
Potential of Surface Enhanced Raman Spectroscopy (SERS) in Therapeutic Drug Monitoring (TDM). A Critical Review


Abstract
:1. Introduction
1.1. Therapeutic Drug Monitoring (TDM)
1.2. Surface Enhanced Raman Spectroscopy (SERS)
1.3. Critical Issues on the Use of SERS for TDM
1.4. Quantification with SERS: The Role of Data Analysis
2. Applications of SERS Relevant to TDM
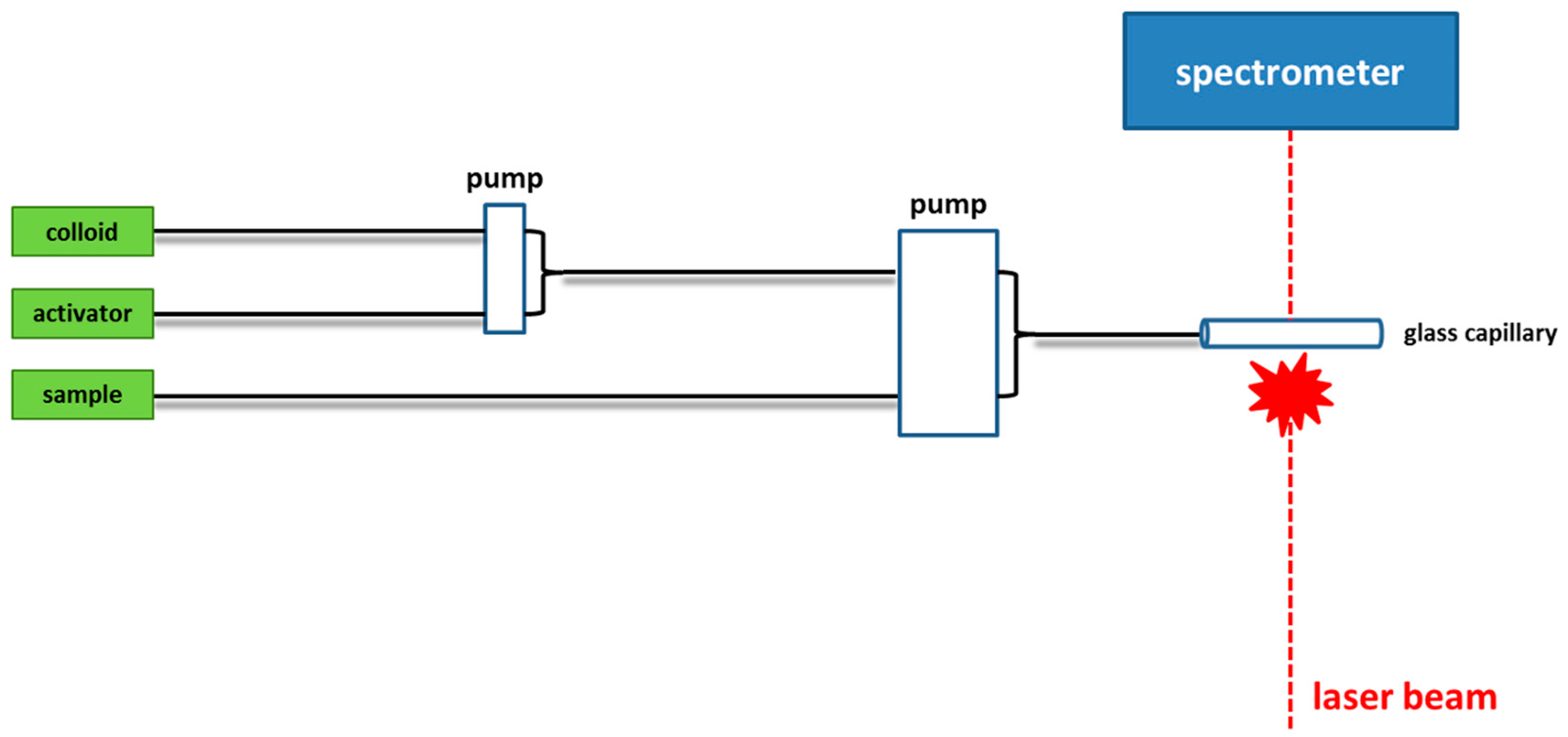
2.1. Colloidal Substrates
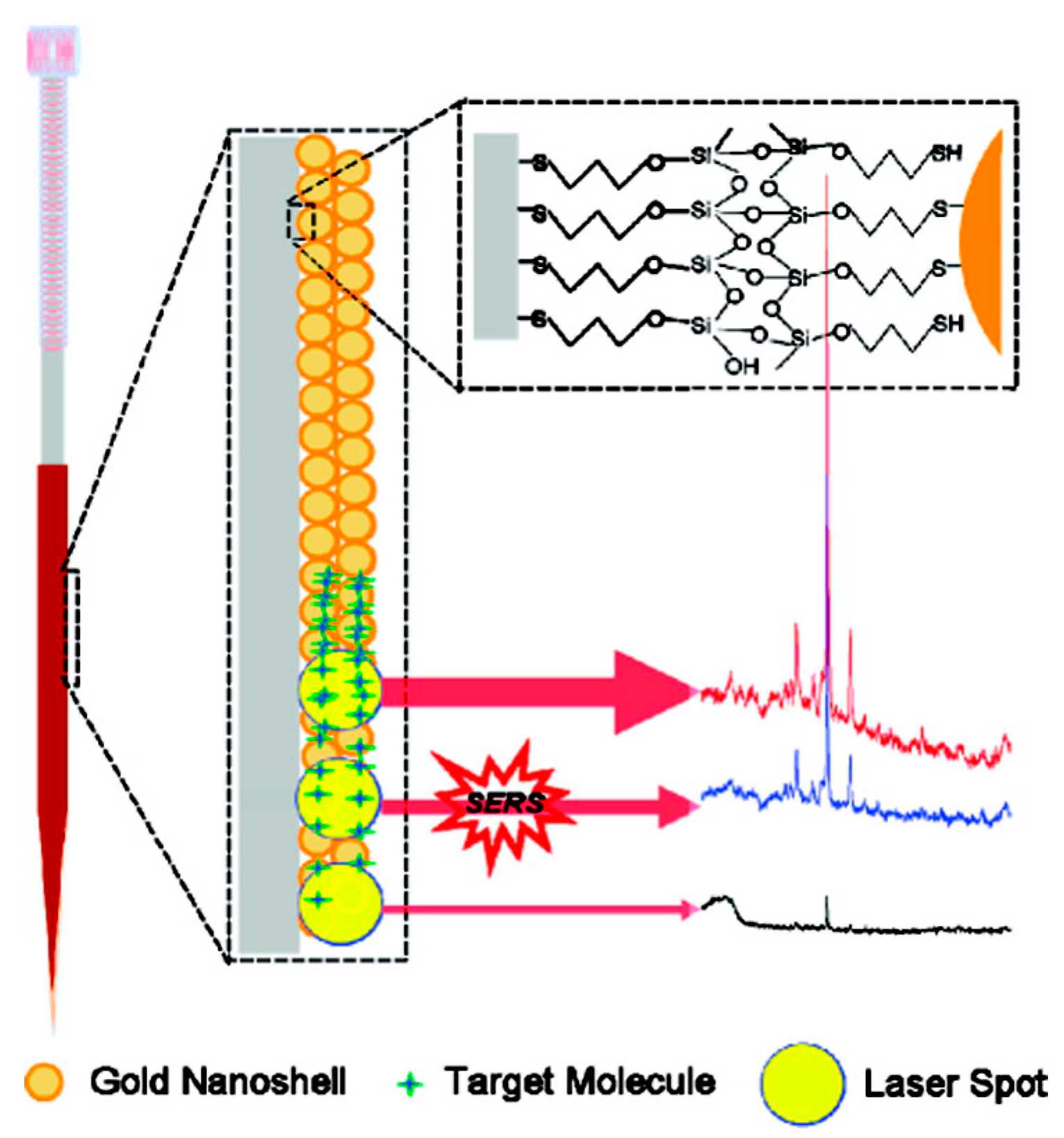
2.2. Non-Colloidal Substrates
2.3. Hybrid Substrates
3. Conclusions and Perspectives
Acknowledgments
Conflicts of Interest
References
- Dasgupta, A. Therapeutic Drug Monitoring: Newer Drugs and Biomarkers; Academic Press: Cambridge, MA, USA, 2012. [Google Scholar]
- Salvati, E.; Stellacci, F.; Krol, S. Nanosensors for early cancer detection and for therapeutic drug monitoring. Nanomedicine 2015, 10, 3495–3512. [Google Scholar] [CrossRef] [PubMed]
- Alnaim, L. Therapeutic drug monitoring of cancer chemotherapy. J. Oncol Pharm. Pract. 2007, 13, 207–221. [Google Scholar] [CrossRef] [PubMed]
- McMahon, G.; O’Connor, R. Therapeutic drug monitoring in oncology: Does it have a future? Bioanalysis 2009, 1, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Bach, D.M.; Straseski, J.A.; Clarke, W. Therapeutic drug monitoring in cancer chemotherapy. Bioanalysis 2010, 2, 863–879. [Google Scholar] [CrossRef] [PubMed]
- Sanavio, B.; Krol, S. On the Slow Diffusion of Point-of-Care Systems in Therapeutic Drug Monitoring. Front. Bioeng. Biotechnol. 2015, 3, 20. [Google Scholar] [CrossRef] [PubMed]
- Ensom, M.H.; Davis, G.A.; Cropp, C.D.; Ensom, R.J. Clinical pharmacokinetics in the 21st century. Does the evidence support definitive outcomes? Clin. Pharmacokinet. 1998, 34, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.S.; Lee, M.H. Overview of therapeutic drug monitoring. Korean J. Intern. Med. 2009, 24, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.Y.; Milton, M.N. The determination and interpretation of the therapeutic index in drug development. Nat. Rev. Drug Discov. 2012, 11, 751–761. [Google Scholar] [CrossRef] [PubMed]
- Neef, C.; Touw, D.J.; Stolk, L.M. Therapeutic drug monitoring in clinical research. Pharm. Med. 2008, 22, 235–244. [Google Scholar] [CrossRef]
- Dasgupta, A. Advances in Chromatographic Techniques for Therapeutic Drug Monitoring; CRC Press: Boca Raton, FL, USA, 2009. [Google Scholar]
