
Animal Models as a Tool to Design Therapeutical Strategies for CMT-like Hereditary Neuropathies

Institute of Experimental Neurology (INSPE), Division of Neuroscience, IRCCS San Raffaele Scientific Institute, 20132 Milan, Italy
*
Author to whom correspondence should be addressed.
Brain Sci. 2021, 11(9), 1237; https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci11091237
Submission received: 25 July 2021
/
Revised: 5 September 2021
/
Accepted: 7 September 2021
/
Published: 18 September 2021
(This article belongs to the Special Issue Advanced Research in Neuromuscular Disorders)
Abstract
:Since ancient times, animal models have provided fundamental information in medical knowledge. This also applies for discoveries in the field of inherited peripheral neuropathies (IPNs), where they have been instrumental for our understanding of nerve development, pathogenesis of neuropathy, molecules and pathways involved and to design potential therapies. In this review, we briefly describe how animal models have been used in ancient medicine until the use of rodents as the prevalent model in present times. We then travel along different examples of how rodents have been used to improve our understanding of IPNs. We do not intend to describe all discoveries and animal models developed for IPNs, but just to touch on a few arbitrary and paradigmatic examples, taken from our direct experience or from literature. The idea is to show how strategies have been developed to finally arrive to possible treatments for IPNs.
1. Introduction and Historical Use of Animal Models in Medicine
Through the course of history, experimentation on non-human vertebrates (hereinafter referred as “animals”) played a pivotal role in the development of biomedical research. At the same time, it also constituted a topic of public debates, raising criticism and controversies of both philosophical and moral nature, depending on the ever-changing perspectives of different ages.
Although the earliest evidence of cranial surgery performed on an animal dates back to the late Neolithic, Ancient Greeks were probably the first to systematically experiment on dissecting animals for anatomical studies [1,2]. During the fifth century BC Alcmaeon of Croton was the first to pioneer the field of comparative anatomy, followed by eclectic personalities such as Aristotle, Herophilus, Erasistratus and Galen, whose works, despite the inevitable anatomical mistakes, remained canonical until the Renaissance [3,4,5].
Notwithstanding the misconception of a period of scientific decadence, animal experimentation never really fell into disuse even during the Middle Ages, carried on by the likes of Leonardo Da Vinci for anatomic discovery. It is during the seventeenth century however that physiology studies marked the dawn of modern scientific research in biomedical sciences, when William Harvey first depicted an accurate description of the circulatory system through the examination of the heart function in eels and several other fishes.
Along with the advances in understanding physiology and pathology, criticisms regarding the use of animals in science emerged, concerning both the validity of conclusions derived from experiments on suffering or dead animals, and the moral question, risen even stronger in light of Darwin’s findings on evolution, which made differences between animals and humans progressively more nuanced [6].
By the beginning of the nineteenth century, scientists such as Francois Magendie and Claude Bernard in physiology, and Louis Pasteur in microbiology, made the use of animal experimentations pivotal towards the validation of the scientific method [7]. In the first half of the 1900s, the discovery and testing of insulin in diabetic dogs, and the development, appraisal and production of a vaccine for poliomyelitis, while requiring the sacrifice of large numbers of monkeys, contributed to saving of millions of human lives [8,9].
These breakthroughs in medical sciences progressively disproved the argument that no medical progress could be obtained through animal research, and the debate shifted towards the need for a regulated environment, aimed at using laboratory subjects in a much more humane, limited and scientifically productive manner [10].
Through the course of the twentieth century, the emergence of rodent species as the preferred laboratory subjects allowed further advancement in biomedical research, due to the multiple additional benefits of such models, including their physiological similarities with humans, their small size, the ease in maintenance, short life cycle and abundant offspring [11].
In 1921 Clarence Cook Little inbred the mouse strain C57BL/6 or “black 6”, which later became the most popular laboratory mouse to date, and whose complete genome was sequenced in 2002 [12].
In 1976 Rudolf Jaenisch proved that foreign DNA could be integrated into the DNA of early mouse embryos through the use of a retrovirus, developing the first transgenic mammals in history, and the development of the first knockout mouse in 1987, granted Mario R. Capecchi, Martin J. Evans, and Oliver Smithies the winning of the Noble Prize in 2007 [13,14]. With the development of the Cre-LoxP and CRISPR-Cas9 recombination technologies, a new era of genome editing sciences began, opening countless possibilities for the understanding of gene function and their influence in several genetic and non-genetic diseases [15,16].
In the last 20 years we witnessed the rise of a large body of literature on the subject, prospects being that it will continue to play a central role in the development of biomedicine in the foreseeable future.
2. Brief Introduction to Human Inherited Peripheral Neuropathies (IPNs)
Inherited peripheral neuropathies (IPNs) comprise a vast and heterogeneous group of disorders of the peripheral nerve ranging from pure motor (hereditary motor neuropathy, HMN) to pure sensory (hereditary sensory, HSN, or hereditary sensory-autonomic neuropathy, HSAN), and including the most frequent sensory-motor Charcot-Marie-Tooth (CMT) disease; for recent review see [17,18,19]. First described as nosological entities by Charcot and Marie in France 1886, and almost independently by Tooth in England at around the same time, they have estimated prevalence ranging from 1 in 8500 to 1 in 1200 [18,20,21].
As described by its discoverers, the disease has a typical onset during late childhood or adolescence, although later onset has been also reported, as for example in axonal myelin protein zero (MPZ) mutations [22,23].
The disease is characterized by distal motor and/or sensory deficits at the four limbs, muscle atrophy, reduced or absent deep tendon reflexes and bone deformities (typically pes cavus and hammertoes). Symptoms slowly progress, with peroneal muscles usually bearing the first signs of the disease, leading the foot to drop during walking or running, and the ankle to become unstable, which can lead to traumatic injury. Later, patients may develop atrophy of the hands and forearms, but the wasting rarely extends proximally to the elbow or above the middle third of the thigh, giving the lower limbs the typical “inverted champagne bottle” or stork appearance. Depending on magnitude of sensory and autonomic involvement, patients seldom develop trophic changes of the skin and bones in the affected limbs, due to repeated injury on analgesic parts and lack of autonomic vascular reflexes [24]. In most cases, despite a variable disability course, life expectancy is not reduced.
For a long time, CMTs have been divided in two groups according to nerve conduction velocities, which are typically slow in type 1 (CMT1; mean motor nerve conduction velocities, MNCV < 38 m/s for upper limb studies) and near normal in CMT2, referred as demyelinating and axonal types, respectively [25]. Due to the great heterogeneity in phenotypes, a broad overlap exists between clinical presentations of CMT1 and CMT2, and thus an intermediate form has been introduced (MNCV >35 and ≤45 m/s).
In recent years, new genetic findings enriched the rapidly evolving landscape of hereditary neuropathies and allowed for a better characterization of the clinical peculiarities in different forms, simplifying in some way the matter of their classification. From the first locus mapped on chromosome 1q22–q23 in 1982 [26], myriads of genes have been attributed to CMTs in the last twenty years, thanks to the development of next generation sequencing tools and genome wide analysis [27]. However, many patients still lack a genetic diagnosis in IPNs, primarily for CMT2, HMN and HSN forms.
The molecular mechanisms at the basis of the pathogenesis of all these many forms of CMTs are therefore multiple and involving almost all the biological functions of a cell [28,29]. In demyelinating CMTs, the original dysfunction resides, or prevails, in Schwann cells leading to abnormal nerve development in the most severe (congenital) cases or to the progressive inability to maintain the myelin sheath in later forms. From the initial findings where most of the causative genes were encoding for glia-specific proteins such as myelin components (PMP22, MPZ), transcription factors (EGR2, SOX10) or adhesion molecules (GJB1/CX32, PRX), it was then clear that the causative genes could perturb almost any aspect of the cell function [28,29]. In fact, there are genes involved in energy production, vesicular/membrane trafficking, cytoskeleton rearrangement and cell signaling [29,30]. Thus, any aspect that affects myelin assembly/maintenance, cells survival or the interaction with surrounding structures, primarily axons, would be responsible for demyelination.
Similarly, axonal CMTs mainly rely on primary deficits in the axon/neuronal compartment. In this case, the causative mechanism mainly relies on the long extension of the axon, which in many cases it can extend over one meter. Any (genetic) event affecting/limiting the survival of this distal portion of the neuron would result in axonal damage/degeneration in so called CMT2 neuropathies [31,32]. Accordingly, causative genes described in CMT2 encode for proteins involved in energy production (mainly for mitochondria function), axonal transport, neuronal survival and synaptic transmission. As a consequence, vital molecules and organelles cannot reach nerve terminals (anterograde) or being removed from the periphery to the neuronal soma (retrograde transport), altering neuronal homeostasis and making axons vulnerable to damage [31,32].
Despite the considerable progresses in diagnostic accuracy and molecular classification, there are currently no disease-modifying therapies for this group of genetic disorders. In recent years we have seen the development and commercialization of many gene-based therapies aimed at changing the natural history of other neuromuscular diseases, providing new hope for the development of specific treatments for hereditary neuropathies, as well.
3. The Importance of Animal Models in Inherited Neuropathies
The development of animal models has been fundamental to study the pathogenesis of IPNs. If we narrow it down to rodents (and primarily mice), which are the most useful models in the field, they not only reproduced (in most of the cases) clinical and pathological aspects of the human neuropathy, but they also shed light on the molecular mechanisms sustaining the disease and opened routes for therapeutical approaches (Figure 1). A list of the rodents developed to mimic IPNs is presented in Table 1.
3.1. Can Animal Models Reproduce Human IPNs?
The most obvious use of animal model is to reproduce human disease. For IPN, the prototype is the rat model of CMT1A, the most frequent form of IPN due to copy number variation of the PMP22 gene [135,136]. CMT1A patients have three copies of the PMP22 gene, due to a genomic duplication spanning around 1.5Mb on chromosome 17 and resulting from unequal meiotic crossover that is mediated by highly homologous repeat sequences flanking the duplicated region. Transgenic rat models overexpressing Pmp22 were generated representing the first direct proof that in humans is the gene dosage of PMP22 responsible for the disease [137] Accordingly, a rat expressing the equivalent of three genetic copies of the Pmp22 gene displayed weakness and gait abnormalities, reduced nerve conduction velocities, and peripheral neuropathy with hypomyelination and onion bulbs. Axonal loss was also observed in subsequent studies [48,138]. Increased overexpression of Pmp22 transcript (obtained by breeding rats in homozygosity) correlates with a dramatic worsening of the disease [139]. Further CMT1A models have been generated in mice, though they needed several extra copies of the Pmp22 gene to induce overexpression of the Pmp22 protein and thus the neuropathy [36,37,45,140].
A subsequent demonstration that the neuropathy is the direct consequence of Pmp22 overexpression and could be rescued by reducing Pmp22 message was shown with the use of an inducible (tetracycline-controlled transcriptional activation) mouse model, in which tetracycline administration was able to switch off Pmp22 overexpression. In this transgenic mouse, tetracycline administration in adult animals, when Pmp22 overexpression had already caused the peripheral neuropathy, was able to ameliorate the disease with a partial correction of hypomyelination [33].
The genetic counterpart of CMT1A is the hereditary neuropathy with liability to pressure palsies (HNPP), in which patients present lower PMP22 copy number (one single allele) and episodic motor and/or sensory deficits [141]. The pathological hallmark of the disease is the presence of focal “sausage-shaped” swellings of the myelin defined tomacula, which are reproduced in mice hemizygous for the Pmp22 gene [142,143]. Similarly, experimental nerve compression in these mutant mice reproduces nerve conduction blocks by neurophysiological analysis [144].
Finally, CMT1E, which is the consequence of PMP22 point mutation, is also reproduced in spontaneous Trembler and Trembler-J mice, both carrying point mutation (respectively Gly160Asp and Leu16Pro) in the Pmp22 gene also found in humans [65,145]. Additionally, these mutants reproduce clinical and pathological findings of human neuropathies (including onion bulbs), whereas the exact pathomechanism remains unclear. Several findings show that the mutated protein forms aggresomes and is retained in the endoplasmic reticulum (ER) [146,147,148,149,150,151,152], eventually triggering an unfolded protein response (UPR) and Schwann cell damage [153] and/or abnormal calcium entry by dysregulation of store operated calcium channel activity [154].
The pathological hallmarks of all the above PMP22 models have been extensively described and compared to human in a recent review article [138]. Overall, rodent models of CMT1A, CMT1E and HNPP, but in general for many of the demyelinating CMTs (see sections below), were able to reproduce most of the clinical and pathological findings of human patients and have been instrumental to elucidate their pathogenetic mechanism. Conversely, this was not the case for axonal CMTs, which minimally reproduced human finding but were anyway useful to reveal their pathogenesis in many cases. These aspects are treated and discussed in the following sections.
3.2. Pathogenesis of IPN Due to Loss- or Gain-of-Function Mechanism
Animal models have been extremely useful also to discriminate between loss- or gain-of-function mechanism in the pathogenesis of IPNs. The clear example is CMT1B, the second most frequent form of CMT due to heterozygous mutations in the MPZ gene. Myelin protein zero (P0) is the glycoprotein encoded by MPZ and the most abundant myelin protein in peripheral nerve [155]. P0 has an immunoglobulin-like structure that assembles to form tetramers emanating from the membrane surface, which interact with tetramers (in trans) on the opposing membrane surface (of the ensheathing Schwann cell) in order to promote myelin sheath compaction [156].
It was quite obvious to postulate that heterozygous mutations in MPZ would have reduced the production of P0, and thus of the component necessary to build and compact myelin, causing the neuropathy with a loss-of-function mechanism, as also recently reported [157]. Accordingly, mice with Mpz disruption by homologous recombination showed neuropathy with hypomyelination in heterozygous mutants [59], which is aggravated in homozygosity and undergoes distal axonal loss in older mice [158].
However, the majority of MPZ mutations do not cause mild phenotypes as reproduced by heterozygous Mpz mice or observed in humans with predicted premature termination and nonsense mediated decay of the mutant allele [23,159,160,161,162,163]. Thus, to definitely prove that most of MPZ mutations could act through a gain-of-function mechanism, transgenic mice bearing specific MPZ human mutation S63del or S63C have been generated. In these mice, an extra copy of the mutated allele (S63del or S63C) was expressed together with two copies of normal Mpz alleles [55]. If the mutated allele had acted through a loss-of-function mechanism, one would have expected no phenotype, as two normal Mpz alleles were present. Conversely, mice exhibited a typical demyelinating neuropathy, including clinical, neurophysiological and histological phenotype, confirming a typical gain-of-function mechanism [55].
Interestingly, animal models also revealed that mutated P0 glycoprotein may act with gain-of-function mechanism by reaching its target site, myelin, or as a consequence of its retention in the ER. For example, transgenic mice expressing a myc-tag at the mature N-terminus of P0 clearly showed that the (artificially) mutated protein arrives in myelin and disrupts the myelin compaction, possibly by the physical displacement of tetramers in myelin sheath [164]. More intriguingly, mice bearing only the S63del mutation showed that the mutated P0 protein does not reach the myelin sheath but is retained in the ER where it elicits a UPR [55]. The UPR is a mechanism generated by cells to respond to stress by activating the transcription of chaperons, attenuate protein translation and/or stimulate protein degradation to eventually reduce the load of improperly folded protein. However, the persistence of this condition becomes maladaptive promoting cell dysfunction or apoptosis. Many evidence showed that attenuation of protein translation is sufficient to ameliorate these forms of peripheral neuropathies, providing a new therapeutic strategy [56,57,165,166,167,168]. Accordingly, Sephin-1, a synthesized drug that prolongs protein translation attenuation, prevented the myelin and motor defects in P0-S63del mice [58].
Finally, the generation of a different transgenic mouse, expressing the P0-S63del and the wild type P0 with a myc epitope tag at the C terminus showed that the mutated P0 can interfere with the transit of the wt-P0 and reduce its amount in myelin with a direct dominant negative mechanism other than UPR [169].
3.3. One Gene but Different Phenotypes
Mutations in CMT genes may also present with a widely heterogeneous phenotype. Sometimes, this variability is caused by mutations in one single gene, as it is the case for MPZ. Mutations in MPZ may range from very severe forms defined as Dejerine-Sottas or with very early onset as congenital hypomyelination, to classical demyelinating CMT with moderate to mild phenotype, axonal CMT or intermediate CMT [170]. In the previous section, we already discussed as mouse models could mimic either milder demyelinating forms with heterozygous Mpz (+/−; loss of function) mice, or more classical moderate to severe demyelinating forms with S63del and S63C (gain of function) mutant mice respectively [55,59]; with the note that in humans, the neuropathy due to S63C is more severe than S63del, while in mice it is the reverse. Interestingly, simple overexpression of normal wild type P0 protein reproduced congenital hypomyelination phenotype [171]. This is partly due to altered trafficking of P0 in promyelin-forming Schwann cells, where the abnormal expression of a “sticky” Ig-like protein such as P0 impairs/arrests axonal sorting and axon ensheathing during development resulting in several Schwann cells arrested in a 1:1 relationship with axons, a typical congenital hypomyelination aspect.
