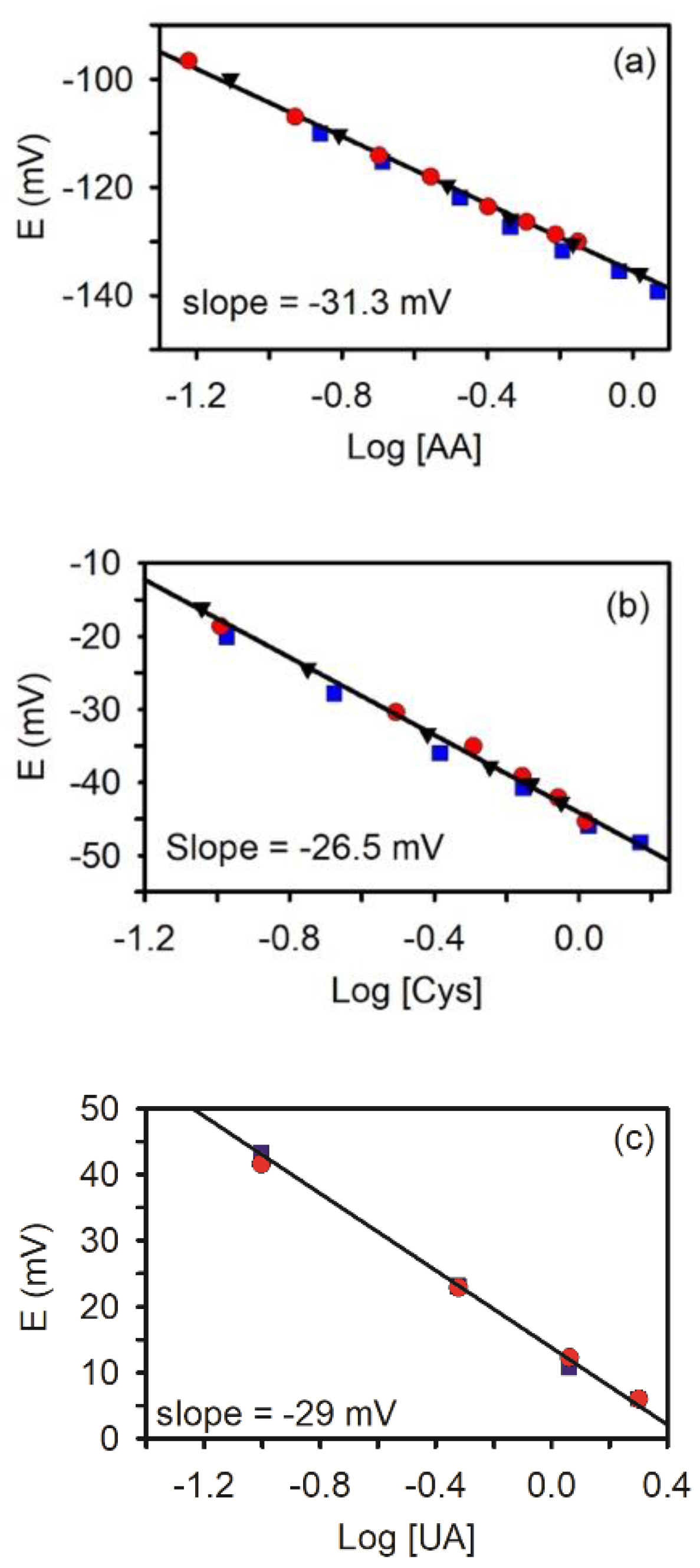{kind=link}
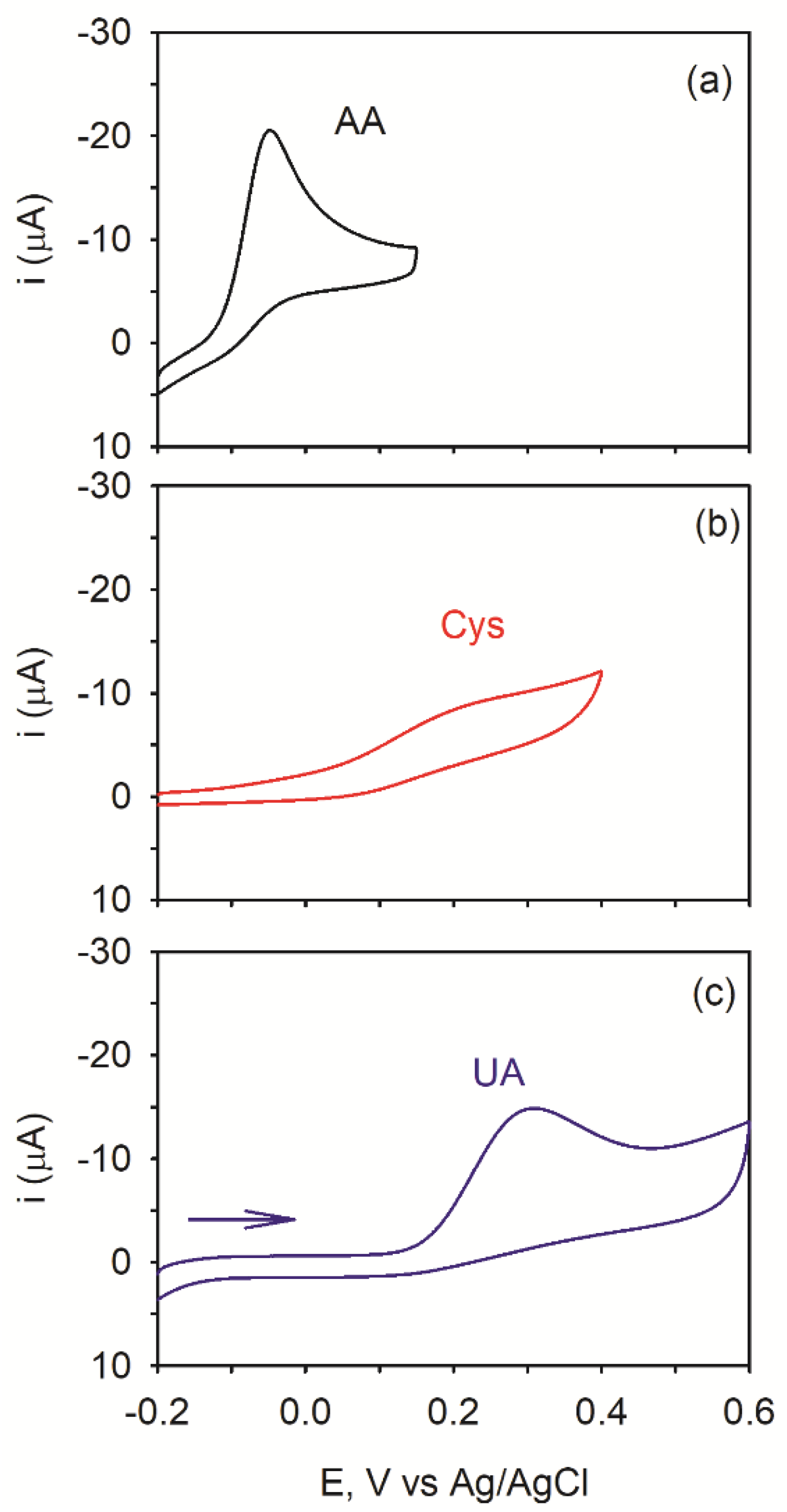{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Electrode Fabrication
3.2. Potentiometric Response
3.3. Potentiometric Response in a Mixed Solution
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bakker, E. Potentiometric Sensors. In Environmental Analysis by Electrochemical Sensors and Biosensors; Nanostructure Science and Technology; Moretto, L., Kalcher, K., Eds.; Springer: New York, NY, USA, 2014. [Google Scholar] [CrossRef]
- Lindner, E.; Pendley, B.D. A tutorial on the application of ion-selective electrode potentiometry: An analytical method with unique qualities, unexplored opportunities and potential pitfalls; Tutorial. Anal. Chim. Acta 2013, 762, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bakker, E.; Pretsch, E. Modern Potentiometry. Angew. Chem. Int. Ed. 2007, 46, 5660–5668. [Google Scholar] [CrossRef] [PubMed]
- Düzgün, A.; Zelada-Guillén, G.A.; Crespo, G.A.; Macho, S.; Riu, J.; Rius, F.X. Nanostructured materials in potentiometry. Anal. Bioanal. Chem. 2011, 399, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Qin, W. Recent advances in potentiometric biosensors. TrAC Trends Anal. Chem. 2020, 124, 115803. [Google Scholar] [CrossRef]
- Zdrachek, E.; Bakker, E. Potentiometric sensing. Anal. Chem. 2018, 91, 2–26. [Google Scholar] [CrossRef] [Green Version]
- Teasdale, P.R.; Minett, A.I.; Dixon, K.; Lewis, T.W.; Batley, G.E. Practical improvements for redox potential (E-H) measurements and the application of a multiple-electrode redox probe (MERP) for characterizing sediment in situ. Anal. Chim. Acta 1998, 367, 201–213. [Google Scholar] [CrossRef]
- Carlsson, T.; Muurinen, A. Practical and Theoretical Basis for Performing Redox-Measurements in Compacted Bentonite: A Literature Survey (POSIVA-WR--08-51); Posiva Oy: Eurajoki, Finland, 2008. [Google Scholar]
- Peiffer, S.; Klemm, O.; Pecher, K.; Hollerung, R. Redox Measurements in Aqueous-Solutions- A Theoretical Approach to Data Interpretation, based on Electrode-Kinetics. J. Contam. Hydrol. 1992, 10, 1–18. [Google Scholar] [CrossRef]
- Spiro, M. Polyelectrodes: The behaviour and applications of mixed redox systems. Chem. Soc. Rev. 1986, 15, 141–165. [Google Scholar] [CrossRef]
- Kodera, T.; Kita, H.; Honda, M. Kinetic analysis of the mixed potential. Electrochim. Acta 1972, 17, 1361–1376. [Google Scholar] [CrossRef]