- Xu, Q.A.; Madden, T.L. Analytical Methods for Therapeutic Drug Monitoring and Toxicology; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- Zhao, S.S.; Bukar, N.; Toulouse, J.L.; Pelechacz, D.; Robitaille, R.; Pelletier, J.N.; Masson, J.-F. Miniature multi-channel SPR instrument for methotrexate monitoring in clinical samples. Biosens. Bioelectron. 2015, 64, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.S.; Bichelberger, M.A.; Colin, D.Y.; Robitaille, R.; Pelletier, J.N.; Masson, J.F. Monitoring methotrexate in clinical samples from cancer patients during chemotherapy with a LSPR-based competitive sensor. Analyst 2012, 137, 4742–4750. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, B.S.; Hoggarth, D.A.; Maliniak, D.; Ploense, K.; White, R.J.; Woodward, N.; Hsieh, K.; Bonham, A.J.; Eisenstein, M.; Kippin, T.E.; et al. Real-time, aptamer-based tracking of circulating therapeutic agents in living animals. Sci. Transl. Med. 2013, 5, 213ra165. [Google Scholar] [CrossRef] [PubMed]
- Ogi, H. Wireless-electrodeless quartz-crystal-microbalance biosensors for studying interactions among biomolecules: A review. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2013, 89, 401–417. [Google Scholar] [CrossRef] [PubMed]
- Blaszykowski, C.; Sheikh, S.; Thompson, M. Surface chemistry to minimize fouling from blood-based fluids. Chem. Soc. Rev. 2012, 41, 5599–5612. [Google Scholar] [CrossRef] [PubMed]
- Ogi, H.; Fukunishi, Y.; Nagai, H.; Okamoto, K.; Hirao, M.; Nishiyama, M. Nonspecific-adsorption behavior of polyethylenglycol and bovine serum albumin studied by 55-MHz wireless–electrodeless quartz crystal microbalance. Biosens. Bioelectron. 2009, 24, 3148–3152. [Google Scholar] [CrossRef] [PubMed]
- Masson, J.-F.; Battaglia, T.M.; Cramer, J.; Beaudoin, S.; Sierks, M.; Booksh, K.S. Reduction of nonspecific protein binding on surface plasmon resonance biosensors. Anal. Bioanal. Chem. 2006, 386, 1951–1959. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.L.; McFarland, A.D.; Duyne, R.P.V. Surface-Enhanced Raman Spectroscopy. Anal. Chem. 2005, 77, 338 A–346 A. [Google Scholar] [CrossRef]
- Schlücker, S. SERS Microscopy: Nanoparticle Probes and Biomedical Applications. In Surface Enhanced Raman Spectroscopy; Schlücker, S., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010; pp. 263–283. [Google Scholar]
- Schlücker, S. Surface-enhanced Raman spectroscopy: Concepts and chemical applications. Angew. Chem. Int. Ed. Engl. 2014, 53, 4756–4795. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, M.; Hendra, P.J.; McQuillan, A.J. Raman spectra of pyridine adsorbed at a silver electrode. Chem. Phys. Lett. 1974, 26, 163–166. [Google Scholar] [CrossRef]
- Moskovits, M. Surface-enhanced spectroscopy. Rev. Mod. Phys. 1985, 57, 783–826. [Google Scholar] [CrossRef]
- Jeanmaire, D.L.; Van Duyne, R.P. Surface raman spectroelectrochemistry. J. Electroanal. Chem. Interfacial Electrochem. 1977, 84, 1–20. [Google Scholar] [CrossRef]
- Albrecht, M.G.; Creighton, J.A. Anomalously intense Raman spectra of pyridine at a silver electrode. J. Am. Chem. Soc. 1977, 99, 5215–5217. [Google Scholar] [CrossRef]
