Anti-CD20 Agents for Multiple Sclerosis: Spotlight on Ocrelizumab and Ofatumumab

 and
and 
Abstract
:1. Introduction
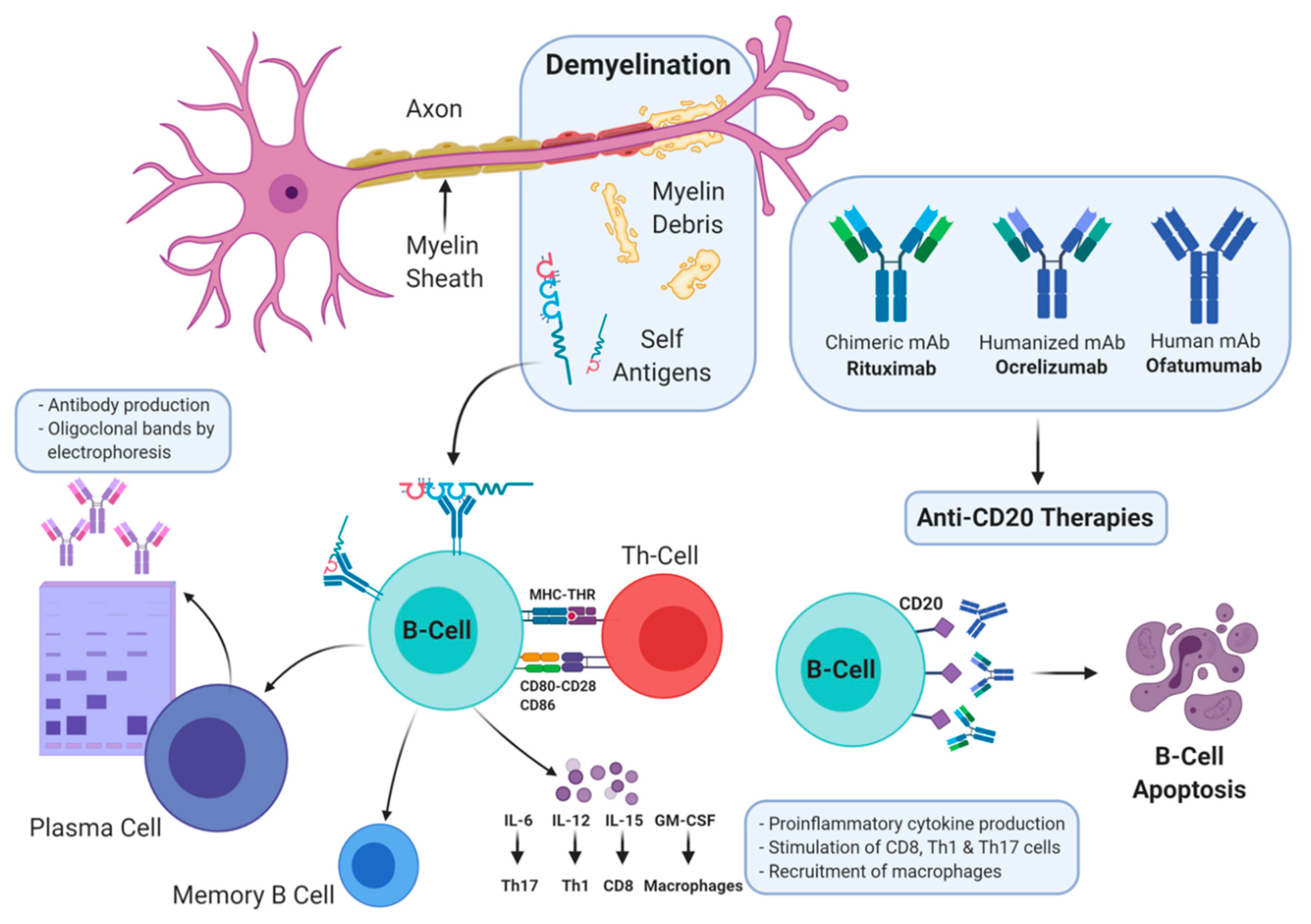
2. T- and B-Cell Hypothesis
3. Anti-CD20 Agents
4. Ocrelizumab
4.1. Dosage and Administration
4.2. Pharmacology and Pharmacokinetics
4.3. Clinical Trials
5. Ofatumumab
5.1. Pharmacology
5.2. Clinical Trials
6. Safety
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Goodin, D.S. The epidemiology of multiple sclerosis: Insights to disease pathogenesis. Handb. Clin. Neurol. 2014, 122, 231–266. [Google Scholar] [CrossRef] [PubMed]
- Lublin, F.D. New multiple sclerosis phenotypic classification. Eur. Neurol. 2014, 72, 1–5. [Google Scholar] [CrossRef]
- Finlayson, M.; Guglielmello, L.; Liefer, K. Describing and predicting the possession of assistive devices among persons with multiple sclerosis. Am. J. Occup. Ther. 2001, 55, 545–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dargahi, N.; Katsara, M.; Tselios, T.; Androutsou, M.E.; de Courten, M.; Matsoukas, J.; Apostolopoulos, V. Multiple Sclerosis: Immunopathology and Treatment Update. Brain Sci. 2017, 7, 78. [Google Scholar] [CrossRef] [Green Version]
- Katsara, M.; Apostolopoulos, V. Editorial: Multiple Sclerosis: Pathogenesis and Therapeutics. Med. Chem. 2018, 14, 104–105. [Google Scholar] [CrossRef] [PubMed]
- Katsara, M.; Deraos, G.; Tselios, T.; Matsoukas, J.; Apostolopoulos, V. Design of novel cyclic altered peptide ligands of myelin basic protein MBP83-99 that modulate immune responses in SJL/J mice. J. Med. Chem. 2008, 51, 3971–3978. [Google Scholar] [CrossRef] [PubMed]
- Bruck, W. The pathology of multiple sclerosis is the result of focal inflammatory demyelination with axonal damage. J. Neurol. 2005, 252, v3–v9. [Google Scholar] [CrossRef] [PubMed]
- Zephir, H. Progress in understanding the pathophysiology of multiple sclerosis. Rev. Neurol. 2018. [Google Scholar] [CrossRef]
- Hersh, C.M.; Cohen, J.A. Alemtuzumab for the treatment of relapsing-remitting multiple sclerosis. Immunotherapy 2014, 6, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Mitsdoerffer, M.; Peters, A. Tertiary Lymphoid Organs in Central Nervous System Autoimmunity. Front. Immunol. 2016, 25, 451. [Google Scholar] [CrossRef]