Moreover, a recently developed mouse model carrying the MPZ-T124M mutation reproduces the axonal phenotype of human CMT2J/I (Maurizio D’Antonio and Shackleford Ghjuvan personal communication and presentation at PNS Society meeting 2021). We may predict that this mouse model will likely reveal the pathomechanism of axonal damage caused by some MPZ mutation, which is still obscure.
3.4. Cell Autonomy in the Pathogenesis of IPN
Animal models have been also fundamental to reveal cell autonomy in the pathogenesis of some CMT forms. In fact, and differently than initially though, many genes mutated in CMT do not encode for specific nerve proteins but for ubiquitously expressed proteins [172]. Whether loss/abnormal function of some proteins could affect both Schwann cells and neurons (axons), or only one of these two compartments was to be demonstrated. In this respect, the use of conditional mouse mutants by means of the Cre/LoxP technology became essential. With this system, the gene of interest to be ablated is flanked by LoxP sites, which are recognized by the Cre-recombinase enzyme expressed by specific transgene only in the cell of interest [173].
CMT4B1 is an autosomal recessive IPN due to mutations in the MTMR2 (myotubularin-related 2) gene, encoding for a phosphoinositide -PtdInsP3 and PtdIns(3,5)P2- phosphatase, characterized by early onset severe polyneuropathy [174]. The disease is characterized by demyelinating and axonal features and typical excessive redundant myelin outfoldings at nerve pathology [175]. Homozygous deletion of the Mtmr2 gene in null mice reproduced the typical myelin outfoldings phenotype [109,110]. Whether myelin outfoldings were generated by a loss of Mtmr2 in Schwann cells or neurons was solved by conditional mouse mutants. Mutants with Schwann cell conditional deletion of Mtmr2, obtained by P0-Cre transgene expressed selectively in Schwann cells [176] revealed nerve pathology and myelin outfoldings identical to the full KO mouse [177]. Conversely, conditional deletion of Mtmr2 in motoneurons, with the HB9 transgene [178], did not result in any overt phenotype suggesting that loss of Mtmr2 in Schwann cells, but not in motor neurons, is both sufficient and necessary to cause CMT4B1 neuropathy [177].
Conversely, conditional mouse models showed that CMT4J is due to defective Fig4 function in both Schwann cells and neurons. Fig4 is a 5-phosphatase involved in the dephosphorylation of PtdIns(3,5)P2. CMT4J is a severe autosomal recessive demyelinating neuropathy with childhood onset [117,179]. When Fig4 was ablated in neurons or motoneurons, respectively by Syn1-Cre or HB9-Cre transgene, mice developed progressive neuronal and axonal degeneration [180,181]. Similarly, when Fig4 was ablated in Schwann cells, by P0-Cre transgene, these cells showed impairment in endosomal trafficking and autophagy resulting in demyelination [181]. Thus, both neurons and Schwann cells need FIG4 to sustain proper nerve function.
Interestingly, as both Mtmr2 and Fig4 have different effects on PtdIns(3,5)P2, respectively; Fig4 has a role in the generation of PtdIns(3,5)P2 and Mtmr2 in its dephosphorylation—double mutant mice showed that this could be a therapeutic target for CMT4B. In fact, reduction of Fig4 in Mtmr2−/−;Fig4+/− mice ameliorated the myelin phenotype [182]. Finally, replacement of Fig4 in neuronal compartment in full Fig4 KO mice was sufficient to rescue mouse survival [180].
3.5. Ex Vivo Models of IPN
Animal models may also facilitate in reproducing in vitro models of the disease, which may reveal further knowledge on the molecular mechanisms that sustain the IPN. One clear example comes from dorsal root ganglia (DRG) explants from the Mtmr2 KO mouse model of CMT4B1. Sensory DRG neurons and Schwann cells can be plated in vitro, either as isolated cells or as a whole in the entire DRG, and upon conditioned media and ascorbic acid they generate myelin segments; recently reviewed in [183]. As mentioned above, CMT4B1 neuropathy is characterized by myelin outfoldings at nerve pathology in both human and mouse models. Strikingly, DRG explants from both full KO and Schwann cell conditional KO mouse reproduced similar findings in vitro [184], and this in vitro model was used to reveal possible interactors and to test potential rescue therapies [111,182,185]. Moreover, this was applied to other mouse models, such as Mtmr13 KO, also reproducing myelin outfoldings typical of CMT4B2 [186].
Another example of in vitro model of IPN was achieved with small heat-shock protein B1 (HspB1) mutant mice reproducing axonal CMT2F and HMN 2B [187,188]. HSPB1 is a ubiquitously expressed chaperone protein preventing aggregation of misfolded or non-native proteins and regulating several events including cytoskeleton stabilization, neurofilament assembly, apoptosis and autophagy [189]. Mutations in heat shock proteins alter their binding affinity to client proteins, in this case for HSPB1 regulating acetylation of α-tubulin in microtubules of peripheral nerve, as shown in the specific mouse mutants and in their DRG neurons explants [95,190]. Microtubule instability and thus deficits in axonal transport was dramatically rescued by drugs inhibiting histone deacetylase 6 (HDAC6) activity as revealed by DRG neurons isolated from symptomatic mutants and then confirmed in mice [95]. As inhibitors of HDAC6 restored axonal transport, including mitochondria transport and (possibly) dynamics, these drugs were then shown to produce benefits also in mouse models of CMT2A [73,191], reproducing the axonal neuropathy characterized by abnormal mitochondria fusion and transport due to mutations in MFN2 gene [191,192].
3.6. Is It Possible to Reproduce Length-Dependent Axonal IPN?
Several other models of IPNs have been generated in the last 20 years, whose description is not the focus of this manuscript, but can be found in Table 1. In many cases, they reproduced most of the clinical and pathological aspects of human neuropathies, whereas in others, namely axonal IPNs, the neuropathy was not induced, or it was very weak.
This was well represented in the case of the NEFL gene, whose mutations cause CMT2E and CMT2F [193,194]. These are axonal neuropathies exhibiting a broad range of phenotype ranging from mild to moderate and severe. NEFL encodes for the light (molecular weight) neurofilament protein (NFL), which associates with subunits of the medium (NFM) and heavy (NFH) molecular weight to generate coiled-coil dimers to form an antiparallel tetramer and then 10-nm filaments [195]. The mutated NFL acts by disrupting the neurofilament network causing protein aggregation and inclusions, and thus affecting axonal transport [196,197]. Surprisingly, the generation of Nefl KO mice did not result in overt phenotype, except for minimal reduction of axon caliber (by 15%), minor abnormality in nerve regeneration after crush [198], and some effect on motor function and spatial orientation revealed by sophisticated analyses [199]. Moreover, no phenotype was also reported in KO mice for Nfm and Nfh [200,201]. Transgenic mice expressing the p.P22S or the p.E396K Nefl mutation and knockin for p.P8R also did not exhibit peripheral neuropathy [90,91,202].
A more consistent phenotype was revealed by knockin mice, harboring heterozygous p.N98S Nefl mutation [91,92], one of the most common mutations in human. These mutants showed abnormal behavioral and nerve conduction studies consistent with neuropathy, and the histology revealed reduced axonal size and paucity of neurofilaments, a modest reduction of the number of myelinated fibers and very rare degenerating fibers [92]. Anyway, even in this case the phenotype was significantly weaker as compared to humans, and this was also observed for other mutants for axonal CMTs such as Gdap1-null mice [98,139], Hint1 [203,204], Mfn2 [74,77,78,79,205] and Mme [206]. This is likely due to many factors. First, the relatively reduced lifespan of mice, which do not live long enough to develop disorders with a very slow progressive mechanism such as axonal neuropathy.
Second, mouse nerves are too short to manifest a length-dependent axonal loss, and this should be searched in very distal axons as those in the hindpaw [207], in the architecture of neuromuscular junctions [84,98,104], or by stressing the system by mechanical nerve injury to reveal regenerating defects [208]. Finally, the exploitation of ex vivo models, such as DRG/primary neuronal explants from rodents modelling axonal CMT, was regardless very successful to reveal the pathogenetic mechanism of the disease and in reproducing the axonal defect, such as for HSPB1, MFN2, GDAP1 and others [95,98,191].
3.7. Can CMT Models Address Complex Schwann Cell-Axon Interactions?
Common fate of either demyelinating or axonal IPNs is the progressive loss of nerve fibers, which results from axonal degeneration independently of the first origin of the disease (axon or Schwann cell). Whether it is intuitive to understand consequences on Schwann cell function/integrity in case of primary axonal degeneration, the reverse is less obvious. In other words, it is unclear why mutant Schwann cells fail to support axonal function and survival, and it still remains a mostly unanswered question.
Secondary axonal damage is reproduced by rodent models of demyelinating CMTs, with the same limits (mild pathological evidence) described above for pure axonal forms [48,127,158].
It is not clear if dys/demyelination by itself is the cause of (or is sufficient to cause) axonal loss, or other Schwann cell functions intervene to support axon integrity and function. Pharmacologically treated rat model of CMT1A revealed that it is possible to uncouple demyelination from axonal loss [50], suggesting that lack of myelin sheath is not per se sufficient to cause axonal degeneration, as similarly described in mouse mutants in which Schwann cells cannot synthesize cholesterol [209,210] or in the shiverer mice (involving CNS myelin) [211].
The prevailing theory is that the lack of energy supply by mutant Schwann cells may weaken axonal integrity/function, thus promoting degeneration [210,212,213]. Alternative or complementary factors may include the transfer of proteins or ribosome to axons, rather than the production of toxic factors by mutant Schwann cells [214]. Accordingly, ER stress resulting from accumulation of mutant proteins in Schwann cells, as previously described in different CMT models [215], would compromise the energy support to axons. Further supporting evidence comes from studies in different experimental settings, as for example the recent discovery that Schwann cells shift to glycolytic activity to protect axons and delay degeneration in injured nerve [216]. Similarly, pyruvate supplementation in CMT1E mouse model supports axonal integrity by supplying (mitochondria) energy loss of the distal axons [217].
In summary, animal models are participating in shedding lights on the complex relationship existing between Schwann cells and axons and on the mechanisms of neuroprotection for long-term axonal integrity.
4. The Importance of Animal Models to Develop Therapies in Inherited Neuropathies
Finally, animal models have been used to test potential therapies since ancient times. The identification of genes responsible for IPNs, the dissection of possible pathomechanisms and the recognition of involved molecular pathways and target molecules allowed to the development of numerous potential therapeutic strategies in the last 10 years. These therapies include classical drugs consisting of chemical products (new molecules or repurposing of old ones), and innovative therapies consisting of nucleic acids as antisense oligonucleotides (ASO), gene therapy and gene editing.
We will not touch on all therapeutic attempts developed for IPNs, but simply report a few examples of potential therapies under investigation in which animal models have been fundamental.
4.1. Conventional Drugs
The identification of the pathogenetic mechanism responsible for specific IPNs, or groups of them, promoted the discovery of new molecules or the repurposing of old ones with the aim to rescue, or ameliorate, clinical and pathological findings of IPNs. Chemical drugs usually have a wider spectrum of use, being useful for different IPN forms with a similar mechanism of action.
Clear examples have been already described above, such as the development of drugs targeting the UPR and tested in mouse models of gain-of-function CMT1B (Mpz) and CMT1E (Pmp22 point mutation; Trembler J) [58,153,165], or HDAC6 inhibitors for CMT2F (Hspb1), CMT2A (Mfn2) and CMT1A [38,73,95,218].
A further example of molecule that may be used in different CMTs and revealed by the discovery of the pathomechanism in animal model is Niaspan. Niaspan is an extended-release formulation of nicotinic acid/niacin, used to treat dyslipidemia. Interestingly, this drug was known to modulate (enhance) TACE activity, which is a negative regulator of neuregulin 1 (NRG1) type III and thus myelination in peripheral nerve [219]. We know from seminal studies by Klaus Nave and Jim Salzer labs that NRG1 type III is the key molecule to dictate which axon must be myelinated and the amount of myelination, reviewed in [220]. This was revealed by in vitro and in vivo studies by generating mice with reduced (NRG1 type III +/−) or increased (transgene) expression of NRG1 type III [221,222]. Some CMT subtypes, in particular HNPP and CMT4B1, are characterized by focal hypermyelination such as tomacula and myelin outfoldings, respectively (see above; [109,142]). Proof of principle studies in DRG explants first revealed that downregulation of NRG1 type III was sufficient to significantly reduce tomacula or myelin outfoldings, and then this was confirmed in vivo in mouse models by Niaspam administration (and thus increased TACE activity and consequently reduced NRG1 type III function) [111]. This strategy might represent as a unifying treatment not only for HNPP and CMT4B1 but also for other hypermyelinating neuropathies including CMT4B2, CMT4B3 and CMT4H.
A further interesting drug with possible generalized function in CMTs is Curcumin. This is a polyphenol extracted from a plant with known multiple cellular targets and biological effects including anti-inflammatory and anti-oxidant function, and activity as a low affinity SERCA inhibitor [223]. As SERCA inhibitors can relieve ER stress, it is expected to ameliorate the phenotype in case of UPR activation. Accordingly, curcumin treatment (with different formulations) improved the peripheral neuropathy of R69C CMT1B mice, a well-known model of activated UPR response [54], as well as mitigated the neuropathy in Trembler-J mice (CMT1E model), which also present ER retention of their mutant protein [224]. However, curcumin presents unfavorable pharmacokinetic. More recently, a new and more stable formulation in nano-crystals showed to have a significant efficacy also in rat models of CMT1A, possibly due to the anti-oxidant effects [225].
4.2. Innovative Drugs: ASO, Gene Therapy and Gene Editing
CMT1A is the consequence of the duplication of one allele of PMP22 resulting in patients having 3 copies of the PMP22 gene (see Section 3.1). Any strategy aimed at reducing the PMP22 gene dosage (or RNA/protein PMP22 production) would constitute and efficacious strategy to treat the neuropathy. The development of antisense oligonucleotides (ASO) targeting PMP22 expression would constitute an ideal and reliable strategy to treat CMT1A, with the main warning to not exceed in PMP22 repression to avoid the induction of a HNPP-like disease [141]. This strategy was achieved by Zhao et al. by treating two rodent models (mouse and rat) of CMT1A with Pmp22-targeting ASO promoting the reduction of 35% of Pmp22 mRNA [39]. They demonstrated that initiation of treatment after the onset of the disease was able to restore myelination and nerve conduction velocities to levels comparable to normal controls. Moreover, they also revealed expression changes in several molecules after treatment that may become useful biomarkers to monitor the disease and the treatment response. One main limitation is that ASO do not cross sufficiently the blood-brain-barrier, as alternative strategies have been used with success to treat spinal muscle atrophy (SMA; reviewed in [226]). However, at least in rodents, Zhao et al. showed that low levels of ASO are sufficient to cross the blood–nerve-barrier and to reach Schwann cells.
Another innovative strategy is gene therapy. In this case a carrier viral vector is used to deliver a gene, a gene silencer or a gene modifier, into the target host. Recent advances in this strategy for CMT have been reviewed in [30]. Again, rodent models of CMT neuropathies have been instrumental to generate proof-of-principle results. Most interesting, although still with limited translatability, are those relating the intraneural injection of an AAV2/9 vector to silence Pmp22 and systemic (intrathecal) AAV9 mediated delivery of Gjb1/Cx32 to treat CMT1X and of Fig4 gene to treat CMT4J. In the first case, a murine Pmp22-targeting small harpin RNA was delivered into the sciatic nerve of CMT1A rats restoring the expression levels of Pmp22 to wild type condition and promoting nerve myelination and preservation of motor and sensory function of the target limb [49]. In the other cases, lumbar intrathecal injection of the carrier vector resulted in widespread distribution of the carried gene in the peripheral nerve of the mouse model and sustained Cx32 expression in these nerves, as well as improved motor functions and nerve conduction velocities [122]. Similarly, intracerebroventricular injection of the human Fig4 gene driven by a ubiquitously expressed chicken β-actin promoter with a CMV enhancer showed increased survival of treated mice, with normal motor functions and amelioration of pathological finding [227]. While intraneural treatment would be difficult to translate in efficient therapy in humans, the other approaches may be favored by the use of a small models as the mouse, and in this case the approach should be revaluated in larger animals to confirm the efficacy of AAV intrathecal/intracerebroventricular administration to reach target cells in peripheral nerves.
