
Epigenetic Small Molecules Rescue Nucleocytoplasmic Transport and DNA Damage Phenotypes in C9ORF72 ALS/FTD

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation and Maintenance of Cell Lines with Constitutive Expression of Biosensors
2.2. RNAi Transfection of U-2 OS Cells
2.3. DPR Transfection of U-2 OS Cells
2.4. iPSC Culturing and Motor Neuron Differentiation
2.5. Immunocytochemistry
2.6. Semi-Automated Image Analysis
2.7. Automated Microscopy and Image Analysis
2.8. High Content Image Data Analysis
2.9. Statistical Analysis
3. Results
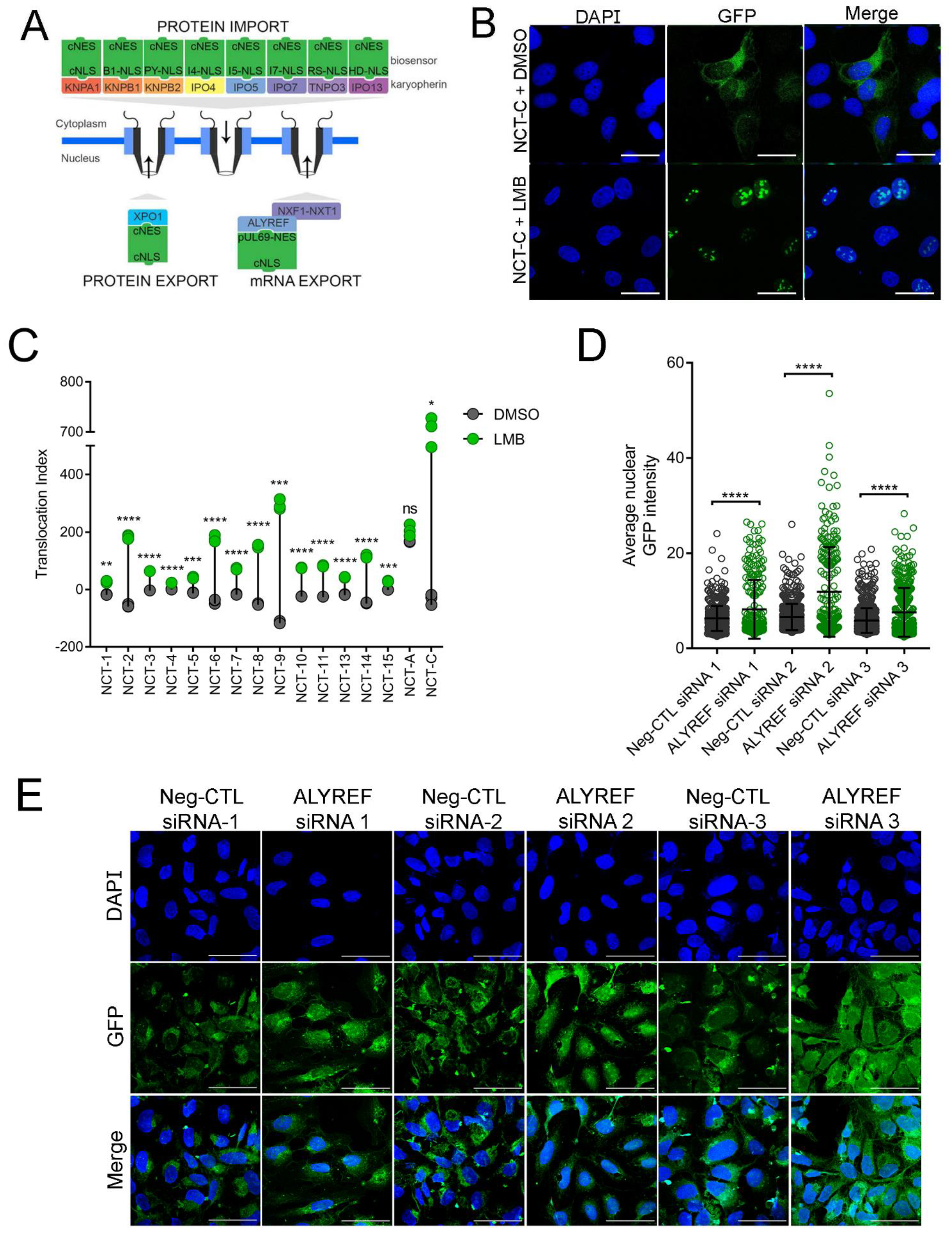
3.1. Nucleocytoplasmic Transport Biosensors Are Functional in Mammalian Cells
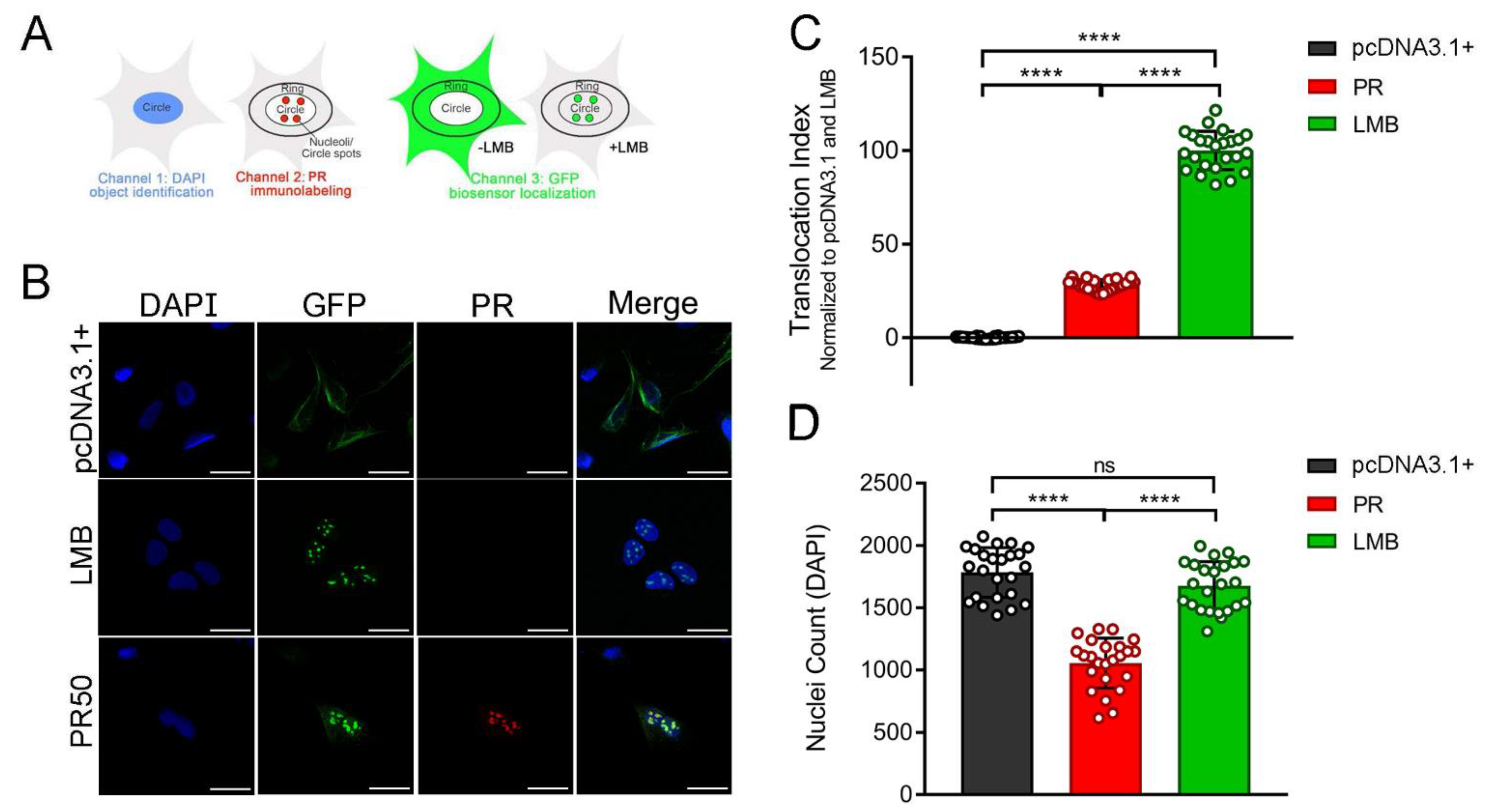
3.2. Proline-Arginine Disrupts the Classical Nucleocytoplasmic Transport Pathway