- Herbelin, J.; Andersen, T.; Eyring, H. Kinetic parameters by the method of mixed potentials. Electrochim. Acta 1970, 15, 1455–1469. [Google Scholar] [CrossRef]
- Fiedler, S.; Vepraskas, M.J.; Richardson, J.L. Soil redox potential: Importance, field measurements, and observations. In Advances in Agronomy; Sparks, D.L., Ed.; Academic Press: Cambridge, MA, USA, 2007; Volume 94, pp. 1–54. [Google Scholar]
- Goncharuk, V.V.; Bagrii, V.A.; Mel’nik, L.A.; Chebotareva, R.D.; Bashtan, S.Y. The use of redox potential in water treatment processes. J. Water Chem. Technol. 2010, 32, 1–9. [Google Scholar] [CrossRef]
- Jang, A.; Lee, J.H.; Bhadri, P.R.; Kumar, S.A.; Timmons, W.; Beyette, F.R.; Papautsky, I.; Bishop, P.L. Miniaturized redox potential probe for in situ environmental monitoring. Environ. Sci. Technol. 2005, 39, 6191–6197. [Google Scholar] [CrossRef] [PubMed]
- Matia, L.; Rauret, G.; Rubio, R. Redox Potential Measurement in Natural-Waters. Fresenius J. Anal. Chem. 1991, 339, 455–462. [Google Scholar] [CrossRef]
- Rabenhorst, M.; Hively, W.; James, B. Measurements of soil redox potential. Soil Sci. Soc. Am. J. 2009, 73, 668–674. [Google Scholar] [CrossRef]
- Lee, J.H.; Jang, A.; Bhadri, P.R.; Myers, R.R.; Timmons, W.; Beyette, F.R.; Bishop, P.L.; Papautsky, I. Fabrication of microelectrode arrays for in situ sensing of oxidation reduction potentials. Sens. Actuators B-Chem. 2006, 115, 220–226. [Google Scholar] [CrossRef]
- Daniels, R.C.; Jun, H.; Tiba, M.H.; McCracken, B.; Herrera-Fierro, P.; Collinson, M.; Ward, K.R. Whole Blood Redox Potential Correlates with Progressive Accumulation of Oxygen Debt and Acts as a Marker of Resuscitation in a Swine Hemorrhagic Shock Model. Shock 2017, 49, 345–351. [Google Scholar] [CrossRef]
- Khan, R.K.; Gadiraju, S.P.; Kumar, M.; Hatmaker, G.A.; Fisher, B.J.; Natarajan, R.; Reiner, J.E.; Collinson, M.M. Redox Potential Measurements in Red Blood Cell Packets Using Nanoporous Gold Electrodes. ACS Sens. 2018, 3, 1601–1608. [Google Scholar] [CrossRef]
- Rao, S.K.; Palazzo, R.S.; Metz, H.N.; Wilson, D.W.; Nikolic, S.D.; Graver, M.; Rao, P.S. Redox potential measurements of plasma in patients undergoing coronary artery bypass graft and its clinical significance. J. Pharmacol. Toxicol. Methods 1997, 38, 151–156. [Google Scholar] [CrossRef]
- Khubutiya, M.S.; Goldin, M.M.; Romasenko, M.V.; Volkov, A.G.; Hall, P.J.; Evseev, A.K.; Levina, O.A.; Aleschenko, E.I.; Krylov, V.V. Redox Potentials of Blood Serum in Patients with Acute Cerebral Pathology. ECS Trans. 2010, 25, 63–71. [Google Scholar] [CrossRef]