Gene editing, using the CRISPR/Cas9 approach, has been tested as a proof-of-concept in animal models of CMT1A. Again, the aim was to reduce the expression of the supernumerary allele(s) responsible for the disease. This was achieved in mouse (C22) model of CMT1A through the direct intraneural delivery of CRISPR/Cas9 designed to target TATA-box of the Pmp22 gene [40]. Although this an interesting and very promising approach, still the translation in clinical practice is limited by off target activity of the gene editing.
5. Conclusions and Warnings
We reported few of many examples of how the use of animal models, particularly rodents, has been useful in the field of CMT neuropathy, ranging from the understanding of developmental processes to the discovery of the pathogenetic mechanisms, till the development of promising future therapies. However, while animal models have been extremely useful to generate this knowledge, we should not forget that their use implies ethical aspects and that not always what is useful in mice or rats it is also working in human. The rule of 3Rs—Replacement, Reduction and Refinement—should constitute the basis for all the scientists working with animal models. Replacement, as the use of other methods whenever possible to replace the use of animals; Reduction, to minimize the number of animals to be used for obtaining a defined result; Refinement, to minimize animal sufferance during experimentation. Moreover, there are cases in which astonishing therapeutic results obtained in animal models (mostly rodents) are not reproduced in humans. Paradigmatic was the use of ascorbic acid to treat CMT1A neuropathy: while treatment in mouse model of CMT1A (C22) significantly ameliorated the neuropathic phenotype and reduced Pmp22 level [41], clinical trials in humans failed to show any effect in patients as well as effect on PMP22 expression [228,229,230,231]. This raises the argument that not always animal models (at least rodents) can reproduce human beings in all its aspects, but also that on occasion, preclinical studies in animal models are not carried out with the same rigor as in humans. In many cases, the number of treated animals is too small, scientists are not blinded to treatment, results are not confirmed to other laboratories, also due to limited funding. This aspect should be better considered by stakeholders as the investments necessary to perform clinical trials in humans are very expansive.
Author Contributions
Conceptualization, S.C.P.; methodology, S.C.P., L.B. and Y.M.F.; data curation, S.C.P., L.B. and Y.M.F.; writing—original draft preparation, S.C.P., L.B. and Y.M.F.; writing—review and editing, S.C.P., L.B. and Y.M.F.; funding acquisition, S.C.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Telethon Foundation, grant number GGP17009; Fondazione Regionale per la Ricerca Biomedica, grant number FRRB CP2_10/2018; Ministry of Health, grant number RF-2016-02361246; and the European Joint Programme on Rare Diseases, grant number EJPRD19-118.
Acknowledgments
We thank Alessandra Bolino and Maurizio D’Antonio for critical reading.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Loew, F.M.; Cohen, B.J. Laboratory Animal Medicine: Historical Perspectives. In Laboratory Animal Medicine; Elsevier: Amsterdam, The Netherlands, 2002; pp. 1–17. ISBN 978-0-12-263951-7. [Google Scholar]
- Rozzi, F.R.; Froment, A. Earliest Animal Cranial Surgery: From Cow to Man in the Neolithic. Sci. Rep. 2018, 8, 5536. [Google Scholar] [CrossRef] [Green Version]
- Perilli, L. Alcmeone di Crotone tra filosofia e scienza. Quad. Urbinati di Cult. Class. 2001, 69, 55. [Google Scholar] [CrossRef]
- Wills, A. Herophilus, Erasistratus, and the Birth of Neuroscience. Lancet 1999, 354, 1719–1720. [Google Scholar] [CrossRef]
- Aufderheide, A.C. The Scientific Study of Mummies; Cambridge University Press: Cambridge, UK, 2003. [Google Scholar]
- Goeke, J.E. The History of the Use of Animals in Research and the Development of the Animal Welfare Concept. J. Am. Coll. Toxicol. 1987, 6, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Franco, N. Animal Experiments in Biomedical Research: A Historical Perspective. Animals 2013, 3, 238–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vecchio, I.; Tornali, C.; Bragazzi, N.L.; Martini, M. The Discovery of Insulin: An Important Milestone in the History of Medicine. Front. Endocrinol. 2018, 9, 613. [Google Scholar] [CrossRef] [Green Version]
- Curtis, T. Monkeys, Viruses, and Vaccines. Lancet 2004, 364, 407–408. [Google Scholar] [CrossRef]
- Montinari, M.R. La Sperimentazione Animale Dall’antichità al Diciannovesimo Secolo. J. Hist. Med. 2017, 29, 467–480. [Google Scholar]
- Bryda, E.C. The Mighty Mouse: The Impact of Rodents on Advances in Biomedical Research. Mo. Med. 2013, 110, 207–211. [Google Scholar]
- Steensma, D.P.; Kyle, R.A.; Shampo, M.A. Abbie Lathrop, the “Mouse Woman of Granby”: Rodent Fancier and Accidental Genetics Pioneer. Mayo Clin. Proc. 2010, 85, e83. [Google Scholar] [CrossRef] [Green Version]
- Jaenisch, R.; Mintz, B. Simian Virus 40 DNA Sequences in DNA of Healthy Adult Mice Derived from Preimplantation Blastocysts Injected with Viral DNA. Proc. Natl. Acad. Sci. USA 1974, 71, 1250–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Nobel Assembly at Karolinska Institutet. The Nobel Prize in Physiology or Medicine. Available online: https://www.nobelprize.org/uploads/2018/06/press.pdf (accessed on 11 May 2021).
- Adli, M. The CRISPR Tool Kit for Genome Editing and Beyond. Nat. Commun. 2018, 9, 1911. [Google Scholar] [CrossRef] [PubMed]
- Orban, P.C.; Chui, D.; Marth, J.D. Tissue- and Site-Specific DNA Recombination in Transgenic Mice. Proc. Natl. Acad. Sci. USA 1992, 89, 6861–6865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pareyson, D.; Saveri, P.; Pisciotta, C. New Developments in Charcot–Marie–Tooth Neuropathy and Related Diseases. Curr. Opin. Neurol. 2017, 30, 471–480. [Google Scholar] [CrossRef]
- Laurá, M.; Pipis, M.; Rossor, A.M.; Reilly, M.M. Charcot–Marie–Tooth Disease and Related Disorders: An Evolving Landscape. Curr. Opin. Neurol. 2019, 32, 641–650. [Google Scholar] [CrossRef]
- Klein, C.J. Charcot-Marie-Tooth Disease and Other Hereditary Neuropathies. Contin. Lifelong Learn. Neurol. 2020, 26, 1224–1256. [Google Scholar] [CrossRef]
- Charcot, J.M.; Marie, P. Sur Une Forme Particulière d’atrophie Musculaire Progressive Souvent Familiale Débutant Par Les Pieds et Les Jambes et Atteignant plus Tard Les Mains. Rev. Med. Fr. 1886, 6, 97–138. [Google Scholar]
- Tooth, H.H. Peroneal Type of Progressive Muscle Atrophy. Ph.D. Thesis, University of Cambridge, Cambridge, UK, 1886. [Google Scholar]
- Marrosu, M.G.; Vaccargiu, S.; Marrosu, G.; Vannelli, A.; Cianchetti, C.; Muntoni, F. Charcot-Marie-Tooth Disease Type 2 Associated with Mutation of the Myelin Protein Zero Gene. Neurology 1998, 50, 1397–1401. [Google Scholar] [CrossRef]
- Shy, M.E.; Jáni, A.; Krajewski, K.; Grandis, M.; Lewis, R.A.; Li, J.; Shy, R.R.; Balsamo, J.; Lilien, J.; Garbern, J.Y.; et al. Phenotypic Clustering in MPZ Mutations. Brain 2004, 127, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Ropper, A.H.; Martin, S.A.; Joshua, K.P.; Sashank, P. Adams and Victor’s Principles of Neurology; McGraw Hill Medical: New York, NY, USA, 2019. [Google Scholar]
- Harding, A.E.; Thomas, P.K. The Clinical Features of Hereditary Motor and Sensory Neuropathy Types I and II. Brain 1980, 103, 259–280. [Google Scholar] [CrossRef] [PubMed]
- Bird, T.D.; Ott, J.; Giblett, E.R. Evidence for Linkage of Charcot-Marie-Tooth Neuropathy to the Duffy Locus on Chromosome 1. Am. J. Hum. Genet. 1982, 34, 388–394. [Google Scholar] [PubMed]
- Timmerman, V.; Strickland, A.; Züchner, S. Genetics of Charcot-Marie-Tooth (CMT) Disease within the Frame of the Human Genome Project Success. Genes 2014, 5, 13–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suter, U.; Scherer, S.S. Disease Mechanisms in Inherited Neuropathies. Nat. Rev. Neurosci. 2003, 4, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Nave, K.-A.; Sereda, M.W.; Ehrenreich, H. Mechanisms of Disease: Inherited Demyelinating Neuropathies—from Basic to Clinical Research. Nat. Rev. Neurol. 2007, 3, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Stavrou, M.; Sargiannidou, I.; Georgiou, E.; Kagiava, A.; Kleopa, K.A. Emerging Therapies for Charcot-Marie-Tooth Inherited Neuropathies. Int. J. Mol. Sci. 2021, 22, 6048. [Google Scholar] [CrossRef] [PubMed]
- Beijer, D.; Sisto, A.; Van Lent, J.; Baets, J.; Timmerman, V. Defects in Axonal Transport in Inherited Neuropathies. J. Neuromuscul. Dis. 2019, 6, 401–419. [Google Scholar] [CrossRef] [Green Version]
- Gentil, B.J.; Cooper, L. Molecular Basis of Axonal Dysfunction and Traffic Impairments in CMT. Brain Res. Bull. 2012, 88, 444–453. [Google Scholar] [CrossRef]
- Perea, J.; Robertson, A.; Tolmachova, T.; Muddle, J.; King, R.H.M.; Ponsford, S.; Thomas, P.K.; Huxley, C. Induced Myelination and Demyelination in a Conditional Mouse Model of Charcot-Marie-Tooth Disease Type 1A. Hum. Mol. Genet. 2001, 10, 1007–1018. [Google Scholar] [CrossRef] [Green Version]
- Boutary, S.; Caillaud, M.; El Madani, M.; Vallat, J.-M.; Loisel-Duwattez, J.; Rouyer, A.; Richard, L.; Gracia, C.; Urbinati, G.; Desmaële, D.; et al. Squalenoyl SiRNA PMP22 Nanoparticles Are Effective in Treating Mouse Models of Charcot-Marie-Tooth Disease Type 1 A. Commun. Biol. 2021, 4, 317. [Google Scholar] [CrossRef]
- Verhamme, C.; King, R.H.M.; ten Asbroek, A.L.M.A.; Muddle, J.R.; Nourallah, M.; Wolterman, R.; Baas, F.; van Schaik, I.N. Myelin and Axon Pathology in a Long-Term Study of PMP22 -Overexpressing Mice. J. Neuropathol. Exp. Neurol. 2011, 70, 386–398. [Google Scholar] [CrossRef] [Green Version]
- Huxley, C.; Passage, E.; Robertson, A.M.; Youl, B.; Huston, S.; Manson, A.; Sabéran-Djoniedi, D.; Figarella-Branger, D.; Pellissier, J.F.; Thomas, P.K.; et al. Correlation between Varying Levels of PMP22 Expression and the Degree of Demyelination and Reduction in Nerve Conduction Velocity in Transgenic Mice. Hum. Mol. Genet. 1998, 7, 449–458. [Google Scholar] [CrossRef] [Green Version]
- Huxley, C.; Passage, E.; Manson, A.; Putzu, G.; Figarella-Branger, D.; Pellissier, J.F.; Fontés, M. Construction of a Mouse Model of Charcot-Marie-Tooth Disease Type 1A by Pronuclear Injection of Human YAC DNA. Hum. Mol. Genet. 1996, 5, 563–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, N.; Choi, Y.I.; Jung, N.; Song, J.Y.; Bae, D.K.; Kim, M.C.; Lee, Y.J.; Song, H.; Kwak, G.; Jeong, S.; et al. A Novel Histone Deacetylase 6 Inhibitor Improves Myelination of Schwann Cells in a Model of Charcot–Marie–Tooth Disease Type 1A. Br. J. Pharmacol. 2020, 177, 5096–5113. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.T.; Damle, S.; Ikeda-Lee, K.; Kuntz, S.; Li, J.; Mohan, A.; Kim, A.; Hung, G.; Scheideler, M.A.; Scherer, S.S.; et al. PMP22 Antisense Oligonucleotides Reverse Charcot-Marie-Tooth Disease Type 1A Features in Rodent Models. J. Clin. Investig. 2017, 128, 359–368. [Google Scholar] [CrossRef]
- Lee, J.-S.; Lee, J.Y.; Song, D.W.; Bae, H.S.; Doo, H.M.; Yu, H.S.; Lee, K.J.; Kim, H.K.; Hwang, H.; Kwak, G.; et al. Targeted PMP22 TATA-Box Editing by CRISPR/Cas9 Reduces Demyelinating Neuropathy of Charcot-Marie-Tooth Disease Type 1A in Mice. Nucleic Acids Res. 2019, 48, 130–140. [Google Scholar] [CrossRef]
- Passage, E.; Norreel, J.C.; Noack-Fraissignes, P.; Sanguedolce, V.; Pizant, J.; Thirion, X.; Robaglia-Schlupp, A.; Pellissier, J.F.; Fontés, M. Ascorbic Acid Treatment Corrects the Phenotype of a Mouse Model of Charcot-Marie-Tooth Disease. Nat. Med. 2004, 10, 396–401. [Google Scholar] [CrossRef]
- Rangaraju, S.; Madorsky, I.; Pileggi, J.G.; Kamal, A.; Notterpek, L. Pharmacological Induction of the Heat Shock Response Improves Myelination in a Neuropathic Model. Neurobiol. Dis. 2008, 32, 105–115. [Google Scholar] [CrossRef] [Green Version]
- Chittoor-Vinod, V.G.; Bazick, H.; Todd, A.G.; Falk, D.; Morelli, K.H.; Burgess, R.W.; Foster, T.C.; Notterpek, L. HSP90 Inhibitor, NVP-AUY922, Improves Myelination in Vitro and Supports the Maintenance of Myelinated Axons in Neuropathic Mice. ACS Chem. Neurosci. 2019, 10, 2890–2902. [Google Scholar] [CrossRef] [PubMed]
- Fledrich, R.; Stassart, R.M.; Klink, A.; Rasch, L.M.; Prukop, T.; Haag, L.; Czesnik, D.; Kungl, T.; Abdelaal, T.A.M.; Keric, N.; et al. Soluble Neuregulin-1 Modulates Disease Pathogenesis in Rodent Models of Charcot-Marie-Tooth Disease 1A. Nat Med 2014, 20, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- Magyar, J.P.; Martini, R.; Ruelicke, T.; Aguzzi, A.; Adlkofer, K.; Dembic, Z.; Zielasek, J.; Toyka, K.V.; Suter, U. Impaired Differentiation of Schwann Cells in Transgenic Mice with Increased PMP22 Gene Dosage. J. Neurosci. 1996, 16, 5351–5360. [Google Scholar] [CrossRef] [Green Version]