- Le Ru, E.C.; Blackie, E.; Meyer, M.; Etchegoin, P.G. Surface Enhanced Raman Scattering Enhancement Factors: A Comprehensive Study. J. Phys. Chem. C 2007, 111, 13794–13803. [Google Scholar] [CrossRef]
- Shiohara, A.; Wang, Y.; Liz-Marzán, L.M. Recent approaches toward creation of hot spots for SERS detection. J. Photochem. Photobiol. C Photochem. Rev. 2014, 21, 2–25. [Google Scholar] [CrossRef]
- Le Ru, E.C.; Etchegoin, P.G. A quick overview of surface-enhanced Raman spectroscopy. In Principles of Surface-Enhanced Raman Spectroscopy; Elsevier: Amsterdam, The Netherlands, 2009; pp. 1–27. [Google Scholar]
- Le Ru, E.C.; Etchegoin, P.G. Metallic colloids and other SERS substrates. In Principles of Surface-Enhanced Raman Spectroscopy; Elsevier: Amsterdam, The Netherlands, 2009; pp. 367–413. [Google Scholar]
- Kämmer, E.; Olschewski, K.; Bocklitz, T.; Rösch, P.; Weber, K.; Cialla, D.; Popp, J. A new calibration concept for a reproducible quantitative detection based on SERS measurements in a microfluidic device demonstrated on the model analyte adenine. Phys. Chem. Chem. Phys. 2014, 16, 9056–9063. [Google Scholar] [CrossRef] [PubMed]
- Schulte, J.P.; Grass, S.; Treuel, L. Adsorption of dicarboxylic acids onto nano-structured silver surfaces—Surface-enhanced raman scattering studies of ph-dependent adsorption geometries. J. Raman Spectrosc. 2013, 44, 247–254. [Google Scholar] [CrossRef]
- Fang, Y.; Seong, N.H.; Dlott, D.D. Measurement of the distribution of site enhancements in surface-enhanced raman scattering. Science 2008, 321, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.J.; Liu, W.H.; Liu, J.M.; Nie, Y.H.; Li, P.; He, C.J. Surface-enhanced raman scattering sensing of trace fenthion coupled with stable silver colloids and oh stretching band of water as an internal standard. J. Anal. Chem. 2016, 71, 891–895. [Google Scholar] [CrossRef]
- Shen, W.; Lin, X.; Jiang, C.; Li, C.; Lin, H.; Huang, J.; Wang, S.; Liu, G.; Yan, X.; Zhong, Q.; et al. Reliable quantitative sers analysis facilitated by core-shell nanoparticles with embedded internal standards. Angew. Chem. Int. Ed. 2015, 54, 7308–7312. [Google Scholar] [CrossRef] [PubMed]
- Kratz, A.; Ferraro, M.; Sluss, P.M.; Lewandrowski, K.B. Case records of the massachusetts general hospital. Weekly clinicopathological exercises. Laboratory reference values. N. Engl. J. Med. 2004, 351, 1548–1563. [Google Scholar] [PubMed]
- Psychogios, N.; Hau, D.D.; Peng, J.; Guo, A.C.; Mandal, R.; Bouatra, S.; Sinelnikov, I.; Krishnamurthy, R.; Eisner, R.; Gautam, B.; et al. The Human Serum Metabolome. PLoS ONE 2011, 6, e16957. [Google Scholar] [CrossRef] [PubMed]