- Greenfield, A.L.; Hauser, S.L. B-cell Therapy for Multiple Sclerosis: Entering an era. Ann. Neurol. 2018, 83, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; Bruck, W.; Lucchinetti, C.F. The immunopathology of multiple sclerosis: An overview. Brain Pathol. 2007, 17, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Bar-Or, A.; Fawaz, L.; Fan, B.; Darlington, P.J.; Rieger, A.; Ghorayeb, C.; Calabresi, P.A.; Waubant, E.; Hauser, S.L.; Zhang, J.; et al. Abnormal B-cell cytokine responses a trigger of T-cell-mediated disease in MS? Ann. Neurol. 2010, 67, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Fraussen, J.; de Bock, L.; Somers, V. B cells and antibodies in progressive multiple sclerosis: Contribution to neurodegeneration and progression. Autoimmun. Rev. 2016, 15, 896–899. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Rezk, A.; Healy, L.M.; Muirhead, G.; Prat, A.; Gommerman, J.L.; Bar-Or, A.; MSSRF Canadian B cells in MS Team. Cytokine-Defined B Cell Responses as Therapeutic Targets in Multiple Sclerosis. Front. Immunol. 2015, 6, 626. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Rezk, A.; Miyazaki, Y.; Hilgenberg, E.; Touil, H.; Shen, P.; Moore, C.S.; Michel, L.; Althekair, F.; Rajasekharan, S.; et al. Proinflammatory GM-CSF-producing B cells in multiple sclerosis and B cell depletion therapy. Sci. Transl. Med. 2015, 7, 310ra166. [Google Scholar] [CrossRef]
- Dobson, R.; Ramagopalan, S.; Davis, A.; Giovannoni, G. Cerebrospinal fluid oligoclonal bands in multiple sclerosis and clinically isolated syndromes: A meta-analysis of prevalence, prognosis and effect of latitude. J. Neurol. Neurosurg. Psychiatry 2013, 84, 909–914. [Google Scholar] [CrossRef]
- Van Langelaar, J.; Rijvers, L.; Janssen, M.; Wierenga-Wolf, A.F.; Melief, M.J.; Siepman, T.A.; de Vries, H.E.; Unger, P.P.A.; van Ham, S.M.; Hintzen, R.Q.; et al. Induction of brain-infiltrating T-bet-expressing B cells in multiple sclerosis. Ann. Neurol. 2019, 86, 264–278. [Google Scholar] [CrossRef]
- Baker, D.; Marta, M.; Pryce, G.; Giovannoni, G.; Schmierer, K. Memory B Cells are Major Targets for Effective Immunotherapy in Relapsing Multiple Sclerosis. EBioMedicine 2017, 16, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Wei, G.; Liu, D. CD19: A biomarker for B cell development, lymphoma diagnosis and therapy. Exp. Hematol. Oncol. 2012, 1, 36. [Google Scholar] [CrossRef] [Green Version]