- Sancho, S.; Magyar, J.P.; Aguzzi, A.; Suter1, U. Distal Axonopathy in Peripheral Nerves of PMP22-Mutant Mice. Brain 1999, 122, 1563–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, A.M.; Perea, J.; McGuigan, A.; King, R.H.M.; Muddle, J.R.; Gabreels-Festen, A.A.; Thomas, P.K.; Huxley, C. Comparison of a New Pmp22 Transgenic Mouse Line with Other Mouse Models and Human Patients with CMT1A*. J Anatomy 2002, 200, 377–390. [Google Scholar] [CrossRef]
- Sereda, M.W.; Meyer zu Hörste, G.; Suter, U.; Uzma, N.; Nave, K.-A. Therapeutic Administration of Progesterone Antagonist in a Model of Charcot-Marie-Tooth Disease (CMT-1A). Nat. Med. 2003, 9, 1533–1537. [Google Scholar] [CrossRef] [PubMed]
- Gautier, B.; Hajjar, H.; Soares, S.; Berthelot, J.; Deck, M.; Abbou, S.; Campbell, G.; Ceprian, M.; Gonzalez, S.; Fovet, C.-M.; et al. AAV2/9-Mediated Silencing of PMP22 Prevents the Development of Pathological Features in a Rat Model of Charcot-Marie-Tooth Disease 1 A. Nat. Commun. 2021, 12, 2356. [Google Scholar] [CrossRef]
- Zu Horste, G.M.; Prukop, T.; Liebetanz, D.; Mobius, W.; Nave, K.-A.; Sereda, M.W. Antiprogesterone Therapy Uncouples Axonal Loss from Demyelination in a Transgenic Rat Model of CMT1A Neuropathy. Ann. Neurol. 2007, 61, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Rünker, A.E.; Kobsar, I.; Fink, T.; Loers, G.; Tilling, T.; Putthoff, P.; Wessig, C.; Martini, R.; Schachner, M. Pathology of a Mouse Mutation in Peripheral Myelin Protein P0 Is Characteristic of a Severe and Early Onset Form of Human Charcot-Marie-Tooth Type 1B Disorder. J. Cell Biol. 2004, 165, 565–573. [Google Scholar] [CrossRef] [Green Version]
- Fratta, P.; Ornaghi, F.; Dati, G.; Zambroni, D.; Saveri, P.; Belin, S.; D’Adamo, P.; Shy, M.; Quattrini, A.; Laura Feltri, M.; et al. A Nonsense Mutation in Myelin Protein Zero Causes Congenital Hypomyelination Neuropathy through Altered P0 Membrane Targeting and Gain of Abnormal Function. Hum. Mol. Genet. 2019, 28, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Saporta, M.A.C.; Shy, B.R.; Patzko, A.; Bai, Y.; Pennuto, M.; Ferri, C.; Tinelli, E.; Saveri, P.; Kirschner, D.; Crowther, M.; et al. MpzR98C Arrests Schwann Cell Development in a Mouse Model of Early-Onset Charcot–Marie–Tooth Disease Type 1B. Brain 2012, 135, 2032–2047. [Google Scholar] [CrossRef]
- Patzkó, Á.; Bai, Y.; Saporta, M.A.; Katona, I.; Wu, X.; Vizzuso, D.; Feltri, M.L.; Wang, S.; Dillon, L.M.; Kamholz, J.; et al. Curcumin Derivatives Promote Schwann Cell Differentiation and Improve Neuropathy in R98C CMT1B Mice. Brain 2012, 135, 3551–3566. [Google Scholar] [CrossRef] [Green Version]
- Wrabetz, L.; D’Antonio, M.; Pennuto, M.; Dati, G.; Tinelli, E.; Fratta, P.; Previtali, S.C.; Imperiale, D.; Zielasek, J.; Toyka, K.; et al. Different Intracellular Pathomechanisms Produce Diverse Myelin Protein Zero Neuropathies in Transgenic Mice. J. Neurosci. 2006, 26, 2358–2368. [Google Scholar] [CrossRef]
- Pennuto, M.; Tinelli, E.; Malaguti, M.; Del Carro, U.; D’Antonio, M.; Ron, D.; Quattrini, A.; Feltri, M.L.; Wrabetz, L. Ablation of the UPR-Mediator CHOP Restores Motor Function and Reduces Demyelination in Charcot-Marie-Tooth 1B Mice. Neuron 2008, 57, 393–405. [Google Scholar] [CrossRef] [Green Version]
- Scapin, C.; Ferri, C.; Pettinato, E.; Bianchi, F.; Del Carro, U.; Feltri, M.L.; Kaufman, R.J.; Wrabetz, L.; D’Antonio, M. Phosphorylation of EIF2α Promotes Schwann Cell Differentiation and Myelination in CMT1B Mice with Activated UPR. J. Neurosci. 2020, 40, 8174–8187. [Google Scholar] [CrossRef]
- Das, I.; Krzyzosiak, A.; Schneider, K.; Wrabetz, L.; D’Antonio, M.; Barry, N.; Sigurdardottir, A.; Bertolotti, A. Preventing Proteostasis Diseases by Selective Inhibition of a Phosphatase Regulatory Subunit. Science 2015, 348, 239–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giese, K.P.; Martini, R.; Lemke, G.; Soriano, P.; Schachner, M. Mouse P0 Gene Disruption Leads to Hypomyelination, Abnormal Expression of Recognition Molecules, and Degeneration of Myelin and Axons. Cell 1992, 71, 565–576. [Google Scholar] [CrossRef]
- Scapin, C.; Ferri, C.; Pettinato, E.; Zambroni, D.; Bianchi, F.; Del Carro, U.; Belin, S.; Caruso, D.; Mitro, N.; Pellegatta, M.; et al. Enhanced Axonal Neuregulin-1 Type-III Signaling Ameliorates Neurophysiology and Hypomyelination in a Charcot–Marie–Tooth Type 1B Mouse Model. Hum. Mol. Genet. 2019, 28, 992–1006. [Google Scholar] [CrossRef]
- Martini, R.; Zielasek, J.; Toyka, K.V.; Giese, K.P.; Schachner, M. Protein Zero (P0)–Deficient Mice Show Myelin Degeneration in Peripheral Nerves Characteristic of Inherited Human Neuropathies. Nat. Genet. 1995, 11, 281–286. [Google Scholar] [CrossRef]
- Lee, S.M.; Sha, D.; Mohammed, A.A.; Asress, S.; Glass, J.D.; Chin, L.-S.; Li, L. Motor and Sensory Neuropathy Due to Myelin Infolding and Paranodal Damage in a Transgenic Mouse Model of Charcot–Marie–Tooth Disease Type 1C. Hum. Mol. Genet. 2013, 22, 1755–1770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Guariglia, S.; Yu, R.Y.L.; Li, W.; Brancho, D.; Peinado, H.; Lyden, D.; Salzer, J.; Bennett, C.; Chow, C.-W. Mutation of SIMPLE in Charcot–Marie–Tooth 1C Alters Production of Exosomes. MBoC 2013, 24, 1619–1637. [Google Scholar] [CrossRef] [PubMed]
- Schneider-Maunoury, S.; Topilko, P.; Seitanidou, T.; Levi, G.; Cohen-Tannoudji, M.; Pournin, S.; Babinet, C.; Charnay, P. Disruption of Krox-20 Results in Alteration of Rhombomeres 3 and 5 in the Developing Hindbrain. Cell 1993, 75, 1199–1214. [Google Scholar] [CrossRef]
- Suter, U.; Moskow, J.J.; Welcher, A.A.; Snipes, G.J.; Kosaras, B.; Sidman, R.L.; Buchberg, A.M.; Shooter, E.M. A Leucine-to-Proline Mutation in the Putative First Transmembrane Domain of the 22-KDa Peripheral Myelin Protein in the Trembler-J Mouse. Proc. Natl. Acad. Sci. USA 1992, 89, 4382–4386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suh, J.-G.; Ichihara, N.; Saigoh, K.; Nakabayashi, O.; Yamanishi, T.; Tanaka, K.; Wada, K.; Kikuchi, T. An In-Frame Deletion in Peripheral Myelin Protein-22 Gene Causes Hypomyelination and Cell Death of the Schwann Cells in the New Trembler Mutant Mice. Neuroscience 1997, 79, 735–744. [Google Scholar] [CrossRef]
- Isaacs, A.M.; Davies, K.E.; Hunter, A.J.; Nolan, P.M.; Vizor, L.; Peters, J.; Gale, D.G.; Kelsell, D.P.; Latham, I.D.; Chase, J.M.; et al. Identification of Two New Pmp22 Mouse Mutants Using Large-Scale Mutagenesis and a Novel Rapid Mapping Strategy. Hum. Mol. Genet. 2000, 9, 1865–1871. [Google Scholar] [CrossRef] [Green Version]
- Sahenk, Z.; Nagaraja, H.N.; McCracken, B.S.; King, W.M.; Freimer, M.L.; Cedarbaum, J.M.; Mendell, J.R. NT-3 Promotes Nerve Regeneration and Sensory Improvement in CMT1A Mouse Models and in Patients. Neurology 2005, 65, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Damián, J.P.; Vázquez Alberdi, L.; Canclini, L.; Rosso, G.; Bravo, S.O.; Martínez, M.; Uriarte, N.; Ruiz, P.; Calero, M.; Di Tomaso, M.V.; et al. Central Alteration in Peripheral Neuropathy of Trembler-J Mice: Hippocampal Pmp22 Expression and Behavioral Profile in Anxiety Tests. Biomolecules 2021, 11, 601. [Google Scholar] [CrossRef] [PubMed]
- Rangaraju, S.; Verrier, J.D.; Madorsky, I.; Nicks, J.; Dunn, W.A.; Notterpek, L. Rapamycin Activates Autophagy and Improves Myelination in Explant Cultures from Neuropathic Mice. J. Neurosci. 2010, 30, 11388–11397. [Google Scholar] [CrossRef] [Green Version]
- Li, J. ACE-083, a Locally-Acting GDF/Activin Ligand Trap, Augments Dorsiflexor Muscle Function in a Murine Model of Charcot- Marie-Tooth (CMT) Disease. In Proceedings of the PNS Meeting, Sitges, Spain, 8 July 2017. [Google Scholar]
- Madorsky, I.; Opalach, K.; Waber, A.; Verrier, J.D.; Solmo, C.; Foster, T.; Dunn, W.A.; Notterpek, L. Intermittent Fasting Alleviates the Neuropathic Phenotype in a Mouse Model of Charcot–Marie–Tooth Disease. Neurobiol. Dis. 2009, 34, 146–154. [Google Scholar] [CrossRef] [Green Version]
- Picci, C.; Wong, V.S.C.; Costa, C.J.; McKinnon, M.C.; Goldberg, D.C.; Swift, M.; Alam, N.M.; Prusky, G.T.; Shen, S.; Kozikowski, A.P.; et al. HDAC6 Inhibition Promotes α-Tubulin Acetylation and Ameliorates CMT2A Peripheral Neuropathy in Mice. Exp. Neurol. 2020, 328, 113281. [Google Scholar] [CrossRef]
- Cartoni, R.; Arnaud, E.; Médard, J.-J.; Poirot, O.; Courvoisier, D.S.; Chrast, R.; Martinou, J.-C. Expression of Mitofusin 2R94Q in a Transgenic Mouse Leads to Charcot–Marie–Tooth Neuropathy Type 2A. Brain 2010, 133, 1460–1469. [Google Scholar] [CrossRef] [Green Version]
- Khajavi, M.; Shiga, K.; Wiszniewski, W.; He, F.; Shaw, C.A.; Yan, J.; Wensel, T.G.; Snipes, G.J.; Lupski, J.R. Oral Curcumin Mitigates the Clinical and Neuropathologic Phenotype of the Trembler-J Mouse: A Potential Therapy for Inherited Neuropathy. Am. J. Hum. Genet. 2007, 81, 438–453. [Google Scholar] [CrossRef] [Green Version]
- Bernard-Marissal, N.; van Hameren, G.; Juneja, M.; Pellegrino, C.; Louhivuori, L.; Bartesaghi, L.; Rochat, C.; El Mansour, O.; Médard, J.-J.; Croisier, M.; et al. Altered Interplay between Endoplasmic Reticulum and Mitochondria in Charcot–Marie–Tooth Type 2A Neuropathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2328–2337. [Google Scholar] [CrossRef] [Green Version]
- Strickland, A.V.; Rebelo, A.P.; Zhang, F.; Price, J.; Bolon, B.; Silva, J.P.; Wen, R.; Züchner, S. Characterization of the Mitofusin 2 R94W Mutation in a Knock-in Mouse Model: Strickland et Al. J. Peripher. Nerv. Syst. 2014, 19, 152–164. [Google Scholar] [CrossRef]
- Bannerman, P.; Burns, T.; Xu, J.; Miers, L.; Pleasure, D. Mice Hemizygous for a Pathogenic Mitofusin-2 Allele Exhibit Hind Limb/Foot Gait Deficits and Phenotypic Perturbations in Nerve and Muscle. PLoS ONE 2016, 11, e0167573. [Google Scholar] [CrossRef] [PubMed]
- Detmer, S.A.; Velde, C.V.; Cleveland, D.W.; Chan, D.C. Hindlimb Gait Defects Due to Motor Axon Loss and Reduced Distal Muscles in a Transgenic Mouse Model of Charcot–Marie–Tooth Type 2A. Hum. Mol. Genet. 2008, 17, 367–375. [Google Scholar] [CrossRef]
- Zhou, Y.; Carmona, S.; Muhammad, A.K.M.G.; Bell, S.; Landeros, J.; Vazquez, M.; Ho, R.; Franco, A.; Lu, B.; Dorn, G.W.; et al. Restoring Mitofusin Balance Prevents Axonal Degeneration in a Charcot-Marie-Tooth Type 2A Model. J. Clin. Investig. 2019, 129, 1756–1771. [Google Scholar] [CrossRef] [PubMed]
- Poitelon, Y.; Kozlov, S.; Devaux, J.; Vallat, J.-M.; Jamon, M.; Roubertoux, P.; Rabarimeriarijaona, S.; Baudot, C.; Hamadouche, T.; Stewart, C.L.; et al. Behavioral and Molecular Exploration of the AR-CMT2A Mouse Model Lmna R298C/R298C. Neuromol. Med. 2012, 14, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Seo, A.J.; Shin, Y.H.; Lee, S.J.; Kim, D.; Park, B.S.; Kim, S.; Choi, K.H.; Jeong, N.Y.; Park, C.; Jang, J.-Y.; et al. A Novel Adenoviral Vector-Mediated Mouse Model of Charcot-Marie-Tooth Type 2D (CMT2D). J. Mol. Hist. 2014, 45, 121–128. [Google Scholar] [CrossRef]
- Achilli, F.; Bros-Facer, V.; Williams, H.P.; Banks, G.T.; AlQatari, M.; Chia, R.; Tucci, V.; Groves, M.; Nickols, C.D.; Seburn, K.L.; et al. An ENU-Induced Mutation in Mouse Glycyl-TRNA Synthetase (GARS) Causes Peripheral Sensory and Motor Phenotypes Creating a Model of Charcot-Marie-Tooth Type 2D Peripheral Neuropathy. Dis. Models Mech. 2009, 2, 359–373. [Google Scholar] [CrossRef] [Green Version]
- Benoy, V.; Van Helleputte, L.; Prior, R.; d’Ydewalle, C.; Haeck, W.; Geens, N.; Scheveneels, W.; Schevenels, B.; Cader, M.Z.; Talbot, K.; et al. HDAC6 Is a Therapeutic Target in Mutant GARS-Induced Charcot-Marie-Tooth Disease. Brain 2018, 141, 673–687. [Google Scholar] [CrossRef]
- Sleigh, J.N.; Mech, A.M.; Aktar, T.; Zhang, Y.; Schiavo, G. Altered Sensory Neuron Development in CMT2D Mice Is Site-Specific and Linked to Increased GlyRS Levels. Front. Cell. Neurosci. 2020, 14, 232. [Google Scholar] [CrossRef] [PubMed]
- Sleigh, J.N.; Mech, A.M.; Schiavo, G. Developmental Demands Contribute to Early Neuromuscular Degeneration in CMT2D Mice. Cell Death Dis. 2020, 11, 564. [Google Scholar] [CrossRef]
- Seburn, K.L.; Nangle, L.A.; Cox, G.A.; Schimmel, P.; Burgess, R.W. An Active Dominant Mutation of Glycyl-TRNA Synthetase Causes Neuropathy in a Charcot-Marie-Tooth 2D Mouse Model. Neuron 2006, 51, 715–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, W.; Bai, G.; Zhou, H.; Wei, N.; White, N.M.; Lauer, J.; Liu, H.; Shi, Y.; Dumitru, C.D.; Lettieri, K.; et al. CMT2D Neuropathy Is Linked to the Neomorphic Binding Activity of Glycyl-TRNA Synthetase. Nature 2015, 526, 710–714. [Google Scholar] [CrossRef] [Green Version]