- Bonifacio, A.; Cervo, S.; Sergo, V. Label-free surface-enhanced Raman spectroscopy of biofluids: Fundamental aspects and diagnostic applications. Anal. Bioanal. Chem. 2014, 407, 8265–8277. [Google Scholar] [CrossRef] [PubMed]
- Bantz, K.C.; Meyer, A.F.; Wittenberg, N.J.; Im, H.; Kurtulus, O.; Lee, S.H.; Lindquist, N.C.; Oh, S.H.; Haynes, C.L. Recent progress in SERS biosensing. Phys. Chem. Chem. Phys. 2011, 13, 11551–11567. [Google Scholar] [CrossRef] [PubMed]
- Vicario, A.; Sergo, V.; Toffoli, G.; Bonifacio, A. Surface-enhanced Raman spectroscopy of the anti-cancer drug irinotecan in presence of human serum albumin. Colloids Surf. B Biointerfaces 2015, 127, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Arráez Román, D.; Efremov, E.; Ariese, F.; Segura Carretero, A.; Gooijer, C. Interfacing capillary electrophoresis and surface-enhanced resonance Raman spectroscopy for the determination of dye compounds. Anal. Bioanal. Chem. 2005, 382, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Danielsson, B. Artificial receptors. Adv. Biochem. Eng. Biotechnol. 2008, 109, 97–122. [Google Scholar] [PubMed]
- Beljebbar, A.; Sockalingum, G.D.; Angiboust, J.F.; Manfait, M. Comparative FT SERS, resonance Raman and SERRS studies of doxorubicin and its complex with DNA. Spectrochim. Acta A Mol. Biomol. Spectrosc. 1995, 51, 2083–2090. [Google Scholar] [CrossRef]
- Yan, Q.; Priebe, W.; Chaires, J.B.; Czernuszewicz, R.S. Interaction of doxorubicin and its derivatives with DNA: Elucidation by resonance Raman and surface-enhanced resonance Raman spectroscopy. Biospectroscopy 1997, 3, 307–316. [Google Scholar] [CrossRef]
- McLaughlin, C.; MacMillan, D.; McCardle, C.; Smith, W.E. Quantitative analysis of mitoxantrone by surface-enhanced resonance Raman scattering. Anal. Chem. 2002, 74, 3160–3167. [Google Scholar] [CrossRef] [PubMed]
- West, M. Bayesian Factor Regression Models in the “Large p, Small n” Paradigm. In Bayesian Statistics; Oxford University Press: Oxford, UK, 2003; pp. 723–732. [Google Scholar]
- Olivieri, A.C. Practical guidelines for reporting results in single- and multi-component analytical calibration: A tutorial. Anal. Chim. Acta 2015, 868, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.V.; Haaland, D.M. Comparison of multivariate calibration methods for quantitative spectral analysis. Anal. Chem. 1990, 62, 1091–1099. [Google Scholar] [CrossRef]
- Martens, H.; Naes, T. Multivariate Calibration; John Wiley & Sons: Hoboken, NJ, USA, 1992. [Google Scholar]
- Varmuza, K.; Filzmoser, P. Introduction to Multivariate Statistical Analysis in Chemometrics; CRC Press: Boca Raton, FL, USA, 2009. [Google Scholar]
- Estienne, F.; Massart, D.L. Mutlivariate calibration with Raman data using fast principal component regression and partial least squares methods. Anal. Chim. Acta 2001, 450, 123–129. [Google Scholar] [CrossRef]