- Noyhouzer, T.; Kohen, R.; Mandler, D. A new approach for measuring the redox state and redox capacity in milk. Anal. Methods 2009, 1, 93–99. [Google Scholar] [CrossRef]
- Chen, P.; Chang, F.; Chen, I.; Lu, F.; Cheng, T.; Chen, R. Redox potential of tea infusion as an index for the degree of fermentation. Anal. Chim. Acta 2007, 594, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Gil, L.; Barat, J.M.; Escriche, I.; Garcia-Breijo, E.; Martínez-Máñez, R.; Soto, J. An electronic tongue for fish freshness analysis using a thick-film array of electrodes. Microchim. Acta 2008, 163, 121–129. [Google Scholar] [CrossRef]
- Gil, L.; Barat, J.M.; Garcia-Breijo, E.; Ibañez, J.; Martínez-Máñez, R.; Soto, J.; Llobet, E.; Brezmes, J.; Aristoy, M.-C.; Toldrá, F. Fish freshness analysis using metallic potentiometric electrodes. Sens. Actuators B Chem. 2008, 131, 362–370. [Google Scholar] [CrossRef]
- Caldeo, V.; McSweeney, P. Changes in oxidation-reduction potential during the simulated manufacture of different cheese varieties. Int. Dairy J. 2012, 25, 16–20. [Google Scholar] [CrossRef]
- Baez, J.F.; Compton, M.; Chahrati, S.; Cánovas, R.; Blondeau, P.; Andrade, F.J. Controlling the mixed potential of polyelectrolyte-coated platinum electrodes for the potentiometric detection of hydrogen peroxide. Anal. Chim. Acta 2020, 1097, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Parrilla, M.; Cánovas, R.; Andrade, F.J. Enhanced potentiometric detection of hydrogen peroxide using a platinum electrode coated with nafion. Electroanalysis 2017, 29, 223–230. [Google Scholar] [CrossRef]
- Meruva, R.K.; Meyerhoff, M.E. Potentiometric oxygen sensing with copper films: Response mechanism and analytical implications. Electroanalysis 1995, 7, 1020–1026. [Google Scholar] [CrossRef]
- Hahn, P.; Johnson, D.; Wechter, M.; Voigt, A. Mixed-potential mechanism for the potentiometric response of the sodium tungsten bronze electrode to dissolved oxygen and in chelometric titrations. Anal. Chem. 1974, 46, 553–558. [Google Scholar] [CrossRef]
- Zeitoun, R.; Biswas, A. Potentiometric Determination of Phosphate Using Cobalt: A Review. J. Electrochem. Soc. 2020, 167, 127507. [Google Scholar] [CrossRef]
- Wei, H.; Li, L.; Jin, J.; Wu, F.; Yu, P.; Ma, F.; Mao, L. Galvanic Redox Potentiometry Based Microelectrode Array for Synchronous Ascorbate and Single-Unit Recordings in Rat Brain. Anal. Chem. 2020, 92, 10177–10182. [Google Scholar] [CrossRef]