- Mo, Z.; Zhao, X.; Liu, H.; Hu, Q.; Chen, X.-Q.; Pham, J.; Wei, N.; Liu, Z.; Zhou, J.; Burgess, R.W.; et al. Aberrant GlyRS-HDAC6 Interaction Linked to Axonal Transport Deficits in Charcot-Marie-Tooth Neuropathy. Nat. Commun. 2018, 9, 1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dequen, F.; Filali, M.; Larivière, R.C.; Perrot, R.; Hisanaga, S.-I.; Julien, J.-P. Reversal of Neuropathy Phenotypes in Conditional Mouse Model of Charcot–Marie–Tooth Disease Type 2E. Hum. Mol. Genet. 2010, 19, 2616–2629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adebola, A.A.; Di Castri, T.; He, C.-Z.; Salvatierra, L.A.; Zhao, J.; Brown, K.; Lin, C.-S.; Worman, H.J.; Liem, R.K.H. Neurofilament Light Polypeptide Gene N98S Mutation in Mice Leads to Neurofilament Network Abnormalities and a Charcot-Marie-Tooth Type 2E Phenotype. Hum. Mol. Genet. 2015, 24, 2163–2174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lancaster, E.; Li, J.; Hanania, T.; Liem, R.; Scheideler, M.A.; Scherer, S.S. Myelinated Axons Fail to Develop Properly in a Genetically Authentic Mouse Model of Charcot-Marie-Tooth Disease Type 2E. Exp. Neurol. 2018, 308, 13–25. [Google Scholar] [CrossRef]
- Morelli, K.H.; Griffin, L.B.; Pyne, N.K.; Wallace, L.M.; Fowler, A.M.; Oprescu, S.N.; Takase, R.; Wei, N.; Meyer-Schuman, R.; Mellacheruvu, D.; et al. Allele-Specific RNA Interference Prevents Neuropathy in Charcot-Marie-Tooth Disease Type 2D Mouse Models. J. Clin. Investig. 2019, 129, 5568–5583. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, A.K.; Renusch, S.R.; Naiman, N.E.; Gu, S.; Sneh, A.; Arnold, W.D.; Sahenk, Z.; Kolb, S.J. Mutant HSPB1 Overexpression in Neurons Is Sufficient to Cause Age-Related Motor Neuronopathy in Mice. Neurobiol. Dis. 2012, 47, 163–173. [Google Scholar] [CrossRef] [Green Version]
- d’Ydewalle, C.; Krishnan, J.; Chiheb, D.M.; Van Damme, P.; Irobi, J.; Kozikowski, A.P.; Berghe, P.V.; Timmerman, V.; Robberecht, W.; Van Den Bosch, L. HDAC6 Inhibitors Reverse Axonal Loss in a Mouse Model of Mutant HSPB1–Induced Charcot-Marie-Tooth Disease. Nat. Med. 2011, 17, 968–974. [Google Scholar] [CrossRef]
- Lee, J.; Jung, S.-C.; Joo, J.; Choi, Y.-R.; Moon, H.W.; Kwak, G.; Yeo, H.K.; Lee, J.-S.; Ahn, H.-J.; Jung, N.; et al. Overexpression of Mutant HSP27 Causes Axonal Neuropathy in Mice. J. Biomed. Sci. 2015, 22, 43. [Google Scholar] [CrossRef] [Green Version]
- Bouhy, D.; Geuens, T.; De Winter, V.; Almeida-Souza, L.; Katona, I.; Weis, J.; Hochepied, T.; Goossens, S.; Haigh, J.J.; Janssens, S.; et al. Characterization of New Transgenic Mouse Models for Two Charcot-Marie-Tooth-Causing HspB1 Mutations Using the Rosa26 Locus. JND 2016, 3, 183–200. [Google Scholar] [CrossRef] [PubMed]
- Barneo-Muñoz, M.; Juárez, P.; Civera-Tregón, A.; Yndriago, L.; Pla-Martin, D.; Zenker, J.; Cuevas-Martín, C.; Estela, A.; Sánchez-Aragó, M.; Forteza-Vila, J.; et al. Lack of GDAP1 Induces Neuronal Calcium and Mitochondrial Defects in a Knockout Mouse Model of Charcot-Marie-Tooth Neuropathy. PLoS Genet. 2015, 11, e1005115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benoy, V.; Vanden Berghe, P.; Jarpe, M.; Van Damme, P.; Robberecht, W.; Van Den Bosch, L. Development of Improved HDAC6 Inhibitors as Pharmacological Therapy for Axonal Charcot–Marie–Tooth Disease. Neurotherapeutics 2017, 14, 417–428. [Google Scholar] [CrossRef] [Green Version]
- Sanbe, A.; Marunouchi, T.; Abe, T.; Tezuka, Y.; Okada, M.; Aoki, S.; Tsumura, H.; Yamauchi, J.; Tanonaka, K.; Nishigori, H.; et al. Phenotype of Cardiomyopathy in Cardiac-Specific Heat Shock Protein B8 K141N Transgenic Mouse. J. Biol. Chem. 2013, 288, 8910–8921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Zhang, F.; Li, X.; Huang, S.; Zi, X.; Liu, T.; Liu, S.; Li, X.; Xia, K.; Pan, Q.; et al. A Novel Transgenic Mouse Model of Chinese Charcot-Marie-Tooth Disease Type 2L. Neural Regen. Res. 2014, 9, 413. [Google Scholar] [CrossRef] [PubMed]
- Bouhy, D.; Juneja, M.; Katona, I.; Holmgren, A.; Asselbergh, B.; De Winter, V.; Hochepied, T.; Goossens, S.; Haigh, J.J.; Libert, C.; et al. A Knock-in/Knock-out Mouse Model of HSPB8-Associated Distal Hereditary Motor Neuropathy and Myopathy Reveals Toxic Gain-of-Function of Mutant Hspb8. Acta Neuropathol. 2018, 135, 131–148. [Google Scholar] [CrossRef]
- Sabblah, T.T.; Nandini, S.; Ledray, A.P.; Pasos, J.; Calderon, J.L.C.; Love, R.; King, L.E.; King, S.J. A Novel Mouse Model Carrying a Human Cytoplasmic Dynein Mutation Shows Motor Behavior Deficits Consistent with Charcot-Marie-Tooth Type 2O Disease. Sci. Rep. 2018, 8, 1739. [Google Scholar] [CrossRef] [Green Version]
- Nandini, S.; Conley Calderon, J.L.; Sabblah, T.T.; Love, R.; King, L.E.; King, S.J. Mice with an Autosomal Dominant Charcot-Marie-Tooth Type 2O Disease Mutation in Both Dynein Alleles Display Severe Moto-Sensory Phenotypes. Sci. Rep. 2019, 9, 11979. [Google Scholar] [CrossRef]
- Bogdanik, L.P.; Sleigh, J.N.; Tian, C.; Samuels, M.E.; Bedard, K.; Seburn, K.L.; Burgess, R.W. Loss of the E3 Ubiquitin Ligase LRSAM1 Sensitizes Peripheral Axons to Degeneration in a Mouse Model of Charcot-Marie-Tooth Disease. Dis. Models Mech. 2013, 6, 780–792. [Google Scholar] [CrossRef] [Green Version]
- Luan, C.; Guo, W.; Chen, L.; Wei, X.; He, Y.; Chen, Y.; Dang, S.; Prior, R.; Li, X.; Kuang, Y.; et al. CMT2Q-Causing Mutation in the Dhtkd1 Gene Lead to Sensory Defects, Mitochondrial Accumulation and Altered Metabolism in a Knock-in Mouse Model. Acta Neuropathol. Commun. 2020, 8, 32. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.-Y.; Zhu, H.; Shen, Y.; Wan, Y.-H.; Tu, X.-D.; Wu, W.-T.; Tang, L.; Zhang, H.-X.; Lu, S.-Y.; Jin, X.-L.; et al. DHTKD1 Deficiency Causes Charcot-Marie-Tooth Disease in Mice. Mol. Cell Biol. 2018, 38. [Google Scholar] [CrossRef] [Green Version]
- Niemann, A.; Huber, N.; Wagner, K.M.; Somandin, C.; Horn, M.; Lebrun-Julien, F.; Angst, B.; Pereira, J.A.; Halfter, H.; Welzl, H.; et al. The Gdap1 Knockout Mouse Mechanistically Links Redox Control to Charcot–Marie–Tooth Disease. Brain 2014, 137, 668–682. [Google Scholar] [CrossRef]
- Bolino, A.; Bolis, A.; Previtali, S.C.; Dina, G.; Bussini, S.; Dati, G.; Amadio, S.; Del Carro, U.; Mruk, D.D.; Feltri, M.L.; et al. Disruption of Mtmr2 Produces CMT4B1-like Neuropathy with Myelin Outfolding and Impaired Spermatogenesis. J. Cell Biol. 2004, 167, 711–721. [Google Scholar] [CrossRef]
- Bonneick, S.; Boentert, M.; Berger, P.; Atanasoski, S.; Mantei, N.; Wessig, C.; Toyka, K.V.; Young, P.; Suter, U. An Animal Model for Charcot–Marie–Tooth Disease Type 4B1. Hum. Mol. Genet. 2005, 14, 3685–3695. [Google Scholar] [CrossRef] [Green Version]
- Bolino, A.; Piguet, F.; Alberizzi, V.; Pellegatta, M.; Rivellini, C.; Guerrero-Valero, M.; Noseda, R.; Brombin, C.; Nonis, A.; D’Adamo, P.; et al. Niacin-mediated Tace Activation Ameliorates CMT Neuropathies with Focal Hypermyelination. EMBO Mol. Med. 2016, 8, 1438–1454. [Google Scholar] [CrossRef] [PubMed]
- Robinson, F.L.; Niesman, I.R.; Beiswenger, K.K.; Dixon, J.E. Loss of the Inactive Myotubularin-Related Phosphatase Mtmr13 Leads to a Charcot-Marie-Tooth 4B2-like Peripheral Neuropathy in Mice. Proc. Natl. Acad. Sci. USA 2008, 105, 4916–4921. [Google Scholar] [CrossRef] [Green Version]
- Arnaud, E.; Zenker, J.; de Preux Charles, A.-S.; Stendel, C.; Roos, A.; Medard, J.-J.; Tricaud, N.; Kleine, H.; Luscher, B.; Weis, J.; et al. SH3TC2/KIAA1985 Protein Is Required for Proper Myelination and the Integrity of the Node of Ranvier in the Peripheral Nervous System. Proc. Natl. Acad. Sci. USA 2009, 106, 17528–17533. [Google Scholar] [CrossRef] [Green Version]
- Cipriani, S.; Phan, V.; Médard, J.-J.; Horvath, R.; Lochmüller, H.; Chrast, R.; Roos, A.; Spendiff, S. Neuromuscular Junction Changes in a Mouse Model of Charcot-Marie-Tooth Disease Type 4C. IJMS 2018, 19, 4072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiza, N.; Georgiou, E.; Kagiava, A.; Médard, J.-J.; Richter, J.; Tryfonos, C.; Sargiannidou, I.; Heslegrave, A.J.; Rossor, A.M.; Zetterberg, H.; et al. Gene Replacement Therapy in a Model of Charcot-Marie-Tooth 4C Neuropathy. Brain 2019, 142, 1227–1241. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, C.S.; Sherman, D.L.; Fleetwood-Walker, S.M.; Cottrell, D.F.; Tait, S.; Garry, E.M.; Wallace, V.C.J.; Ure, J.; Griffiths, I.R.; Smith, A.; et al. Peripheral Demyelination and Neuropathic Pain Behavior in Periaxin-Deficient Mice. Neuron 2000, 26, 523–531. [Google Scholar] [CrossRef] [Green Version]
- Chow, C.Y.; Zhang, Y.; Dowling, J.J.; Jin, N.; Adamska, M.; Shiga, K.; Szigeti, K.; Shy, M.E.; Li, J.; Zhang, X.; et al. Mutation of FIG4 Causes Neurodegeneration in the Pale Tremor Mouse and Patients with CMT4J. Nature 2007, 448, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Wu, T.; Hu, X.; Wang, T. Efficacy and Safety of Botulinum Toxin Type A for Upper Limb Spasticity after Stroke or Traumatic Brain Injury: A Systematic Review with Meta-Analysis and Trial Sequential Analysis. Eur. J. Phys. Rehabil. Med. 2017, 53, 256–267. [Google Scholar] [CrossRef]
- Hu, B.; McCollum, M.; Ravi, V.; Arpag, S.; Moiseev, D.; Castoro, R.; Mobley, B.; Burnette, B.; Siskind, C.; Day, J.; et al. Myelin Abnormality in Charcot-Marie-Tooth Type 4J Recapitulates Features of Acquired Demyelination: Demyelination in CMT4J. Ann. Neurol. 2018, 83, 756–770. [Google Scholar] [CrossRef]
- Mones, S.; Bordignon, B.; Fontes, M. Connexin 32 Is Involved in Mitosis. Glia 2012, 60, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Mones, S.; Bordignon, B.; Peiretti, F.; Landrier, J.F.; Gess, B.; Bourguignon, J.J.; Bihel, F.; Fontés, M. CamKII Inhibitors Reduce Mitotic Instability, Connexon Anomalies and Progression of the in Vivo Behavioral Phenotype in Transgenic Animals Expressing a Mutated Gjb1 Gene. Front. Neurosci. 2014, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagiava, A.; Karaiskos, C.; Richter, J.; Tryfonos, C.; Jennings, M.J.; Heslegrave, A.J.; Sargiannidou, I.; Stavrou, M.; Zetterberg, H.; Reilly, M.M.; et al. AAV9-Mediated Schwann Cell-Targeted Gene Therapy Rescues a Model of Demyelinating Neuropathy. Gene Ther. 2021, 1–17. [Google Scholar] [CrossRef]
- Nelles, E.; Butzler, C.; Jung, D.; Temme, A.; Gabriel, H.D.; Dahl, U.; Traub, O.; Stumpel, F.; Jungermann, K.; Zielasek, J.; et al. Defective Propagation of Signals Generated by Sympathetic Nerve Stimulation in the Liver of Connexin32-Deficient Mice. Proc. Natl. Acad. Sci. USA 1996, 93, 9565–9570. [Google Scholar] [CrossRef] [Green Version]
- Anzini, P.; Neuberg, D.H.-H.; Schachner, M.; Nelles, E.; Willecke, K.; Zielasek, J.; Toyka, K.V.; Suter, U.; Martini, R. Structural Abnormalities and Deficient Maintenance of Peripheral Nerve Myelin in Mice Lacking the Gap Junction Protein Connexin 32. J. Neurosci. 1997, 17, 4545–4551. [Google Scholar] [CrossRef] [PubMed]
- Kagiava, A.; Sargiannidou, I.; Theophilidis, G.; Karaiskos, C.; Richter, J.; Bashiardes, S.; Schiza, N.; Nearchou, M.; Christodoulou, C.; Scherer, S.S.; et al. Intrathecal Gene Therapy Rescues a Model of Demyelinating Peripheral Neuropathy. Proc. Natl. Acad. Sci. USA 2016, 113, E2421–E2429. [Google Scholar] [CrossRef] [Green Version]
- Ozes, B.; Myers, M.; Moss, K.; Mckinney, J.; Ridgley, A.; Chen, L.; Bai, S.; Abrams, C.K.; Freidin, M.M.; Mendell, J.R.; et al. AAV1.NT-3 Gene Therapy for X-Linked Charcot–Marie–Tooth Neuropathy Type 1. Gene Ther 2021. [Google Scholar] [CrossRef]
- Kobsar, I.; Berghoff, M.; Samsam, M.; Wessig, C.; Mäurer, M.; Toyka, K.V.; Martini, R. Preserved Myelin Integrity and Reduced Axonopathy in Connexin32-Deficient Mice Lacking the Recombination Activating Gene-1. Brain 2003, 126, 804–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherer, S.S.; Xu, Y.-T.; Nelles, E.; Fischbeck, K.; Willecke, K.; Bone, L.J. Connexin32-Null Mice Develop Demyelinating Peripheral Neuropathy. Glia 1998, 24, 8–20. [Google Scholar] [CrossRef]
- Song, G.J.; Gupta, D.P.; Rahman, M.H.; Park, H.T.; Al Ghouleh, I.; Bisello, A.; Lee, M.; Park, J.; Park, H.H.; Jun, J.H.; et al. Loss-of-function of EBP50 Is a New Cause of Hereditary Peripheral Neuropathy: EBP50 Functions in Peripheral Nerve System. Glia 2020, 68, s344–s359. [Google Scholar] [CrossRef]
- Huang, C.; Shen, Z.R.; Huang, J.; Sun, S.C.; Ma, D.; Li, M.Y.; Wang, Z.K.; Zheng, Y.C.; Zheng, Z.J.; He, F.; et al. C1orf194 Deficiency Leads to Incomplete Early Embryonic Lethality and Dominant Intermediate Charcot–Marie–Tooth Disease in a Knockout Mouse Model. Hum. Mol. Genet. 2020, 29, 2471–2480. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-C.; Ma, D.; Li, M.-Y.; Zhang, R.-X.; Huang, C.; Huang, H.-J.; Xie, Y.; Wang, Z.-J.; Liu, J.; Cai, D.-C.; et al. Mutations in C1orf194, Encoding a Calcium Regulator, Cause Dominant Charcot-Marie-Tooth Disease. Brain 2019, 142, 2215–2229. [Google Scholar] [CrossRef]
- Pereira, J.A.; Gerber, J.; Ghidinelli, M.; Gerber, D.; Tortola, L.; Ommer, A.; Bachofner, S.; Santarella, F.; Tinelli, E.; Lin, S.; et al. Mice Carrying an Analogous Heterozygous Dynamin 2 K562E Mutation That Causes Neuropathy in Humans Develop Predominant Characteristics of a Primary Myopathy. Hum. Mol. Genet. 2020, 29, 1253–1273. [Google Scholar] [CrossRef]