- Gryniewicz, C.M.; Kauffman, J.F. Multivariate Calibration of Covalent Aggregate Fraction to the Raman Spectrum of Regular Human Insulin. J. Pharm. Sci. 2008, 97, 3727–3734. [Google Scholar] [CrossRef] [PubMed]
- Wold, S.; Sjöström, M.; Eriksson, L. PLS-regression: A basic tool of chemometrics. Chemom. Intell. Lab. Syst. 2001, 58, 109–130. [Google Scholar] [CrossRef]
- Fornasaro, S.; Marta, S.D.; Rabusin, M.; Bonifacio, A.; Sergo, V. Toward SERS-based point-of-care approaches for therapeutic drug monitoring: The case of methotrexate. Faraday Discuss. 2016, 187, 485–499. [Google Scholar] [CrossRef] [PubMed]
- Hidi, I.J.; Jahn, M.; Pletz, M.W.; Weber, K.; Cialla-May, D.; Popp, J. Toward Levofloxacin Monitoring in Human Urine Samples by Employing the LoC-SERS Technique. J. Phys. Chem. C 2016. [Google Scholar] [CrossRef]
- Lasch, P. Spectral pre-processing for biomedical vibrational spectroscopy and microspectroscopic imaging. Chemom. Intell. Lab. Syst. 2012, 117, 100–114. [Google Scholar] [CrossRef]
- Bocklitz, T.; Walter, A.; Hartmann, K.; Rösch, P.; Popp, J. How to pre-process Raman spectra for reliable and stable models? Anal. Chim. Acta 2011, 704, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Valderrama, P.; Braga, J.W.B.; Poppi, R.J. Variable Selection, Outlier Detection, and Figures of Merit Estimation in a Partial Least-Squares Regression Multivariate Calibration Model. A Case Study for the Determination of Quality Parameters in the Alcohol Industry by Near-Infrared Spectroscopy. J. Agric. Food Chem. 2007, 55, 8331–8338. [Google Scholar] [CrossRef] [PubMed]
- López, M.I.; Ruisánchez, I.; Callao, M.P. Figures of merit of a SERS method for Sudan I determination at traces levels. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2013, 111, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Broadhurst, D.I.; Kell, D.B. Statistical strategies for avoiding false discoveries in metabolomics and related experiments. Metabolomics 2006, 2, 171–196. [Google Scholar] [CrossRef]
- Filzmoser, P.; Liebmann, B.; Varmuza, K. Repeated double cross validation. J. Chemom. 2009, 23, 160–171. [Google Scholar] [CrossRef]
- Krstajic, D.; Buturovic, L.J.; Leahy, D.E.; Thomas, S. Cross-validation pitfalls when selecting and assessing regression and classification models. J. Cheminform. 2014, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- 2002/657/EC: Commission Decision of 12 August 2002 Implementing Council Directive 96/23/EC Concerning the Performance of Analytical Methods and the Interpretation of Results (notified under document number C(2002) 3044) (1). Available online: http://eur-lex.europa.eu/legal-content/EN/TXT/?uri=OJ:L:2002:221:TOC (accessed on 3 July 2016).