- Wu, F.; Cheng, H.; Wei, H.; Xiong, T.; Yu, P.; Mao, L. Galvanic Redox Potentiometry for Self-Driven in Vivo Measurement of Neurochemical Dynamics at Open-Circuit Potential. Anal. Chem. 2018, 90, 13021–13029. [Google Scholar] [CrossRef] [PubMed]
- Nishitani, S.; Sakata, T. Enhancement of Signal-to-Noise Ratio for Serotonin Detection with Well-Designed Nanofilter-Coated Potentiometric Electrochemical Biosensor. ACS Appl. Mater. Interfaces 2020, 12, 14761–14769. [Google Scholar] [CrossRef] [PubMed]
- Nishitani, S.; Sakata, T. Polymeric nanofilter biointerface for potentiometric small-biomolecule recognition. ACS Appl. Mater. Interfaces 2019, 11, 5561–5569. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Zhou, H.; Percival, S.J.; Zhang, B.; Fan, F.-R.F.; Bard, A.J. Open circuit (mixed) potential changes upon contact between different inert electrodes–size and kinetic effects. Anal. Chem. 2013, 85, 964–970. [Google Scholar] [CrossRef] [PubMed]
- Percival, S.J.; Bard, A.J. Ultra-Sensitive Potentiometric Measurements of Dilute Redox Molecule Solutions and Determination of Sensitivity Factors at Platinum Ultramicroelectrodes. Anal. Chem. 2017, 89, 9843–9849. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Park, J.H.; Fan, F.-R.F.; Bard, A.J. Observation of single metal nanoparticle collisions by open circuit (mixed) potential changes at an ultramicroelectrode. J. Am. Chem. Soc. 2012, 134, 13212–13215. [Google Scholar] [CrossRef]
- Dasari, R.; Tai, K.; Robinson, D.A.; Stevenson, K.J. Electrochemical monitoring of single nanoparticle collisions at mercury-modified platinum ultramicroelectrodes. ACS Nano 2014, 8, 4539–4546. [Google Scholar] [CrossRef]
- Figueiredo, P.G.; Grob, L.; Rinklin, P.; Krause, K.J.; Wolfrum, B. On-chip stochastic detection of silver nanoparticles without a reference electrode. ACS Sens. 2018, 3, 93–98. [Google Scholar] [CrossRef] [Green Version]
- Trojánek, A.; Mareček, V.; Samec, Z. Open circuit potential transients associated with single emulsion droplet collisions at an interface between two immiscible electrolyte solutions. Electrochem. Commun. 2018, 86, 113–116. [Google Scholar] [CrossRef]
- Trojánek, A.; Samec, Z. Study of the emulsion droplet collisions with the polarizable water/1, 2-dichloroethane interface by the open circuit potential measurements. Electrochim. Acta 2019, 299, 875–885. [Google Scholar] [CrossRef]