- Lee, S.; Panthi, S.; Jo, H.; Cho, J.; Kim, M.-S.; Jeong, N.; Jung, J.; Huh, Y. Anatomical Distributional Defects in Mutant Genes Associated with Dominant Intermediate Charcot-Marie-Tooth Disease Type C in an Adenovirus-Mediated Mouse Model. Neural Regen. Res. 2017, 12, 486. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Sarute, N.; Lancaster, E.; Otkiran-Clare, G.; Fagla, B.M.; Ross, S.R.; Scherer, S.S. A Recessive Trim2 Mutation Causes an Axonal Neuropathy in Mice. Neurobiol. Dis. 2020, 140, 104845. [Google Scholar] [CrossRef] [PubMed]
- Lupski, J.R.; de Oca-Luna, R.M.; Slaugenhaupt, S.; Pentao, L.; Guzzetta, V.; Trask, B.J.; Saucedo-Cardenas, O.; Barker, D.F.; Killian, J.M.; Garcia, C.A.; et al. DNA Duplication Associated with Charcot-Marie-Tooth Disease Type 1A. Cell 1991, 66, 219–232. [Google Scholar] [CrossRef]
- Raeymaekers, P.; Timmerman, V.; Nelis, E.; De Jonghe, P.; Hoogenduk, J.E.; Baas, F.; Barker, D.F.; Martin, J.J.; De Visser, M.; Bolhuis, P.A.; et al. Duplication in Chromosome 17p11.2 in Charcot-Marie-Tooth Neuropathy Type 1a (CMT 1a). Neuromuscul. Disord. 1991, 1, 93–97. [Google Scholar] [CrossRef]
- Sereda, M.; Griffiths, I.; Pühlhofer, A.; Stewart, H.; Rossner, M.J.; Zimmermann, F.; Magyar, J.P.; Schneider, A.; Hund, E.; Meinck, H.-M.; et al. A Transgenic Rat Model of Charcot-Marie-Tooth Disease. Neuron 1996, 16, 1049–1060. [Google Scholar] [CrossRef] [Green Version]
- Jouaud, M.; Mathis, S.; Richard, L.; Lia, A.-S.; Magy, L.; Vallat, J.-M. Rodent Models with Expression of PMP22: Relevance to Dysmyelinating CMT and HNPP. J. Neurol. Sci. 2019, 398, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Niemann, S.; Sereda, M.W.; Suter, U.; Griffiths, I.R.; Nave, K.-A. Uncoupling of Myelin Assembly and Schwann Cell Differentiation by Transgenic Overexpression of Peripheral Myelin Protein 22. J. Neurosci. 2000, 20, 4120–4128. [Google Scholar] [CrossRef]
- Robertson, A.M.; Huxley, C.; King, R.H.M.; Thomas, P.K. Development of Early Postnatal Peripheral Nerve Abnormalities in Trembler-J and PMP22 Transgenic Mice. J. Anat. 1999, 195, 331–339. [Google Scholar] [CrossRef]
- Chance, P.F.; Alderson, M.K.; Leppig, K.A.; Lensch, M.W.; Matsunami, N.; Smith, B.; Swanson, P.D.; Odelberg, S.J.; Disteche, C.M.; Bird, T.D. DNA Deletion Associated with Hereditary Neuropathy with Liability to Pressure Palsies. Cell 1993, 72, 143–151. [Google Scholar] [CrossRef]
- Adlkofer, K.; Martini, R.; Aguzzi, A.; Zielasek, J.; Toyka, K.V.; Suter, U. Hypermyelination and Demyelinating Peripheral Neuropathy in Pmp22-Deficient Mice. Nat. Genet. 1995, 11, 274–280. [Google Scholar] [CrossRef]
- Adlkofer, K.; Frei, R.; Neuberg, D.H.-H.; Zielasek, J.; Toyka, K.V.; Suter, U. Heterozygous Peripheral Myelin Protein 22-Deficient Mice Are Affected by a Progressive Demyelinating Tomaculous Neuropathy. J. Neurosci. 1997, 17, 4662–4671. [Google Scholar] [CrossRef]
- Bai, Y.; Zhang, X.; Katona, I.; Saporta, M.A.; Shy, M.E.; O’Malley, H.A.; Isom, L.L.; Suter, U.; Li, J. Conduction Block in PMP22 Deficiency. J. Neurosci. 2010, 30, 600–608. [Google Scholar] [CrossRef] [Green Version]
- Suter, U.; Welcher, A.A.; Özcelik, T.; Snipes, G.J.; Kosaras, B.; Francke, U.; Billings-Gagliardi, S.; Sidman, R.L.; Shooter, E.M. Trembler Mouse Carries a Point Mutation in a Myelin Gene. Nature 1992, 356, 241–244. [Google Scholar] [CrossRef]
- D’Urso, D.; Prior, R.; Greiner–Petter, R.; Gabreëls–Festen, A.A.W.M.; Müller, H.W. Overloaded Endoplasmic Reticulum–Golgi Compartments, a Possible Pathomechanism of Peripheral Neuropathies Caused by Mutations of the Peripheral Myelin Protein PMP22. J. Neurosci. 1998, 18, 731–740. [Google Scholar] [CrossRef]
- Naef, R.; Suter, U. Impaired Intracellular Trafficking Is a Common Disease Mechanism OfPMP22Point Mutations in Peripheral Neuropathies. Neurobiol. Dis. 1999, 6, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Colby, J.; Nicholson, R.; Dickson, K.M.; Orfali, W.; Naef, R.; Suter, U.; Snipes, G.J. PMP22 Carrying the Trembler or Trembler-J Mutation Is Intracellularly Retained in Myelinating Schwann Cells. Neurobiol. Dis. 2000, 7, 561–573. [Google Scholar] [CrossRef] [Green Version]
- Tobler, A.R.; Liu, N.; Mueller, L.; Shooter, E.M. Differential Aggregation of the Trembler and Trembler J Mutants of Peripheral Myelin Protein 22. Proc. Natl. Acad. Sci. USA 2002, 99, 483–488. [Google Scholar] [CrossRef] [Green Version]
- Myers, J.K.; Mobley, C.K.; Sanders, C.R. The Peripheral Neuropathy-Linked Trembler and Trembler-J Mutant Forms of Peripheral Myelin Protein 22 Are Folding-Destabilized. Biochemistry 2008, 47, 10620–10629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakakura, M.; Hadziselimovic, A.; Wang, Z.; Schey, K.L.; Sanders, C.R. Structural Basis for the Trembler-J Phenotype of Charcot-Marie-Tooth Disease. Structure 2011, 19, 1160–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlebach, J.P.; Narayan, M.; Alford, C.; Mittendorf, K.F.; Carter, B.D.; Li, J.; Sanders, C.R. Conformational Stability and Pathogenic Misfolding of the Integral Membrane Protein PMP22. J. Am. Chem. Soc. 2015, 137, 8758–8768. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, Y.; Pehlivan, D.; Wiszniewski, W.; Beck, C.R.; Snipes, G.J.; Lupski, J.R.; Khajavi, M. Curcumin Facilitates a Transitory Cellular Stress Response in Trembler-J Mice. Hum. Mol. Genet. 2013, 22, 4698–4705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanoye, C.G.; Sakakura, M.; Follis, R.M.; Trevisan, A.J.; Narayan, M.; Li, J.; Sanders, C.R.; Carter, B.D. Peripheral Myelin Protein 22 Modulates Store-Operated Calcium Channel Activity, Providing Insights into Charcot-Marie-Tooth Disease Etiology. J. Biol. Chem. 2019, 294, 12054–12065. [Google Scholar] [CrossRef]
- Greenfield, S.; Brostoff, S.; Eylar, E.H.; Morell, P. Protein Composition of Myelin of the Peripheral Nervous System. J. Neurochem. 1973, 20, 1207–1216. [Google Scholar] [CrossRef]
- Shapiro, L.; Doyle, J.P.; Hensley, P.; Colman, D.R.; Hendrickson, W.A. Crystal Structure of the Extracellular Domain from P0, the Major Structural Protein of Peripheral Nerve Myelin. Neuron 1996, 17, 435–449. [Google Scholar] [CrossRef] [Green Version]
- Howard, P.; Feely, S.M.E.; Grider, T.; Bacha, A.; Scarlato, M.; Fazio, R.; Quattrini, A.; Shy, M.E.; Previtali, S.C. Loss of Function MPZ Mutation Causes Milder CMT1B Neuropathy. J. Peripher. Nerv. Syst. 2021, 26, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Frei, R.; Mötzing, S.; Kinkelin, I.; Schachner, M.; Koltzenburg, M.; Martini, R. Loss of Distal Axons and Sensory Merkel Cells and Features Indicative of Muscle Denervation in Hindlimbs of P0-Deficient Mice. J. Neurosci. 1999, 19, 6058–6067. [Google Scholar] [CrossRef] [Green Version]
- Baets, J.; Timmerman, V. Inherited Peripheral Neuropathies: A Myriad of Genes and Complex Phenotypes. Brain 2011, 134, 1587–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prada, V.; Passalacqua, M.; Bono, M.; Luzzi, P.; Scazzola, S.; Nobbio, L.A.; Capponi, S.; Bellone, E.; Mandich, P.; Mancardi, G.; et al. Gain of Glycosylation: A New Pathomechanism of Myelin Protein Zero Mutations. Ann. Neurol. 2012, 71, 427–431. [Google Scholar] [CrossRef] [Green Version]
- Grandis, M.; Vigo, T.; Passalacqua, M.; Jain, M.; Scazzola, S.; La Padula, V.; Brucal, M.; Benvenuto, F.; Nobbio, L.; Cadoni, A.; et al. Different Cellular and Molecular Mechanisms for Early and Late-Onset Myelin Protein Zero Mutations. Hum. Mol. Genet. 2008, 17, 1877–1889. [Google Scholar] [CrossRef] [PubMed]
- Callegari, I.; Gemelli, C.; Geroldi, A.; Veneri, F.; Mandich, P.; D’Antonio, M.; Pareyson, D.; Shy, M.E.; Schenone, A.; Prada, V.; et al. Mutation Update for Myelin Protein Zero-Related Neuropathies and the Increasing Role of Variants Causing a Late-Onset Phenotype. J. Neurol. 2019, 266, 2629–2645. [Google Scholar] [CrossRef]
- Sanmaneechai, O.; Feely, S.; Scherer, S.S.; Herrmann, D.N.; Burns, J.; Muntoni, F.; Li, J.; Siskind, C.E.; Day, J.W.; Laura, M.; et al. Genotype–Phenotype Characteristics and Baseline Natural History of Heritable Neuropathies Caused by Mutations in the MPZ Gene. Brain 2015, 138, 3180–3192. [Google Scholar] [CrossRef] [Green Version]
- Previtali, S.C.; Quattrini, A.; Fasolini, M.; Panzeri, M.C.; Villa, A.; Filbin, M.T.; Li, W.; Chiu, S.-Y.; Messing, A.; Wrabetz, L.; et al. Epitope-Tagged P0Glycoprotein Causes Charcot-Marie-Tooth–Like Neuropathy in Transgenic Mice. J. Cell Biol. 2000, 151, 1035–1046. [Google Scholar] [CrossRef]
- D’Antonio, M.; Musner, N.; Scapin, C.; Ungaro, D.; Del Carro, U.; Ron, D.; Feltri, M.L.; Wrabetz, L. Resetting Translational Homeostasis Restores Myelination in Charcot-Marie-Tooth Disease Type 1B Mice. J. Exp. Med. 2013, 210, 821–838. [Google Scholar] [CrossRef] [Green Version]
- Miller, L.J.; Patzko, A.; Lewis, R.A.; Shy, M.E. Phenotypic Presentation of the Ser63Del MPZ Mutation. J. Peripher. Nerv. Syst. 2012, 17, 197–200. [Google Scholar] [CrossRef] [Green Version]
- Sidoli, M.; Musner, N.; Silvestri, N.; Ungaro, D.; D’Antonio, M.; Cavener, D.R.; Feltri, M.L.; Wrabetz, L. Ablation of Perk in Schwann Cells Improves Myelination in the S63del Charcot-Marie-Tooth 1B Mouse. J. Neurosci. 2016, 36, 11350–11361. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Wu, X.; Brennan, K.M.; Wang, D.S.; D’Antonio, M.; Moran, J.; Svaren, J.; Shy, M.E. Myelin Protein Zero Mutations and the Unfolded Protein Response in Charcot Marie Tooth Disease Type 1B. Ann. Clin. Transl. Neurol. 2018, 5, 445–455. [Google Scholar] [CrossRef]
- Fratta, P.; Saveri, P.; Zambroni, D.; Ferri, C.; Tinelli, E.; Messing, A.; D’Antonio, M.; Feltri, M.L.; Wrabetz, L. P0S63del Impedes the Arrival of Wild-Type P0 Glycoprotein to Myelin in CMT1B Mice. Hum. Mol. Genet. 2011, 20, 2081–2090. [Google Scholar] [CrossRef] [Green Version]
- Shy, M.E. Peripheral Neuropathies Caused by Mutations in the Myelin Protein Zero. J. Neurol. Sci. 2006, 242, 55–66. [Google Scholar] [CrossRef]
- Wrabetz, L.; Feltri, M.L.; Quattrini, A.; Imperiale, D.; Previtali, S.; D’Antonio, M.; Martini, R.; Yin, X.; Trapp, B.D.; Zhou, L.; et al. P0 Glycoprotein Overexpression Causes Congenital Hypomyelination of Peripheral Nerves. J. Cell Biol. 2000, 148, 1021–1034. [Google Scholar] [CrossRef] [Green Version]
- Rossor, A.M.; Polke, J.M.; Houlden, H.; Reilly, M.M. Clinical Implications of Genetic Advances in Charcot–Marie–Tooth Disease. Nat. Rev. Neurol. 2013, 9, 562–571. [Google Scholar] [CrossRef]
- McLellan, M.A.; Rosenthal, N.A.; Pinto, A.R. Cre-Lox P-Mediated Recombination: General Principles and Experimental Considerations: Cre- Lox P-Mediated Recombination. Curr. Protoc. Mouse Biol. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Previtali, S.C.; Quattrini, A.; Bolino, A. Charcot–Marie–Tooth Type 4B Demyelinating Neuropathy: Deciphering the Role of MTMR Phosphatases. Expert Rev. Mol. Med. 2007, 9, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, A.; Murai, Y.; Ikeda, M.; Fujita, T.; Furuya, H.; Kuroiwa, Y. Autosomal Recessive Motor and Sensory Neuropathy with Excessive Myelin Outfolding. Muscle Nerve 1989, 12, 568–575. [Google Scholar] [CrossRef]
- Feltri, M.L.; D’Antonio, M.; Previtali, S.; Fasolini, M.; Messing, A.; Wrabetz, L. P0-Cre Transgenic Mice for Inactivation of Adhesion Molecules in Schwann Cells. Ann. N. Y. Acad. Sci. 1999, 883, 116–123. [Google Scholar] [CrossRef]
- Bolis, A.; Coviello, S.; Bussini, S.; Dina, G.; Pardini, C.; Previtali, S.C.; Malaguti, M.; Morana, P.; Del Carro, U.; Feltri, M.L.; et al. Loss of Mtmr2 Phosphatase in Schwann Cells But Not in Motor Neurons Causes Charcot-Marie-Tooth Type 4B1 Neuropathy with Myelin Outfoldings. J. Neurosci. 2005, 25, 8567–8577. [Google Scholar] [CrossRef] [PubMed]
- Arber, S.; Han, B.; Mendelsohn, M.; Smith, M.; Jessell, T.M.; Sockanathan, S. Requirement for the Homeobox Gene Hb9 in the Consolidation of Motor Neuron Identity. Neuron 1999, 23, 659–674. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Chow, C.Y.; Sahenk, Z.; Shy, M.E.; Meisler, M.H.; Li, J. Mutation of FIG4 Causes a Rapidly Progressive, Asymmetric Neuronal Degeneration. Brain 2008, 131, 1990–2001. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, C.J.; Lenk, G.M.; Jones, J.M.; Grant, A.E.; Winters, J.J.; Dowling, J.J.; Giger, R.J.; Meisler, M.H. Neuronal Expression of Fig4 Is Both Necessary and Sufficient to Prevent Spongiform Neurodegeneration. Hum. Mol. Genet. 2012, 21, 3525–3534. [Google Scholar] [CrossRef] [Green Version]
- Vaccari, I.; Carbone, A.; Previtali, S.C.; Mironova, Y.A.; Alberizzi, V.; Noseda, R.; Rivellini, C.; Bianchi, F.; Del Carro, U.; D’Antonio, M.; et al. Loss of Fig4 in Both Schwann Cells and Motor Neurons Contributes to CMT4J Neuropathy. Hum. Mol. Genet. 2015, 24, 383–396. [Google Scholar] [CrossRef]
- Vaccari, I.; Dina, G.; Tronchère, H.; Kaufman, E.; Chicanne, G.; Cerri, F.; Wrabetz, L.; Payrastre, B.; Quattrini, A.; Weisman, L.S.; et al. Genetic Interaction between MTMR2 and FIG4 Phospholipid Phosphatases Involved in Charcot-Marie-Tooth Neuropathies. PLoS Genet. 2011, 7, e1002319. [Google Scholar] [CrossRef]