- International Organization for Standardization, ISO 5725-1:2003, Accuracy (Trueness and Precision) of Measurement Methods and Results—Part 1: General Principles and Definitions. Available online: http://www.iso.org/iso/home.htm (accessed on 3 July 2016).
- Olivieri, A.C.; Faber, N.M.; Ferré, J.; Boqué, R.; Kalivas, J.H.; Mark, H. Uncertainty estimation and figures of merit for multivariate calibration (IUPAC Technical Report). Pure Appl. Chem. 2009, 78, 633–661. [Google Scholar] [CrossRef]
- Allegrini, F.; Olivieri, A.C. IUPAC-Consistent Approach to the Limit of Detection in Partial Least-Squares Calibration. Anal. Chem. 2014, 86, 7858–7866. [Google Scholar] [CrossRef] [PubMed]
- Farquharson, S.; Gift, A.D.; Shende, C.; Maksymiuk, P.; Inscore, F.E.; Murran, J. Detection of 5-fluorouracil in saliva using surface-enhanced Raman spectroscopy. Vib. Spectrosc. 2005, 38, 79–84. [Google Scholar] [CrossRef]
- Farquharson, S.; Gift, A.; Shende, C.; Inscore, F.; Ordway, B.; Farquharson, C.; Murren, J. Surface-enhanced Raman spectral measurements of 5-fluorouracil in saliva. Molecules 2008, 13, 2608–2627. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Chen, Q.; Rong, C.; Li, D.; Rao, Y. Minimally invasive surface-enhanced Raman scattering detection with depth profiles based on a surface-enhanced Raman scattering-active acupuncture needle. Anal. Chem. 2011, 83, 6191–6195. [Google Scholar] [CrossRef] [PubMed]
- Lorén, A.; Eliasson, C.; Josefson, M.; Murty, K.V.G.K.; Käll, M.; Abrahamsson, J.; Abrahamsson, K. Feasibility of quantitative determination of doxorubicin with surface-enhanced Raman spectroscopy. J. Raman Spectrosc. 2001, 32, 971–974. [Google Scholar] [CrossRef]
- Rath, S.; Sahu, A.; Gota, V.; Martínez-Torres, P.G.; Pichardo-Molina, J.L.; Murali, K. Raman spectroscopy for detection of imatinib in plasma: A proof of concept. J. Innov. Opt. Health Sci. 2015, 8, 1550019. [Google Scholar] [CrossRef]
- Yuen, C.; Zheng, W.; Huang, Z. Low-level detection of anti-cancer drug in blood plasma using microwave-treated gold-polystyrene beads as surface-enhanced Raman scattering substrates. Biosens. Bioelectron. 2010, 26, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Tan, X.; Shih, W.-C.; Cheng, M.M.-C. A sandwich substrate for ultrasensitive and label-free SERS spectroscopic detection of folic acid / methotrexate. Biomed. Microdevices 2014, 16, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Gu, H.; Yuan, X.; Wu, J.; Wei, H. Surface-enhanced Raman spectroscopy study of the interaction of antitumoral drug Paclitaxel with human serum albumin. Proc. SPIE 2008, 7280. [Google Scholar] [CrossRef]
- Yang, L.; Chen, Y.; Li, H.; Luo, L.; Zhao, Y.; Zhang, H.; Tian, Y. Application of silver nanoparticles decorated with β-cyclodextrin in determination of 6-mercaptopurine by surface-enhanced Raman spectroscopy. Anal. Methods 2015, 7, 6520–6527. [Google Scholar] [CrossRef]
- Li, Y.-T.; Qu, L.-L.; Li, D.-W.; Song, Q.-X.; Fathi, F.; Long, Y.-T. Rapid and sensitive in-situ detection of polar antibiotics in water using a disposable Ag–graphene sensor based on electrophoretic preconcentration and surface-enhanced Raman spectroscopy. Biosens. Bioelectron. 2013, 43, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-Y.; Cunningham, B.T. Point-of-care detection and real-time monitoring of intravenously delivered drugs via tubing with an integrated SERS sensor. Nanoscale 2014, 6, 5162–5171. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.L.; Zhen, S.J.; Huang, C.Z. Controllable preparation of graphene oxide/metal nanoparticle hybrids as surface-enhanced Raman scattering substrates for 6-mercaptopurine detection. RSC Adv. 2014, 4, 16327–16332. [Google Scholar] [CrossRef]