- Riley, J.K.; Sliwkowski, M.X. CD20: A gene in search of a function. Semin. Oncol. 2000, 27, 17–24. [Google Scholar] [PubMed]
- Uchida, J.; Lee, Y.; Hasegawa, M.; Liang, Y.; Bradney, A.; Oliver, J.A.; Bowen, K.; Steeber, D.A.; Haas, K.M.; Poe, J.C.; et al. Mouse CD20 expression and function. Int. Immunol. 2004, 16, 119–129. [Google Scholar] [CrossRef] [Green Version]
- Gingele, S.; Jacobus, T.L.; Konen, F.F.; Hümmert, M.W.; Sühs, K.-W.; Schwenkenbecher, P.; Ahlbrecht, J.; Möhn, N.; Müschen, L.H.; Bönig, L. Ocrelizumab depletes CD20+ T cells in multiple sclerosis patients. Cells 2019, 8, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno Torres, I.; Garcia-Merino, A. Anti-CD20 monoclonal antibodies in multiple sclerosis. Expert Rev. Neurother. 2017, 17, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Sellebjerg, F.; Blinkenberg, M.; Sorensen, P.S. Anti-CD20 Monoclonal Antibodies for Relapsing and Progressive Multiple Sclerosis. CNS Drugs 2020, 34, 1–12. [Google Scholar] [CrossRef]
- Cambridge, G.; Leandro, M.J.; Teodorescu, M.; Manson, J.; Rahman, A.; Isenberg, D.A.; Edwards, J.C. B cell depletion therapy in systemic lupus erythematosus: Effect on autoantibody and antimicrobial antibody profiles. Arthritis Rheum. 2006, 54, 3612–3622. [Google Scholar] [CrossRef]
- Cornec, D.; Avouac, J.; Youinou, P.; Saraux, A. Critical analysis of rituximab-induced serological changes in connective tissue diseases. Autoimmun. Rev. 2009, 8, 515–519. [Google Scholar] [CrossRef]
- World Health Organization. Proposed INN: List 101. WHO Drug Inf. 2009, 23, 176. [Google Scholar]
- Srinivasan, A.; Mukherji, S.K. Tositumomab and iodine I 131 tositumomab (Bexaar). Am. J. Neuroradiol. 2011, 32, 637–638. [Google Scholar] [CrossRef] [Green Version]
- Lehmann-Horn, K.; Kinzel, S.; Weber, M.S. Deciphering the Role of B Cells in Multiple Sclerosis-Towards Specific Targeting of Pathogenic Function. Int. J. Mol. Sci. 2017, 18, 2048. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization: WHO Model List of Essential Medicines (19th List). Available online: https://www.who.int/medicines/publications/essentialmedicines/en/ (accessed on 16 December 2018).