- Smith, L.A.; Glasscott, M.W.; Vannoy, K.J.; Dick, J.E. Enzyme Kinetics via Open Circuit Potentiometry. Anal. Chem. 2019, 92, 2266–2273. [Google Scholar] [CrossRef] [PubMed]
- Farcas, M.; Cosman, N.P.; Ting, D.K.; Roscoe, S.G.; Omanovic, S. A comparative study of electrochemical techniques in investigating the adsorption behaviour of fibrinogen on platinum. J. Electroanal. Chem. 2010, 649, 206–218. [Google Scholar] [CrossRef]
- Roscoe, S.G.; Fuller, K.L. Interfacial Behavior of Globular-Proteins at A Platinum-Electrode. J. Colloid Interface Sci. 1992, 152, 429–441. [Google Scholar] [CrossRef]
- Farghaly, A.A.; Lam, M.; Freeman, C.J.; Uppalapati, B.; Collinson, M.M. Potentiometric measurements in biofouling solutions: Comparison of nanoporous gold to planar gold. J. Electrochem. Soc. 2015, 163, H3083. [Google Scholar] [CrossRef] [Green Version]
- Bae, J.H.; Han, J.-H.; Chung, T.D. Electrochemistry at nanoporous interfaces: New opportunity for electrocatalysis. Phys. Chem. Chem. Phys. 2012, 14, 448–463. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Kim, H.C.; Chung, T.D. Electrochemical analysis based on nanoporous structures. Analyst 2012, 137, 3891–3903. [Google Scholar] [CrossRef]
- Seo, M.; Chung, T.D. Nanoconfinement effects in electrochemical reactions. Curr. Opin. Electrochem. 2019, 13, 47–54. [Google Scholar] [CrossRef]
- Patel, J.; Radhakrishnan, L.; Zhao, B.; Uppalapati, B.; Daniels, R.C.; Ward, K.R.; Collinson, M.M. Electrochemical properties of nanostructured porous gold electrodes in biofouling solutions. Anal. Chem. 2013, 85, 11610–11618. [Google Scholar] [CrossRef]
- Khan, R.K.; Yadavalli, V.K.; Collinson, M.M. Flexible Nanoporous Gold Electrodes for Electroanalysis in Complex Matrices. ChemElectroChem 2019, 6, 4660–4665. [Google Scholar] [CrossRef]
- Haensch, M.; Balboa, L.; Graf, M.; Silva Olaya, A.R.; Weissmüller, J.; Wittstock, G. Mass Transport in Porous Electrodes Studied by Scanning Electrochemical Microscopy: Example of Nanoporous Gold. ChemElectroChem 2019, 6, 3160–3166. [Google Scholar] [CrossRef]
- Collinson, M.M. Nanoporous gold electrodes and their applications in analytical chemistry. ISRN Anal. Chem. 2013, 2013, 21. [Google Scholar] [CrossRef]
- Kim, S.H. Nanoporous gold: Preparation and applications to catalysis and sensors. Curr. Appl. Phys. 2018, 18, 810–818. [Google Scholar] [CrossRef]