- Litti, L.; Amendola, V.; Toffoli, G.; Meneghetti, M. Detection of low-quantity anticancer drugs by surface-enhanced Raman scattering. Anal. Bioanal. Chem. 2016, 408, 2123–2131. [Google Scholar] [CrossRef] [PubMed]
- Hidi, I.J.; Mühlig, A.; Jahn, M.; Liebold, F.; Cialla, D.; Weber, K.; Popp, J. LOC-SERS: Towards point-of-care diagnostic of methotrexate. Anal. Methods 2014, 6, 3943–3947. [Google Scholar] [CrossRef]
- Cottat, M.; Lidgi-Guigui, N.; Hamouda, F.; Bartenlian, B.; Venkataraman, D.; Marks, R.S.; Steele, T.W.J.; de la Chapelle, M.L. Highly sensitive detection of paclitaxel by surface-enhanced Raman scattering. J. Opt. 2015, 17, 114019. [Google Scholar] [CrossRef]
- Ackermann, K.R.; Henkel, T.; Popp, J. Quantitative online detection of low-concentrated drugs via a SERS microfluidic system. ChemPhysChem 2007, 8, 2665–2670. [Google Scholar] [CrossRef] [PubMed]
- März, A.; Mönch, B.; Rösch, P.; Kiehntopf, M.; Henkel, T.; Popp, J. Detection of thiopurine methyltransferase activity in lysed red blood cells by means of lab-on-a-chip surface enhanced Raman spectroscopy (LOC-SERS). Anal. Bioanal. Chem. 2011, 400, 2755–2761. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Liu, R.; Han, M.Y.; Jiang, C.; Wang, J.; Du, S.; Liu, B.; Zhang, Z. Label-free surface-enhanced Raman scattering imaging to monitor the metabolism of antitumor drug 6-mercaptopurine in living cells. Anal. Chem. 2014, 86, 11503–11507. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Cui, Y.; Zong, S.; Zhang, R.; Song, C.; Wang, Z. Tracking Multiplex Drugs and Their Dynamics in Living Cells Using the Label-Free Surface-Enhanced Raman Scattering Technique. Mol. Pharm. 2012, 9, 842–849. [Google Scholar] [CrossRef] [PubMed]
- Prakash, J.; Harris, R.A.; Swart, H.C. Embedded plasmonic nanostructures: Synthesis, fundamental aspects and their surface enhanced Raman scattering applications. Int. Rev. Phys. Chem. 2016, 35, 353–398. [Google Scholar] [CrossRef]
- Gamelin, E.C.; Danquechin-Dorval, E.M.; Dumesnil, Y.F.; Maillart, P.J.; Goudier, M.J.; Burtin, P.C.; Delva, R.G.; Lortholary, A.H.; Gesta, P.H.; Larra, F.G. Relationship between 5-fluorouracil (5-FU) dose intensity and therapeutic response in patients with advanced colorectal cancer receiving infusional therapy containing 5-FU. Cancer 1996, 77, 441–451. [Google Scholar] [CrossRef]
- Sarkar, S.; Dutta, S.; Pal, T. Tailored “Sandwich” Strategy in Surface Enhanced Raman Scattering: Case Study with para-Phenylenediamine and Application in Femtomolar Detection of Melamine. J. Phys. Chem. C 2014, 118, 28152–28161. [Google Scholar] [CrossRef]
- Park, S.M.; Sabour, A.F.; Son, J.H.; Lee, S.H.; Lee, L.P. Toward Integrated Molecular Diagnostic System (iMDx): Principles and Applications. IEEE Trans. Biomed. Eng. 2014, 61, 1506–1521. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technique | Description | Pros | Cons |
|---|---|---|---|
| Chromatography-based methods | Chromatography separates individual compounds by their physical or chemical interaction with an immobile material. Once separated, combined selective MS techniques provide mass-based identification of the compounds | Gold standard; Robust methods with superior sensitivity and sensibility; Relatively free from interferences; Multiplexing capabilities; Reduced drug class/metabolites cross reactivity | Time consuming; Laboratory-developed tests; Interlaboratory variability; Matrix effects; High technical expertise required; High costs for installation, personnel training, and method validation; Need for sample clean-up |
| GC-MS/MS LC-MS/MS | |||