- Greenwald, M.; Tesser, J.; Sewell, K.L. Biosimilars Have Arrived: Rituximab. Arthritis 2018, 2018, 3762864. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.L.; Waubant, E.; Arnold, D.L.; Vollmer, T.; Antel, J.; Fox, R.J.; Bar-Or, A.; Panzara, M.; Sarkar, N.; Agarwal, S.; et al. B-cell depletion with rituximab in relapsing–remitting multiple sclerosis. N. Engl. J. Med. 2008, 358, 676–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawker, K.; O’Connor, P.; Freedman, M.S.; Calabresi, P.A.; Antel, J.; Simon, J.; Hauser, S.; Waubant, E.; Vollmer, T.; Panitch, H.; et al. Rituximab in patients with primary progressive multiple sclerosis: Results of a randomized double-blind placebo-controlled multicenter trial. Ann. Neurol. 2009, 66, 460–471. [Google Scholar] [CrossRef] [PubMed]
- De Flon, P.; Gunnarsson, M.; Laurell, K.; Söderström, L.; Birgander, R.; Lindqvist, T.; Krauss, W.; Dring, A.; Bergman, J.; Sundström, P.; et al. Reduced inflammation in relapsing-remitting multiple sclerosis after therapy switch to rituximab. Neurology 2016, 87, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Salzer, J.; Svenningsson, R.; Alping, P.; Novakova, L.; Björck, A.; Fink, K.; Islam-Jakobsson, P.; Malmeström, C.; Axelsson, M.; Vågberg, M.; et al. Rituximab in multiple sclerosis: A retrospective observational study on safety and efficacy. Neurology 2016, 87, 2074–2081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zecca, C.; Bovis, F.; Novi, G.; Capobianco, M.; Lanzillo, R.; Frau, J.; Repice, A.M.; Hakiki, B.; Realmuto, S.; Bonavita, S.; et al. Treatment of multiple sclerosis with rituximab: A multicentric Italian–Swiss experience. Mult. Scler. J. 2019, 12, 1352458519872889. [Google Scholar] [CrossRef]
- Naegelin, Y.; Naegelin, P.; von Felten, S.; Lorscheider, J.; Sonder, J.; Uitdehaag, B.M.; Scotti, B.; Zecca, C.; Gobbi, C.; Kappos, L.; et al. Association of rituximab treatment with disability progression among patients with secondary progressive multiple sclerosis. JAMA Neurol. 2019, 76, 274–281. [Google Scholar] [CrossRef]
- Linden, J.; Granåsen, G.; Salzer, J.; Svenningsson, A.; Sundström, P. Inflammatory activity and vitamin D levels in an MS population treated with rituximab. Mult. Scler. J. Exp. Transl. Clin. 2019, 5, 2055217319826598. [Google Scholar] [CrossRef] [Green Version]
- Sheridan, C. Genentech’s Ocrevus heralds new chapter in MS treatment. Nat. Biotechnol. 2017, 35, 393–394. [Google Scholar] [CrossRef]
- Ocrelizumab (Ocrevus) for MS. Med. Lett. Drugs Ther. 2017, 59, 98–101.
- Forbes. How Much Can Roche’s Share Price Grow If Ocrevus Doubles Its Share in Multiple Sclerosis Market? Available online: https://www.forbes.com/sites/greatspeculations/2019/03/13/how-much-can-roches-share-price-grow-if-ocrevus-doubles-its-share-in-multiple-sclerosis-market/#25c78a3c4c98 (accessed on 1 August 2020).