- Bhattarai, J.K.; Neupane, D.; Nepal, B.; Mikhaylov, V.; Demchenko, A.V.; Stine, K.J. Preparation, modification, characterization, and biosensing application of nanoporous gold using electrochemical techniques. Nanomaterials 2018, 8, 171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Meek, R. Metal nanoparticles-grafted functionalized graphene coated with nanostructured polyaniline ‘hybrid’ nanocomposites as high-performance biosensors. Sens. Actuators B Chem. 2018, 274, 85–101. [Google Scholar] [CrossRef]
- Kumar, A.; Furtado, V.L.; Gonçalves, J.M.; Bannitz-Fernandes, R.; Netto, L.E.S.; Araki, K.; Bertotti, M. Amperometric microsensor based on nanoporous gold for ascorbic acid detection in highly acidic biological extracts. Anal. Chim. Acta 2020, 1095, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Bai, J.; Wang, L.; XuHong, E.; Huang, P.; Wang, H.; Zhang, L. Simultaneous electrochemical determination of uric acid and ascorbic acid using L-cysteine self-assembled gold electrode. Int. J. Electrochem. Sci. 2006, 1, 363–371. [Google Scholar]
- Sun, C.-L.; Lee, H.-H.; Yang, J.-M.; Wu, C.-C. The simultaneous electrochemical detection of ascorbic acid, dopamine, and uric acid using graphene/size-selected Pt nanocomposites. Biosens. Bioelectron. 2011, 26, 3450–3455. [Google Scholar] [CrossRef]
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef]
- Shi, L.; Rong, X.; Wang, Y.; Ding, S.; Tang, W. High-performance and versatile electrochemical aptasensor based on self-supported nanoporous gold microelectrode and enzyme-induced signal amplification. Biosens. Bioelectron. 2018, 102, 41–48. [Google Scholar] [CrossRef]
- Xiao, C.; Liu, Y.-L.; Xu, J.-Q.; Lv, S.-W.; Guo, S.; Huang, W.-H. Real-time monitoring of H2O2 release from single cells using nanoporous gold microelectrodes decorated with platinum nanoparticles. Analyst 2015, 140, 3753–3758. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo, D.X.O.; Sukeri, A.; Saravia, L.P.; Espinoza-Montero, P.J.; Bertotti, M. Nanoporous gold microelectrode: A novel sensing platform for highly sensitive and selective determination of arsenic (III) using anodic stripping voltammetry. Electroanalysis 2017, 29, 2316–2322. [Google Scholar] [CrossRef]
- Freeman, C.J.; Farghaly, A.A.; Choudhary, H.; Chavis, A.E.; Brady, K.T.; Reiner, J.E.; Collinson, M.M. Microdroplet-Based Potentiometric Redox Measurements on Gold Nanoporous Electrodes. Anal. Chem. 2016, 88, 3768–3774. [Google Scholar] [CrossRef] [PubMed]