| Immunoassay platforms | Analyte is detected by its binding with a specific binding molecule, which in most cases is an analyte-specific antibody | Small amount of sample (<100 μL); Run on automated, continuous, random access systems; No need for sample clean-up; Multiplexing capabilities | Several steps to achieve quantification of the analyte; Reduced specificity and sensitivity; Often show significant bias; Antibody cross-reactivity; Interferences from bilirubin, hemoglobin, high lipid content, very high or very low protein content, endogenous antibodies, various drugs and metabolites |
| ACMIA, CEDIA, CMIA, ELISA, EMIT, FPIA, MEIA, PETNIA | |||
| SERS-based methods | Inelastic light scattering on molecule adsorbed on the roughened metal surface is measured | No need for sample preparation; Fast measurement; Multiplexing capabilities; Availability of portable Raman spectrometer | Very often high RSD of the SERS substrates; method optimization needed for each drug; |
| Sample | Drug | SERS Substrate | Laser Line (nm) | LOD (M) | Calibration | Refs. |
|---|---|---|---|---|---|---|
| Clinical sample | ||||||
| serum | Mitoxantrone | Ag colloid FLOW | 514 633 | 4 × 10−11 b | U | [45] |
| Spiked body fluid | ||||||
| saliva | 5-FU | SERS-active capillaries | 785 | 1.15 × 10−06 a | U | [67,68] |
| blood | 6-MP | Si-AuNPs needles | 785 | n.r. | n.r. | [69] |
| bovine plasma | Doxorubicin | Ag colloid | 488 | n.r. | M | [70] |
| plasma | Imatinib | Au on glass with Al | 785 | n.r. | n.r | [71] |
| human serum | MTX | Au colloid on paper | 785 | n.r. | M | [54] |
| blood plasma | Paclitaxel | Au-polystyrene beads | 785 | n.r. | M | [72] |
| Surrogate matrix | ||||||
| 1.5% HSA-PBS | Irinotecan | Ag and Au colloid on TLC plate | 514 785 | n.r. | n.r. | [40] |
| 1% BSA-PBS | MTX | Sandwich substrate | 532 | 10−09 b | n.r. | [73] |
| 0.6% HSA | Paclitaxel | Ag colloid | 532 | n.r. | n.r. | [74] |
| Other solutions | ||||||
| water | 6-MP | β-CD AgNPs Ag colloid | 785 | 2.4 × 10−09 a 1.4 × 10−08 a | U | [75] |
| water | MTX | Ag-graphene | 785 | 6 × 10−10 b | U | [76] |
| water | Imatinib | Au on glass with Al | 785 | n.r. | n.r. | [71] |
| water | Mitoxantrone | PNA | 785 | 4.18 × 10−08 c | U | [77] |
| BRB (pH 2.0) | 6-MP | GO/AgNP hybrids | 532 | 1.05 × 10−07 b | U | [78] |
| MeOH | Irinotecan SN-38 Sunitinib | Klarite™ | 633 | 34–40 *,e 11–28 *,e 11–15 *,e | U | [79] |
| 150 nM KOH | MTX | Ag colloid FLOW CELL | 514 785 | 1.70 × 10−07 b | U | [80] |
| DMSO | Paclitaxel | GNC UV-NIL | 633 | 10−09 f | U | [81] |
| concentration of mitoxantrone (ng/mL) | ||
|---|---|---|
| Time | HPLC | SERS * |
| 0 | 0 | nd |
| 5 | 3.1 | 2.9 |
| 10 | 1.9 | 1.0 |
| 15 | 252.3 | 247.3 |
| 20 | 186.9 | 183.2 |
| 30 | 53.7 | 54.1 |
| 45 | 23.7 | 20.3 |
| 60 | 11.6 | 9.2 |
| 90 | 9.4 | 8.9 |
| 120 | 6.2 | 6.0 |
| 180 | 5.7 | 4.8 |
| 240 | 1.8 | 1.0 |
| 360 | 3.2 | 3.0 |
| 720 | 1.8 | 1.3 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaworska, A.; Fornasaro, S.; Sergo, V.; Bonifacio, A. Potential of Surface Enhanced Raman Spectroscopy (SERS) in Therapeutic Drug Monitoring (TDM). A Critical Review. Biosensors 2016, 6, 47. https://0-doi-org.brum.beds.ac.uk/10.3390/bios6030047
Jaworska A, Fornasaro S, Sergo V, Bonifacio A. Potential of Surface Enhanced Raman Spectroscopy (SERS) in Therapeutic Drug Monitoring (TDM). A Critical Review. Biosensors. 2016; 6(3):47. https://0-doi-org.brum.beds.ac.uk/10.3390/bios6030047
Chicago/Turabian StyleJaworska, Aleksandra, Stefano Fornasaro, Valter Sergo, and Alois Bonifacio. 2016. "Potential of Surface Enhanced Raman Spectroscopy (SERS) in Therapeutic Drug Monitoring (TDM). A Critical Review" Biosensors 6, no. 3: 47. https://0-doi-org.brum.beds.ac.uk/10.3390/bios6030047