- Klein, C.; Lammens, A.; Schafer, W.; Georges, G.; Schwaiger, M.; Mossner, E.; Hopfner, K.P.; Umana, P.; Niederfellner, G. Epitope interactions of monoclonal antibodies targeting CD20 and their relationship to functional properties. MAbs 2013, 5, 22–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelfand, J.M.; Cree, B.A.C.; Hauser, S.L. Ocrelizumab and Other CD20(+) B-Cell-Depleting Therapies in Multiple Sclerosis. Neurotherapeutics 2017, 14, 835–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kappos, L.; Li, D.; Calabresi, P.A.; O’Connor, P.; Bar-Or, A.; Barkhof, F.; Yin, M.; Leppert, D.; Glanzman, R.; Tinbergen, J.; et al. Ocrelizumab in relapsing-remitting multiple sclerosis: A phase 2, randomised, placebo-controlled, multicentre trial. Lancet 2011, 378, 1779–1787. [Google Scholar] [CrossRef]
- Hauser, S.L.; Bar-Or, A.; Comi, G.; Giovannoni, G.; Hartung, H.P.; Hemmer, B.; Lublin, F.; Montalban, X.; Rammohan, K.W.; Selmaj, K.; et al. Ocrelizumab versus Interferon Beta-1a in Relapsing Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Montalban, X.; Hauser, S.L.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Comi, G.; de Seze, J.; Giovannoni, G.; Hartung, H.P.; Hemmer, B.; et al. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Polman, C.H.; Reingold, S.C.; Banwell, B.; Clanet, M.; Cohen, J.A.; Filippi, M.; Fujihara, K.; Havrdova, E.; Hutchinson, M.; Kappos, L. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann. Neurol. 2011, 69, 292–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Food and Drug Administration. BLA Approval Letter; Department of Health and Human Services: Washington, DC, USA, 2017. [Google Scholar]
- AlDallal, S.M. Ofatumumab—A valid treatment option for chronic lymphocytic leukemia patients. Ther. Clin. Risk Manag. 2017, 13, 905–907. [Google Scholar] [CrossRef] [Green Version]
- Masoud, S.; McAdoo, S.P.; Bedi, R.; Cairns, T.D.; Lightstone, L. Ofatumumab for B cell depletion in patients with systemic lupus erythematosus who are allergic to rituximab. Rheumatology 2018. [Google Scholar] [CrossRef] [Green Version]
- Babiker, H.M.; Glode, A.E.; Cooke, L.S.; Mahadevan, D. Ublituximab for the treatment of CD20 positive B-cell malignancies. Expert Opin. Investig. Drugs 2018, 27, 407–412. [Google Scholar] [CrossRef]
- Ostergaard, M.; Baslund, B.; Rigby, W.; Rojkovich, B.; Jorgensen, C.; Dawes, P.T.; Wiell, C.; Wallace, D.J.; Tamer, S.C.; Kastberg, H.; et al. Ofatumumab, a human anti-CD20 monoclonal antibody, for treatment of rheumatoid arthritis with an inadequate response to one or more disease-modifying antirheumatic drugs: Results of a randomized, double-blind, placebo-controlled, phase I/II study. Arthritis Rheum. 2010, 62, 2227–2238. [Google Scholar] [CrossRef]
- Reagan, J.L.; Castillo, J.J. Ofatumumab for newly diagnosed and relapsed/refractory chronic lymphocytic leukemia. Expert Rev. Anticancer. Ther. 2011, 11, 151–160. [Google Scholar] [CrossRef]
- Sorensen, P.S.; Lisby, S.; Grove, R.; Derosier, F.; Shackelford, S.; Havrdova, E.; Drulovic, J.; Filippi, M. Safety and efficacy of ofatumumab in relapsing-remitting multiple sclerosis: A phase 2 study. Neurology 2014, 82, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Bar-Or, A.; Grove, R.A.; Austin, D.J.; Tolson, J.M.; VanMeter, S.A.; Lewis, E.W.; Derosier, F.J.; Lopez, M.C.; Kavanagh, S.T.; Miller, A.E.; et al. Subcutaneous ofatumumab in patients with relapsing-remitting multiple sclerosis: The MIRROR study. Neurology 2018. [Google Scholar] [CrossRef]
- Hauser, S.L.; Bar-Or, A.; Cohen, J.A.; Comi, G.; Correale, J.; Coyle, P.K.; Cross, A.H.; de Seze, J.; Leppert, D.; Montalban, X.; et al. Ofatumumab versus teriflunomide in multiple sclerosis. N. Engl. J. Med. 2020, 383, 546–557. [Google Scholar] [CrossRef] [PubMed]