- Wittstock, A.; Neumann, B.r.; Schaefer, A.; Dumbuya, K.; Kübel, C.; Biener, M.M.; Zielasek, V.; Steinrück, H.-P.; Gottfried, J.M.; Biener, J.r. Nanoporous Au: An unsupported pure gold catalyst? J. Phys. Chem. C 2009, 113, 5593–5600. [Google Scholar] [CrossRef]
- Kuhlmann, J.; Dzugan, L.C.; Heineman, W.R. Comparison of the effects of biofouling on voltammetric and potentiometric measurements. Electroanalysis 2012, 24, 1732–1738. [Google Scholar] [CrossRef]
- Arrigoni, O.; De Tullio, M.C. Ascorbic acid: Much more than just an antioxidant. Biochim. Biophys. Acta (BBA) Gen. Subj. 2002, 1569, 1–9. [Google Scholar] [CrossRef]
- Lakshmi, D.; Whitcombe, M.J.; Davis, F.; Sharma, P.S.; Prasad, B.B. Electrochemical detection of uric acid in mixed and clinical samples: A review. Electroanalysis 2011, 23, 305–320. [Google Scholar] [CrossRef]
- Clemente Plaza, N.; Reig García-Galbis, M.; Martínez-Espinosa, R.M. Effects of the Usage of l-Cysteine (l-Cys) on Human Health. Molecules 2018, 23, 575. [Google Scholar] [CrossRef] [Green Version]
- Noyhouzer, T.; Valdinger, I.; Mandler, D. Enhanced Potentiometry by Metallic Nanoparticles. Anal. Chem. 2013, 85, 8347–8353. [Google Scholar] [CrossRef]
- Porter, R.S.; Kaplan, J.L.; Lynn, R.B.; Reddy, M.T. The Merck Manual of Diagnosis and Therapy; Merck Sharp & Dohme Corp.: Kenilworth, NJ, USA, 2018. [Google Scholar]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications, 2nd ed.; John Wiley & Sons: New York, NY, USA, 2000. [Google Scholar]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Freeman, C.J.; Ullah, B.; Islam, M.S.; Collinson, M.M. Potentiometric Biosensing of Ascorbic Acid, Uric Acid, and Cysteine in Microliter Volumes Using Miniaturized Nanoporous Gold Electrodes. Biosensors 2021, 11, 10. https://0-doi-org.brum.beds.ac.uk/10.3390/bios11010010
Freeman CJ, Ullah B, Islam MS, Collinson MM. Potentiometric Biosensing of Ascorbic Acid, Uric Acid, and Cysteine in Microliter Volumes Using Miniaturized Nanoporous Gold Electrodes. Biosensors. 2021; 11(1):10. https://0-doi-org.brum.beds.ac.uk/10.3390/bios11010010
Chicago/Turabian StyleFreeman, Christopher J., Borkat Ullah, Md. Shafiul Islam, and Maryanne M. Collinson. 2021. "Potentiometric Biosensing of Ascorbic Acid, Uric Acid, and Cysteine in Microliter Volumes Using Miniaturized Nanoporous Gold Electrodes" Biosensors 11, no. 1: 10. https://0-doi-org.brum.beds.ac.uk/10.3390/bios11010010