- Taveggia, C.; Bolino, A. DRG Neuron/Schwann Cells Myelinating Cocultures. In Myelin, Methods in Molecular Biology; Woodhoo, A., Ed.; Springer: New York, NY, USA, 2018; Volume 1791, pp. 115–129. ISBN 978-1-4939-7861-8. [Google Scholar]
- Bolis, A.; Coviello, S.; Visigalli, I.; Taveggia, C.; Bachi, A.; Chishti, A.H.; Hanada, T.; Quattrini, A.; Previtali, S.C.; Biffi, A.; et al. Dlg1, Sec8, and Mtmr2 Regulate Membrane Homeostasis in Schwann Cell Myelination. J. Neurosci. 2009, 29, 8858–8870. [Google Scholar] [CrossRef]
- Guerrero-Valero, M.; Grandi, F.; Cipriani, S.; Alberizzi, V.; Di Guardo, R.; Chicanne, G.; Sawade, L.; Bianchi, F.; Del Carro, U.; De Curtis, I.; et al. Dysregulation of Myelin Synthesis and Actomyosin Function Underlies Aberrant Myelin in CMT4B1 Neuropathy. Proc. Natl. Acad. Sci. USA 2021, 118, e2009469118. [Google Scholar] [CrossRef]
- Robinson, D.C.; Mammel, A.E.; Logan, A.M.; Larson, A.A.; Schmidt, E.J.; Condon, A.F.; Robinson, F.L. An In Vitro Model of Charcot-Marie-Tooth Disease Type 4B2 Provides Insight Into the Roles of MTMR13 and MTMR2 in Schwann Cell Myelination. ASN Neuro 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Evgrafov, O.V.; Mersiyanova, I.; Irobi, J.; Van Den Bosch, L.; Dierick, I.; Leung, C.L.; Schagina, O.; Verpoorten, N.; Van Impe, K.; Fedotov, V.; et al. Mutant Small Heat-Shock Protein 27 Causes Axonal Charcot-Marie-Tooth Disease and Distal Hereditary Motor Neuropathy. Nat. Genet. 2004, 36, 602–606. [Google Scholar] [CrossRef] [Green Version]
- Rossor, A.M.; Kalmar, B.; Greensmith, L.; Reilly, M.M. The Distal Hereditary Motor Neuropathies. J. Neurol. Neurosurg. Psychiatry 2012, 83, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Vendredy, L.; Adriaenssens, E.; Timmerman, V. Small Heat Shock Proteins in Neurodegenerative Diseases. Cell Stress Chaperones 2020, 25, 679–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida-Souza, L.; Asselbergh, B.; D’Ydewalle, C.; Moonens, K.; Goethals, S.; de Winter, V.; Azmi, A.; Irobi, J.; Timmermans, J.-P.; Gevaert, K.; et al. Small Heat-Shock Protein HSPB1 Mutants Stabilize Microtubules in Charcot-Marie-Tooth Neuropathy. J. Neurosci. 2011, 31, 15320–15328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misko, A.; Jiang, S.; Wegorzewska, I.; Milbrandt, J.; Baloh, R.H. Mitofusin 2 Is Necessary for Transport of Axonal Mitochondria and Interacts with the Miro/Milton Complex. J. Neurosci. 2010, 30, 4232–4240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Züchner, S.; Mersiyanova, I.V.; Muglia, M.; Bissar-Tadmouri, N.; Rochelle, J.; Dadali, E.L.; Zappia, M.; Nelis, E.; Patitucci, A.; Senderek, J.; et al. Mutations in the Mitochondrial GTPase Mitofusin 2 Cause Charcot-Marie-Tooth Neuropathy Type 2A. Nat. Genet. 2004, 36, 449–451. [Google Scholar] [CrossRef]
- Mersiyanova, I.V.; Perepelov, A.V.; Polyakov, A.V.; Sitnikov, V.F.; Dadali, E.L.; Oparin, R.B.; Petrin, A.N.; Evgrafov, O.V. A New Variant of Charcot-Marie-Tooth Disease Type 2 Is Probably the Result of a Mutation in the Neurofilament-Light Gene. Am. J. Hum. Genet. 2000, 67, 37–46. [Google Scholar] [CrossRef] [Green Version]
- De Jonghe, P.; Mersivanova, I.; Nelis, E.; Del Favero, J.; Martin, J.J.; Van Broeckhoven, C.; Evgrafov, O.; Timmerman, V. Further Evidence That Neurofilament Light Chain Gene Mutations Can Cause Charcot-Marie-Tooth Disease Type 2E. Ann. Neurol. 2001, 49, 245–249. [Google Scholar] [CrossRef]
- Herrmann, H.; Aebi, U. Intermediate Filaments: Molecular Structure, Assembly Mechanism, and Integration into Functionally Distinct Intracellular Scaffolds. Annu. Rev. Biochem. 2004, 73, 749–789. [Google Scholar] [CrossRef]
- Brownlees, J.; Ackerley, S.; Grierson, A.J.; Jacobsen, N.J.; Shea, K.; Anderton, B.H.; Leigh, P.N.; Shaw, C.E.; Miller, C.C. Charcot-Marie-Tooth Disease Neurofilament Mutations Disrupt Neurofilament Assembly and Axonal Transport. Hum. Mol. Genet. 2002, 11, 2837–2844. [Google Scholar] [CrossRef] [Green Version]
- Perez-Olle, R.; Leung, C.L.; Liem, R.K.H. Effects of Charcot-Marie-Tooth-Linked Mutations of the Neurofilament Light Subunit on Intermediate Filament Formation. J. Cell Sci. 2002, 115, 4937–4946. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Couillard-Després, S.; Julien, J.-P. Delayed Maturation of Regenerating Myelinated Axons in Mice Lacking Neurofilaments. Exp. Neurol. 1997, 148, 299–316. [Google Scholar] [CrossRef]
- Dubois, M.; Strazielle, C.; Julien, J.-P.; Lalonde, R. Mice with the Deleted Neurofilament of Low Molecular Weight (Nefl) Gene: 2. Effects on Motor Functions and Spatial Orientation. J. Neurosci. Res. 2005, 80, 751–758. [Google Scholar] [CrossRef]
- Elder, G.A.; Friedrich, V.L.; Bosco, P.; Kang, C.; Gourov, A.; Tu, P.-H.; Lee, V.M.-Y.; Lazzarini, R.A. Absence of the Mid-Sized Neurofilament Subunit Decreases Axonal Calibers, Levels of Light Neurofilament (NF-L), and Neurofilament Content. J. Cell Biol. 1998, 141, 727–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elder, G.A.; Friedrich, V.L.; Kang, C.; Bosco, P.; Gourov, A.; Tu, P.-H.; Zhang, B.; Lee, V.M.-Y.; Lazzarini, R.A. Requirement of Heavy Neurofilament Subunit in the Development of Axons with Large Calibers. J. Cell Biol. 1998, 143, 195–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, H.; Barry, D.M.; Dale, J.M.; Garcia, V.B.; Calcutt, N.A.; Garcia, M.L. Muscle Pathology without Severe Nerve Pathology in a New Mouse Model of Charcot–Marie–Tooth Disease Type 2E. Hum. Mol. Genet. 2011, 20, 2535–2548. [Google Scholar] [CrossRef] [Green Version]
- Jackson, K.J.; Wang, J.B.; Barbier, E.; Chen, X.; Damaj, M.I. Acute Behavioral Effects of Nicotine in Male and Female HINT1 Knockout Mice: HINT1 and Acute Nicotine. Genes Brain Behav. 2012, 11, 993–1000. [Google Scholar] [CrossRef]
- Seburn, K.L.; Morelli, K.H.; Jordanova, A.; Burgess, R.W. Lack of Neuropathy-Related Phenotypes in Hint1 Knockout Mice. J. Neuropathol. Exp. Neurol. 2014, 73, 693–701. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 Coordinately Regulate Mitochondrial Fusion and Are Essential for Embryonic Development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Auer-Grumbach, M.; Toegel, S.; Schabhüttl, M.; Weinmann, D.; Chiari, C.; Bennett, D.L.H.; Beetz, C.; Klein, D.; Andersen, P.M.; Böhme, I.; et al. Rare Variants in MME, Encoding Metalloprotease Neprilysin, Are Linked to Late-Onset Autosomal-Dominant Axonal Polyneuropathies. Am. J. Hum. Genet. 2016, 99, 607–623. [Google Scholar] [CrossRef] [Green Version]
- Dacci, P.; Dina, G.; Cerri, F.; Previtali, S.C.; Lopez, I.D.; Lauria, G.; Feltri, M.L.; Bolino, A.; Comi, G.; Wrabetz, L.; et al. Foot Pad Skin Biopsy in Mouse Models of Hereditary Neuropathy. Glia 2010, 58, 2005–2016. [Google Scholar] [CrossRef] [Green Version]
- Villalón, E.; Dale, J.M.; Jones, M.; Shen, H.; Garcia, M.L. Exacerbation of Charcot–Marie–Tooth Type 2E Neuropathy Following Traumatic Nerve Injury. Brain Res. 2015, 1627, 143–153. [Google Scholar] [CrossRef] [Green Version]
- Saher, G.; Quintes, S.; Mobius, W.; Wehr, M.C.; Kramer-Albers, E.-M.; Brugger, B.; Nave, K.-A. Cholesterol Regulates the Endoplasmic Reticulum Exit of the Major Membrane Protein P0 Required for Peripheral Myelin Compaction. J. Neurosci. 2009, 29, 6094–6104. [Google Scholar] [CrossRef] [Green Version]
- Nave, K.-A. Myelination and the Trophic Support of Long Axons. Nat. Rev. Neurosci. 2010, 11, 275–283. [Google Scholar] [CrossRef]
- Griffiths, I.; Klugmann, M.; Anderson, T.; Yool, D.; Thomson, C.; Schwab, M.H.; Schneider, A.; Zimmermann, F.; McCulloch, M.; Nadon, N.; et al. Axonal Swellings and Degeneration in Mice Lacking the Major Proteolipid of Myelin. Science 1998, 280, 1610–1613. [Google Scholar] [CrossRef] [PubMed]
- Beirowski, B.; Babetto, E.; Wrabetz, L. Axon Degeneration: Linking Axonal Bioenergetics to Myelin. J. Cell Biol. 2016, 215, 437–440. [Google Scholar] [CrossRef] [PubMed]
- Bouçanova, F.; Chrast, R. Metabolic Interaction between Schwann Cells and Axons under Physiological and Disease Conditions. Front. Cell. Neurosci. 2020, 14, 148. [Google Scholar] [CrossRef]
- Moss, K.R.; Bopp, T.S.; Johnson, A.E.; Höke, A. New Evidence for Secondary Axonal Degeneration in Demyelinating Neuropathies. Neurosci. Lett. 2021, 744, 135595. [Google Scholar] [CrossRef] [PubMed]
- Volpi, V.G.; Touvier, T.; D’Antonio, M. Endoplasmic Reticulum Protein Quality Control Failure in Myelin Disorders. Front. Mol. Neurosci. 2017, 9, 162. [Google Scholar] [CrossRef] [Green Version]
- Babetto, E.; Wong, K.M.; Beirowski, B. Publisher Correction: A Glycolytic Shift in Schwann Cells Supports Injured Axons. Nat. Neurosci. 2021, 24, 150–153. [Google Scholar] [CrossRef] [PubMed]
- Sahenk, Z.; Yalvac, M.E.; Amornvit, J.; Arnold, W.D.; Chen, L.; Shontz, K.M.; Lewis, S. Efficacy of Exogenous Pyruvate in Trembler J Mouse Model of Charcot-Marie-Tooth Neuropathy. Brain Behav. 2018, 8, e01118. [Google Scholar] [CrossRef]
- Adalbert, R.; Kaieda, A.; Antoniou, C.; Loreto, A.; Yang, X.; Gilley, J.; Hoshino, T.; Uga, K.; Makhija, M.T.; Coleman, M.P. Novel HDAC6 Inhibitors Increase Tubulin Acetylation and Rescue Axonal Transport of Mitochondria in a Model of Charcot–Marie–Tooth Type 2F. ACS Chem. Neurosci. 2020, 11, 258–267. [Google Scholar] [CrossRef] [PubMed]
- La Marca, R.; Cerri, F.; Horiuchi, K.; Bachi, A.; Feltri, M.L.; Wrabetz, L.; Blobel, C.P.; Quattrini, A.; Salzer, J.L.; Taveggia, C. TACE (ADAM17) Inhibits Schwann Cell Myelination. Nat. Neurosci. 2011, 14, 857–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nave, K.-A.; Salzer, J.L. Axonal Regulation of Myelination by Neuregulin 1. Curr. Opin. Neurobiol. 2006, 16, 492–500. [Google Scholar] [CrossRef]
- Taveggia, C.; Zanazzi, G.; Petrylak, A.; Yano, H.; Rosenbluth, J.; Einheber, S.; Xu, X.; Esper, R.M.; Loeb, J.A.; Shrager, P.; et al. Neuregulin-1 Type III Determines the Ensheathment Fate of Axons. Neuron 2005, 47, 681–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michailov, G.V.; Sereda, M.W.; Brinkmann, B.G.; Fischer, T.M.; Haug, B.; Birchmeier, C.; Role, L.; Lai, C.; Schwab, M.H.; Nave, K.-A. Axonal Neuregulin-1 Regulates Myelin Sheath Thickness. Science 2004, 304, 700–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epstein, J.; Sanderson, I.R.; Macdonald, T.T. Curcumin as a Therapeutic Agent: The Evidence from in Vitro, Animal and Human Studies. Br. J. Nutr. 2010, 103, 1545–1557. [Google Scholar] [CrossRef] [Green Version]
- Khajavi, M.; Inoue, K.; Wiszniewski, W.; Ohyama, T.; Snipes, G.J.; Lupski, J.R. Curcumin Treatment Abrogates Endoplasmic Reticulum Retention and Aggregation-Induced Apoptosis Associated with Neuropathy-Causing Myelin Protein Zero-Truncating Mutants. Am. J. Hum. Genet. 2005, 77, 841–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caillaud, M.; Msheik, Z.; Ndong-Ntoutoume, G.M.-A.; Vignaud, L.; Richard, L.; Favreau, F.; Faye, P.-A.; Sturtz, F.; Granet, R.; Vallat, J.-M.; et al. Curcumin–Cyclodextrin/Cellulose Nanocrystals Improve the Phenotype of Charcot-Marie-Tooth-1A Transgenic Rats through the Reduction of Oxidative Stress. Free. Radic. Biol. Med. 2020, 161, 246–262. [Google Scholar] [CrossRef]
- Ludolph, A.C.; Wurster, C.D. Therapeutic Advances in SMA. Curr. Opin. Neurol. 2019, 32, 777–781. [Google Scholar] [CrossRef] [PubMed]
- Presa, M.; Bailey, R.M.; Davis, C.; Murphy, T.; Cook, J.; Walls, R.; Wilpan, H.; Bogdanik, L.; Lenk, G.M.; Burgess, R.W.; et al. AAV9-Mediated FIG4 Delivery Prolongs Life Span in Charcot-Marie-Tooth Disease Type 4J Mouse Model. J. Clin. Investig. 2021, 131, e137159. [Google Scholar] [CrossRef]
- Burns, J.; Ouvrier, R.A.; Yiu, E.M.; Joseph, P.D.; Kornberg, A.J.; Fahey, M.C.; Ryan, M.M. Ascorbic Acid for Charcot–Marie–Tooth Disease Type 1A in Children: A Randomised, Double-Blind, Placebo-Controlled, Safety and Efficacy Trial. Lancet Neurol. 2009, 8, 537–544. [Google Scholar] [CrossRef]
- Verhamme, C.; de Haan, R.J.; Vermeulen, M.; Baas, F.; de Visser, M.; van Schaik, I.N. Oral High Dose Ascorbic Acid Treatment for One Year in Young CMT1A Patients: A Randomised, Double-Blind, Placebo-Controlled Phase II Trial. BMC Med. 2009, 7, 70. [Google Scholar] [CrossRef] [Green Version]
- Pareyson, D.; Reilly, M.M.; Schenone, A.; Fabrizi, G.M.; Cavallaro, T.; Santoro, L.; Vita, G.; Quattrone, A.; Padua, L.; Gemignani, F.; et al. Ascorbic Acid in Charcot–Marie–Tooth Disease Type 1A (CMT-TRIAAL and CMT-TRAUK): A Double-Blind Randomised Trial. Lancet Neurol. 2011, 10, 320–328. [Google Scholar] [CrossRef]
- Lewis, R.A. High-Dosage Ascorbic Acid Treatment in Charcot-Marie-Tooth Disease Type 1A: Results of a Randomized, Double-Masked, Controlled Trial. JAMA Neurol. 2013, 70, 981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Use of rodent models in IPNs.