- Auricchio, F.; Scavone, C.; Cimmaruta, D.; Di Mauro, G.; Capuano, A.; Sportiello, L.; Rafaniello, C. Drugs approved for the treatment of multiple sclerosis: Review of their safety profile. Expert Opin. Drug Saf. 2017, 16, 1359–1371. [Google Scholar] [CrossRef] [PubMed]
- Tran, V.; Miller, P.; Olson, J.; Miller, T.; Miravalle, A. The Effect of Ocrelizumab Therapy on Immunoglobulin Levels in Patients with Multiple Sclerosis (P4. 2-042); AAN Enterprises: Haryana, India, 2019. [Google Scholar]
- Clifford, D.; Gass, A.; Richert, N.; Tornatore, C.; Vermersch, P.; Hughs, R. Cases reported as progressive multifocal leukoencephalopathy in ocrelizumab-treated patients with multiple sclerosis. In Proceedings of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS), Stockholm, Sweden, 11–13 September 2019. [Google Scholar]
- Byrd, J.C.; Brown, J.R.; O’Brien, S.; Barrientos, J.C.; Kay, N.E.; Reddy, N.M.; Coutre, S.; Tam, C.S.; Mulligan, S.P.; Jaeger, U.; et al. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N. Engl. J. Med. 2014, 371, 213–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osterborg, A.; Udvardy, M.; Zaritskey, A.; Andersson, P.O.; Grosicki, S.; Mazur, G.; Kaplan, P.; Steurer, M.; Schuh, A.; Montillo, M.; et al. Phase III, randomized study of ofatumumab versus physicians’ choice of therapy and standard versus extended-length ofatumumab in patients with bulky fludarabine-refractory chronic lymphocytic leukemia. Leuk. Lymphoma 2016, 57, 2037–2046. [Google Scholar] [CrossRef]
- Avila, J.; Han, J.; Zaydan, I. Progressive Multifocal Leukoencephalopathy Associated with Ofatumumab Presenting as Alexia without Agraphia: A Case Report. Neurol. 2014, 82, 4–319. [Google Scholar]
- Gross, R.H.; Corboy, J.R. Monitoring, switching, and stopping multiple sclerosis disease-modifying therapies. CONTINUUM Lifelong Learn. Neurol. 2019, 25, 715–735. [Google Scholar] [CrossRef]
- Van der Kolk, L.E.; Grillo-Lopez, A.J.; Baars, J.W.; Hack, C.E.; van Oers, M.H. Complement activation plays a key role in the side-effects of rituximab treatment. Br. J. Haematol 2001, 115, 807–811. [Google Scholar] [CrossRef]
- Ontaneda, D.; Thompson, A.J.; Fox, R.J.; Cohen, J.A. Progressive multiple sclerosis: Prospects for disease therapy, repair, and restoration of function. Lancet 2017, 389, 1357–1366. [Google Scholar] [CrossRef]


{kind=link}
| Author | Participants | Trial Design | Clinical Findings | MRI Findings | Adverse Effects |
|---|---|---|---|---|---|
| Hauser et al., 2008 [33] | 104 participants with RRMS | 48 wk phase 2, randomized, parallel, double-blind, placebo-controlled study | Decreased number of patients with relapse (24 and 48 wks) and decreased ARR (24 wks only) | Decreased total and new GAD-enhancing lesions with rituximab | Infusion-associated AEs common in Rituximab-treated patients (78.3%). Infection rate similar between groups |
| Hawker et al., 2009 [34] | 439 patients with PPMS | 96 wk, phase 2/3, randomized, double-blind, placebo-controlled study | No change in time to disease progression, or the MSFC (with exception of the 25-foot walk) | Decrease T2 lesion volume | 8.6% of rituximab patients experienced severe or disabling AEs. 67% of rituximab patients experienced infusion- associated symptoms. |
| De Flon et al., 2016 [35] | 75 patients with clinically stable RRMS | Open Label, uncontrolled phase 2 study | Nil | Decreased number of GAD-enhancing lesions per patient, decreased new or enlarged T2 lesions | Not fully reported. 8% of patients reported severe AEs. |
| Salzer et al., 2016 [36] | 822 patients with MS; 557 RRMS, 198 SPMS, 67 PPMS | Retrospective uncontrolled observational study | EDSS unchanged in RRMS, slight decreases in SPMS and PPMS | Lower disease activity suggested on rituximab | AEs likely underreported; however, 10.8% of patients reported to experience grade 2 or greater AEs. |
| Zecca et al., 2019 [37] | 355 patients with MS | Retrospective uncontrolled observational study | ARR decreased compared to year before rituximab treatment in RRMS and SPMS | Descriptive only | 34.5% of patients treated with rituximab had infection-related AEs, 22.7% had non-infectious AEs. |
| Naeglin et al., 2019 [38] | 113 patients with SPMS | Retrospective cohort study | Lower EDSS scores in rituximab cohort | Nil | 9% of patients experienced serious AEs |
| Linden et al., 2019 [39] | 272 patients with MS of all subtypes | Retrospective Cohort Study with 43 months mean follow-up | No change in EDSS | T2 lesions correlated with vitamin D levels with rituximab. | 38.7% of patients experienced non-infusion AEs, with 7.1% experiencing severe AEs (mostly infectious). |
| Ocrelizumab | |||||
|---|---|---|---|---|---|
| Author | Participants | Trial Design | Clinical Findings | MRI Findings | Adverse Effects |
| Kappos et al., 2011 [45] | 220 participants with RRMS | 48 wk, phase 2, randomized, parallel, double-blind, placebo-controlled study | Lower ARR with ocrelizumab. Reduction in disease activity in placebo and control when crossed over to ocrelizumab. | Decreased GAD-enhancing lesions with 600 & 2000 mg ocrelizumab. | Infusion-associated AEs more common in ocrelizumab. Serious AEs were also reported. |
| Hauser et al., 2017 [46] | Two studies of 821 and 835 patients with RRMS. | 96 wk, phase 3, randomized, double-blind, active-controlled, parallel group studies | ARR reduced in ocrelizumab compared to IFN-β1a | Decrease in new or newly expanding T2 lesions and GAD-enhancing lesions with ocrelizumab | Mild-to-moderate infusion-associated AEs were reported, equally across ocrelizumab and IFN-β1a. |
| Montalban et al., 2017 [47] | 732 patients with RRMS | 120 wk, phase 3, double-blind, randomized, placebo-controlled, parallel group study | Reduction in disability at 12 and 24 weeks with ocrelizumab | 34% reduction in brain T2 lesions and percentage loss of brain volume decreased with ocrelizumab | Infusion-associated AEs, upper respiratory infections and neoplasms |
| Ofatumumab | |||||
|---|---|---|---|---|---|
| Author | Participants | Trial Design | Clinical Findings | MRI Findings | Adverse Effects |
| Sorensen et al., 2014 [55] | 38 patients with RRMS | 48 wk, phase 2, double-blind, randomized, placebo-controlled study | Decreasing relapse incidence versus placebo (19% vs. 25%) | Decreased new and expanding MRI lesions & total number of T1 GAD-enhancing lesions | Mostly mild-to-moderate-severity AEs |
| Bar-Or et al., 2014 [56] | 231 patients with RRMS | 48 wk, phase 2, double-blind, randomized, placebo-controlled study | Nil | Dose dependent decrease in GAD-enhancing lesions with ofatumumab | Increased number of infection-associated AEs in ofatumumab |
| Hauser et al., 2019 [57] | 900 patients with RRMS | 30 month maximum parallel Phase 3, double-blind, randomized, placebo-controlled studies (ASCLEPIOS I & II) | Improved relapse rate, and confirmed disease improvement compared to teriflunomide | Decreased number of T1 and T2 gadolinium-enhancing lesions | Equivalent AEs compared to teriflunomide |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Florou, D.; Katsara, M.; Feehan, J.; Dardiotis, E.; Apostolopoulos, V. Anti-CD20 Agents for Multiple Sclerosis: Spotlight on Ocrelizumab and Ofatumumab. Brain Sci. 2020, 10, 758. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci10100758
Florou D, Katsara M, Feehan J, Dardiotis E, Apostolopoulos V. Anti-CD20 Agents for Multiple Sclerosis: Spotlight on Ocrelizumab and Ofatumumab. Brain Sciences. 2020; 10(10):758. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci10100758
Chicago/Turabian StyleFlorou, Despoina, Maria Katsara, Jack Feehan, Efthimios Dardiotis, and Vasso Apostolopoulos. 2020. "Anti-CD20 Agents for Multiple Sclerosis: Spotlight on Ocrelizumab and Ofatumumab" Brain Sciences 10, no. 10: 758. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci10100758