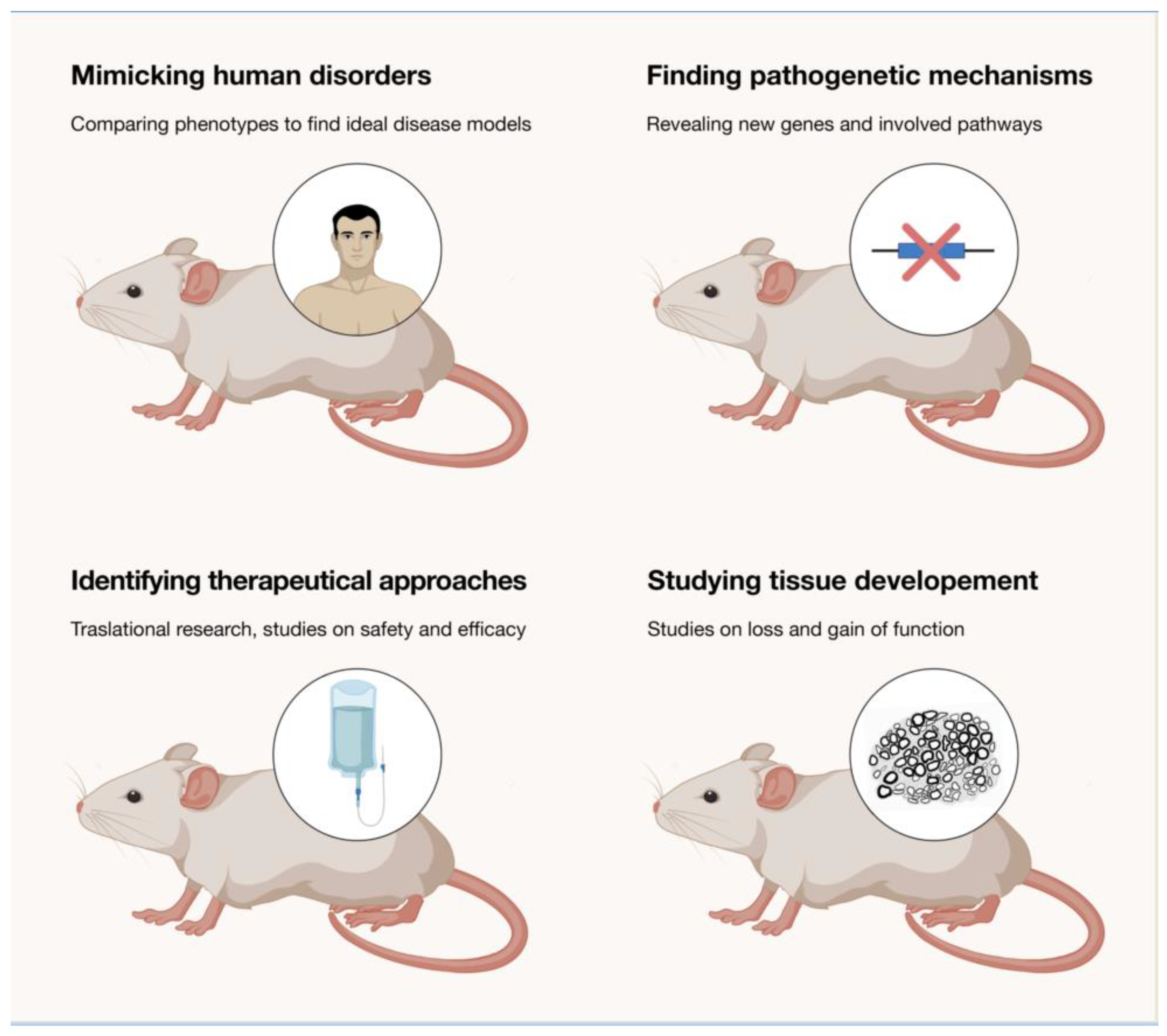{kind=link}
Table 1.
Overview of rodent models for IPNs.
| CMT Subtype | Gene | Model | Mutation | Phenotype (Neuropathy) | Conduction Studies | Nerve Pathology | Experimental Therapy | References |
|---|---|---|---|---|---|---|---|---|
| CMT1A | PMP22 | Transgenic mouse | PMP22 JP18/JY13tg; tTA | Mild | Demyelinating | Mild demyelination | siRNA | [33,34] |
| CMT1A | PMP22 | Transgenic mouse | PMP22 duplication (three–four copies) C3-PMP | Mild | Demyelinating | Mild hypomyelination | [35] | |
| CMT1A | PMP22 | Transgenic mouse | PMP22 duplication (four copies) C61 Het | Normal | Demyelinating | Mild hypomyelination | [36] | |
| CMT1A | PMP22 | Transgenic mouse | PMP22 duplication (seven–eight copies) C22 Het | Present | Demyelinating | Severe hypo/dysmyelination | Ascorbic acid; geldanamycin; HSP90 inhibitor (NVP-AUT922); ASO treatment; CKD-504-HDAC6 nhibitor; CRISPR/Cas9-mediated downregulation of PMP22 via targeting TATA-box; miR-381; soluble NRG1 | [37,38,39,40,41,42,43,44] |
| CMT1A | PMP22 | Transgenic mouse | PMP22 duplication (16 copies) TgN248 | Present | Demyelinating | Severe hypo/dysmyelination | [45,46] | |
| CMT1A | PMP22 | Transgenic mouse | PMP22 duplication (unknown copies) MY41 | Present | N/A | Severe hypo/dysmyelination | [47] | |
| CMT1A | PMP22 | Transgenic rat | PMP22 duplication (three copies)—CMT rat | Present | Demyelinating | Severe hypo/dysmyelination and onion bulbs | Progesterone antagonist; AAV2/9; soluble neuregulin1 | [44,48,49,50] |
| CMT1B | MPZ | Transgenic mouse | I106L; Mpz+/+ | Present | Demyelinating | Severe hypo/dysmyelination and onion bulbs | [51] | |
| CMT1B | MPZ | Knock-in mouse | MPZ Q215X/+ | Normal | N/A | Mild hypo/demyelination | [52] | |
| CMT1B | MPZ | Knock-in mouse | MPZ R98C/+ and R98C/R98C | Mild | Demyelinating | Mild hypo/demyelination | Curcumin derivates | [53,54] |
| CMT1B | MPZ | Transgenic mouse | MPZ S63C | Present | Demyelinating | Mild hypo/demyelination | [55] | |
| CMT1B | MPZ | Transgenic mouse | MPZ S63del | Present | Demyelinating | Severe hypo/demyelination and onion bulbs | Chop ablation; sephin 1; over-expression of Nrg1 type-III; inhibition of eIF2α phosphorylation | [54,55,56,57,58] |
| CMT1B | MPZ | Knock-out mouse | MPZ−/− | Present | Demyelinating | Severe hypo/demyelination | [59,60] | |
| CMT1B | MPZ | Knock-out mouse | MPZ−/+ | Mild | Demyelinating | Mild hypo/demyelination | [60] | |
| CMT1C | LITAF | Transgenic mouse | Simple W116G/W116G; Simple W116G/+ | Mild | Demyelinating | Mild dysmyelination | [61] | |
| CMT1C | LITAF | Knock-in mouse | SimpleT115N/+; SimpleT115N/T115N | Mild | Normal | N/A | [62] | |
| CMT1D | EGR2 | Knock-out mouse | Egr2+/+ and −/− | Present | N/A | Normal in Het, lethal in Homo | [63] | |
| CMT1E | PMP22 | Mutant mouse | Exon V deletion (trembler-Ncnp) Het | Present | N/A | Demyelination | [64] | |
| CMT1E | PMP22 | Mutant mouse | Exon V deletion (trembler-Ncnp) Hom | Present | N/A | Hypomyelination/amyelination | [64] | |
| CMT1E | PMP22 | Mutant mouse | PMP22 point mutation (G150D) (Trembler) Het | Present | N/A | Severe dysmyelination | [65] | |
| CMT1E | PMP22 | Mutant mouse | Pmp22 point mutation (H12R) (Trembler-m1H) Het | Present | N/A | Severe hypomyelination and reduced axon number | [66] | |
| CMT1E | PMP22 | Mutant mouse | Pmp22 point mutation (Y153X) (Trembler-m2H) Het | Present | N/A | Severe hypomyelination and reduced axon number | [66] | |
| CMT1E | PMP22 | Mutant mouse | PMP22 spontaneous point mutation (L16P) (Trembler-J) | Mild | N/A | Severe hypo/demyelination and onion bulbs | HSP90 inhibitor (NVP-AUT922); Neurotrophin-3; Rapamicyn (in vitro); ACE-083; intermittent fasting; curcumin | [43,65,67,68,69,70,71,72] |
| CMT2A | MFN2 | Transgenic mouse | hMFN2 R94QL51/+ (Mitocharc 1) | Mild | Normal | Mild axon atrophy | SW100 (HDAC6 inhibitor); salubrinal | [73,74,75] |
| CMT2A | MFN2 | Transgenic mouse | MFN2 R94Q/+ | Present | N/A | Mild axon atrophy | transgenic MFN1 overexpression | [76] |
| CMT2A | MFN2 | Knock-in mouse | MFN2 R94W/+ | Mild | N/A | Normal | [77] | |
| CMT2A | MFN2 | Knock-in mouse | MFN2 T105M/− | Mild | N/A | Normal | [78] | |
| CMT2A | MFN2 | Transgenic mouse | MFN2 T105M/+ | Mild | N/A | Normal | [79] | |
| CMT2A | MFN2 | Transgenic mouse | MFN2 T105M/T105M | Present | N/A | Reduced axon number | [79] | |
| CMT2A | MFN2 | Transgenic mouse | MFN2R94QL87/R94QL87 (Mitocharc 2) | Mild | N/A | Reduced axon number | [74] | |
| CMT2B1 | LMNA | Knock-in mouse | LMNA R298C/R298C | Normal | Normal | Normal | [80] | |
| CMT2D | GARS | Transgenic mouse | AdhGARS G240R | N/A | N/A | Normal | [81] | |
| CMT2D | GARS | Mutant mouse | GARS C201R/+ | Present | Mixed | Mild axon atrophy | HDAC6 inhibitors | [82,83,84,85] |
| CMT2D | GARS | Mutant mouse | GARS NMF249/+ | Present | Mixed | Mild axon atrophy | [86] | |
| CMT2D | GARS | Mutant mouse | GARS P234KY/+ | Present | N/A | Normal | VEGF; HDAC6 inhibitors | [87,88] |
| CMT2D | GARS | Knock-in mouse | ΔETAQ/+ | Present | Mixed | Reduced axon number and axon atrophy | AAV9-mediated allele specific knockdown | [89] |
| CMT2E | NEFL | Transgenic mouse | hNF-LP22S; tTa | Mild | N/A | Normal | [90] | |
| CMT2E | NEFL | Transgenic mouse | hNF-LE397K (NF-L−/−) | Mild | Demyelinating | Mild axon atrophy | [91] | |
| CMT2E | NEFL | Knock-in mouse | Nefl P8R/+; P8R/P8R | Normal | N/A | Normal | [92] | |
| CMT2E | NEFL | Knock-in mouse | NeflN98S/+ | Present | N/A | Mild axon atrophy | [92] | |
| CMT2F | HSPB1 | Transgenic mouse | hHSPB1 R136W | Normal | Axonal | Mild axon atrophy | [93] | |
| CMT2F | HSPB1 | Transgenic mouse | HSP27-S135F | Mild | Axonal | Reduced axon number | [94] | |
| CMT2F | HSPB1 | Transgenic mouse | P182L | Mild | Axonal | Reduced axon number | [95] | |
| CMT2F | HSPB1 | Transgenic mouse | R127W, P182L in ROSA26 locus | Normal | Normal | Normal | [96] | |
| CMT2F | HSPB1 | Transgenic mouse | S135F | Mild | Axonal | Reduced axon number | HDAC6 inhibitors | [95,97] |
| CMT2K | GDAP | Knock-out mouse | GDAP−/− | Present | Axonal | Reduced axon number | [98] | |
| CMT2L | HSPB8 | Transgenic mouse | HSPB8 K141N | Present | Axonal | Reduced axon number | [99,100] | |
| CMT2L | HSPB8 | Knock-in mouse | HSPB8 K141N/+ | Normal | Normal | Normal | [101] | |
| CMT2L | HSPB8 | Knock-in mouse | HSPB8 K141N/K141N | Mild | Axonal | Reduced axon number | [102] | |
| CMT2O | DYNC1H1 | Knock-in mouse | H304R/+ | Mild | N/A | Alteration of NMJ | [103] | |
| CMT2O | DYNC1H1 | Knock-in mouse | H304R/H304R | Present | N/A | Alteration of NMJ | [104] | |
| CMT2P | LRSAM1 | Knock-out (gene trap) mouse | RRK239, RRK461Null mutation | Normal | Normal | Reduced axon number | [105] | |
| CMT2Q | DHTKD1 | Knock-in mouse | DHTKD1 Y486*/Y486* | Mild | Normal | Mild axon atrophy | [106] | |
| CMT2Q | DHTKD1 | Knock-out mouse | DHTKD1−/− | Present | Mixed | Reduced axon number | [107] | |
| CMT4A | GDAP1 | Knock-out mouse | GDAP1−/− | Normal | Demyelinating | Normal | [108] | |
| CMT4B1 | MTMR2 | Knock-out mouse | MTMR2−/− | Mild | Demyelinating | Aberrant myelin folding | Niacin | [109,110,111] |
| CMT4B2 | MTMR13 | Knock-out mouse | MTMR13−/− | Mild | Demyelinating | Aberrant myelin folding | [112] | |
| CMT4C | SH3TC2 | Knock-out mouse | SH3TC2−/− | Mild | Demyelinating | Mild hypo/demyelination | Gene replacement therapy | [113,114,115] |
| CMT4F | PRX | Knock-out mouse | PRX−/− | Present | Demyelinating | Segmental demyelination | [116] | |
| CMT4J | FIG4 | Mutant mouse | Fig4−/− (Pale tremor mouse) | Present | Mixed | Severe hypo/demyelination and onion bulbs | AAV gene therapy | [117,118,119] |
| CMTX | CX32 | Transgenic mouse | S26L (S3) hCX32 | Present | N/A | N/A | CamKII inhibitor | [120,121] |
| CMTX | CX32 | Transgenic mouse | G12S (G1,G2); S26L (S1,S2) hCX32 | Mild | N/A | N/A | CamKII inhibitor | [120,121] |
| CMTX | CX32 | Knock-out mouse | CX32−/−; CX32y/- | Mild | Mixed | Demyelination and onion bulbs | Intratecal gene delivery (early); AAV9-Mpz.GJB1 therapy; scAAV1. tMCK.NT-3 | [122,123,124,125,126,127,128] |
| CMT-DI | EBP50 | Knock-out mouse | EBP50+/− | Mild | Demyelinating | Mild axon atrophy | [129] | |
| CMT-DI | C1orf194 | Knock-out mouse | C1orf194−/+; C1orf194−/− | Present | Mixed | Demyelination | [130] | |
| CMT-DI | C1orf194 | Knock-in mouse | I121N/I121N; I121N/+ | Present | Axonal | Demyelination | [131] | |
| CMTDIB | DNM1 | Knock-in mouse | DNM2 K552E/+ | Mild | CMAP decrease (Myopatic) | Normal | [132] | |
| CMTDIC | YARS | Transgenic mouse | AdhYARS E196K/ChAT mice | N/A | N/A | N/A | [133] | |
| ARCMT2 | TRIM2 | Knock-out mouse | TRIM2A/A | Present | Axonal | Reduced axon number | [134] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bosco, L.; Falzone, Y.M.; Previtali, S.C. Animal Models as a Tool to Design Therapeutical Strategies for CMT-like Hereditary Neuropathies. Brain Sci. 2021, 11, 1237. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci11091237
AMA Style
Bosco L, Falzone YM, Previtali SC. Animal Models as a Tool to Design Therapeutical Strategies for CMT-like Hereditary Neuropathies. Brain Sciences. 2021; 11(9):1237. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci11091237
Chicago/Turabian StyleBosco, Luca, Yuri Matteo Falzone, and Stefano Carlo Previtali. 2021. "Animal Models as a Tool to Design Therapeutical Strategies for CMT-like Hereditary Neuropathies" Brain Sciences 11, no. 9: 1237. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci11091237
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.