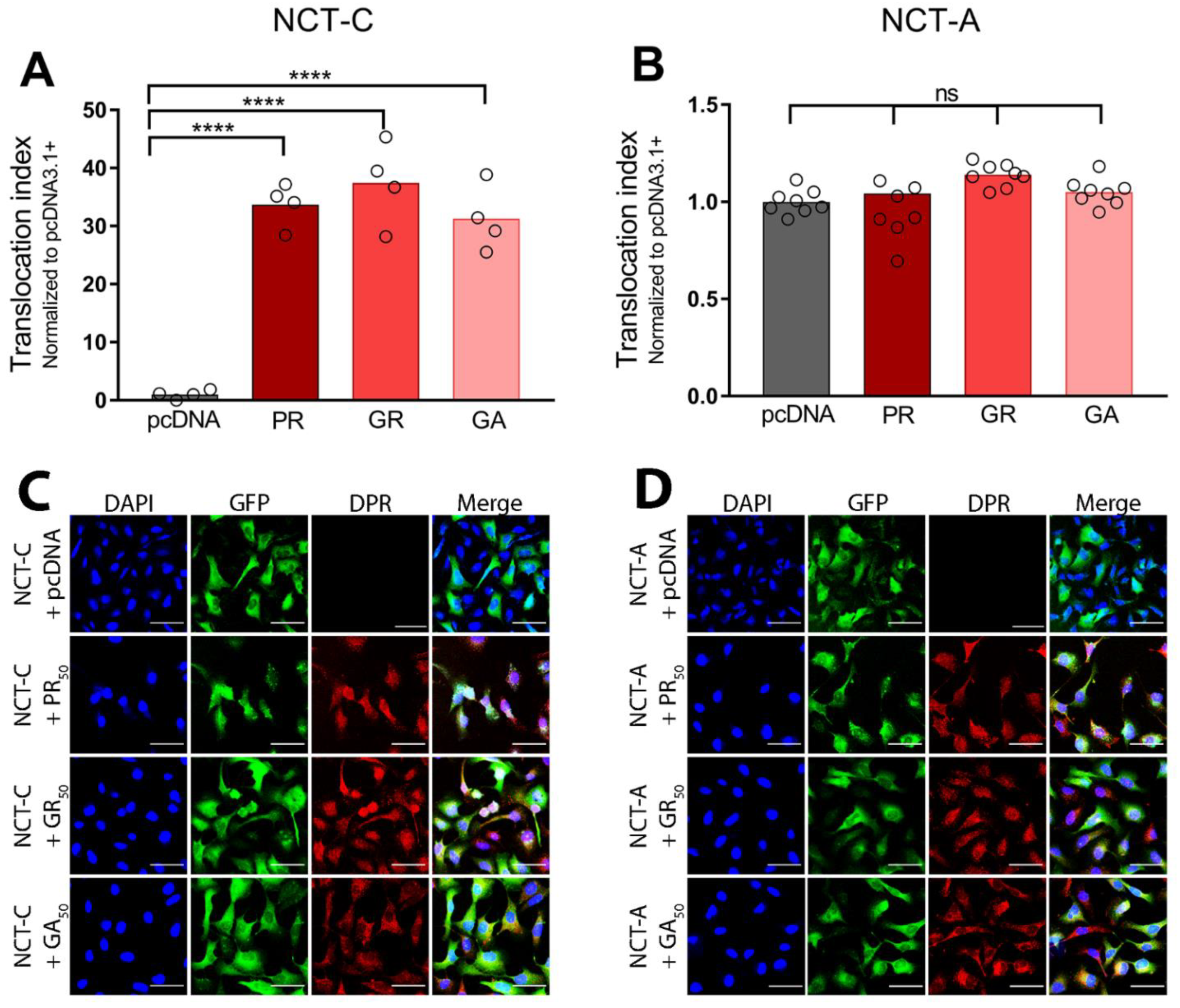
3.3. Poly-Dipeptide Repeat Proteins Disrupt the Nuclear Export of Proteins but Not mRNA
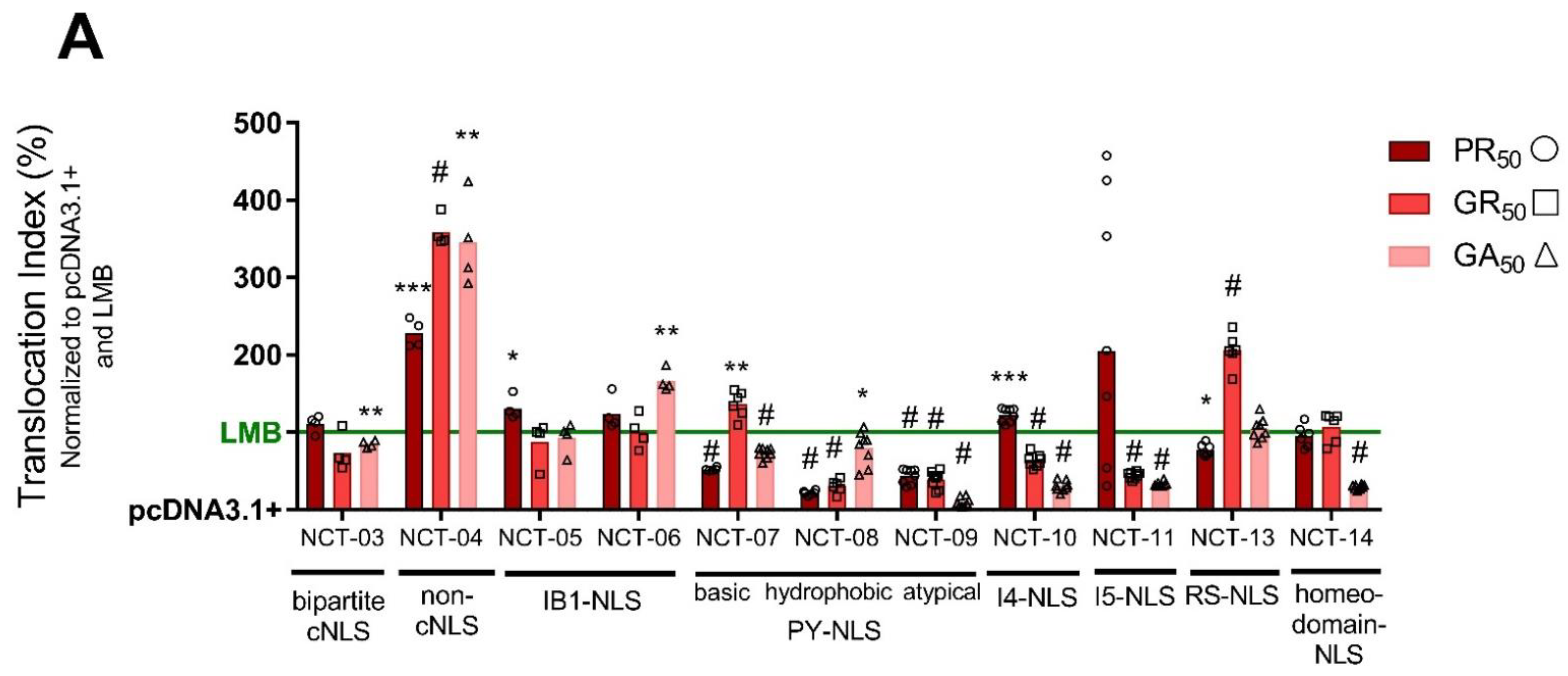
3.4. Poly-Dipeptide Repeat Proteins Inhibit Multiple Nuclear Import Pathways
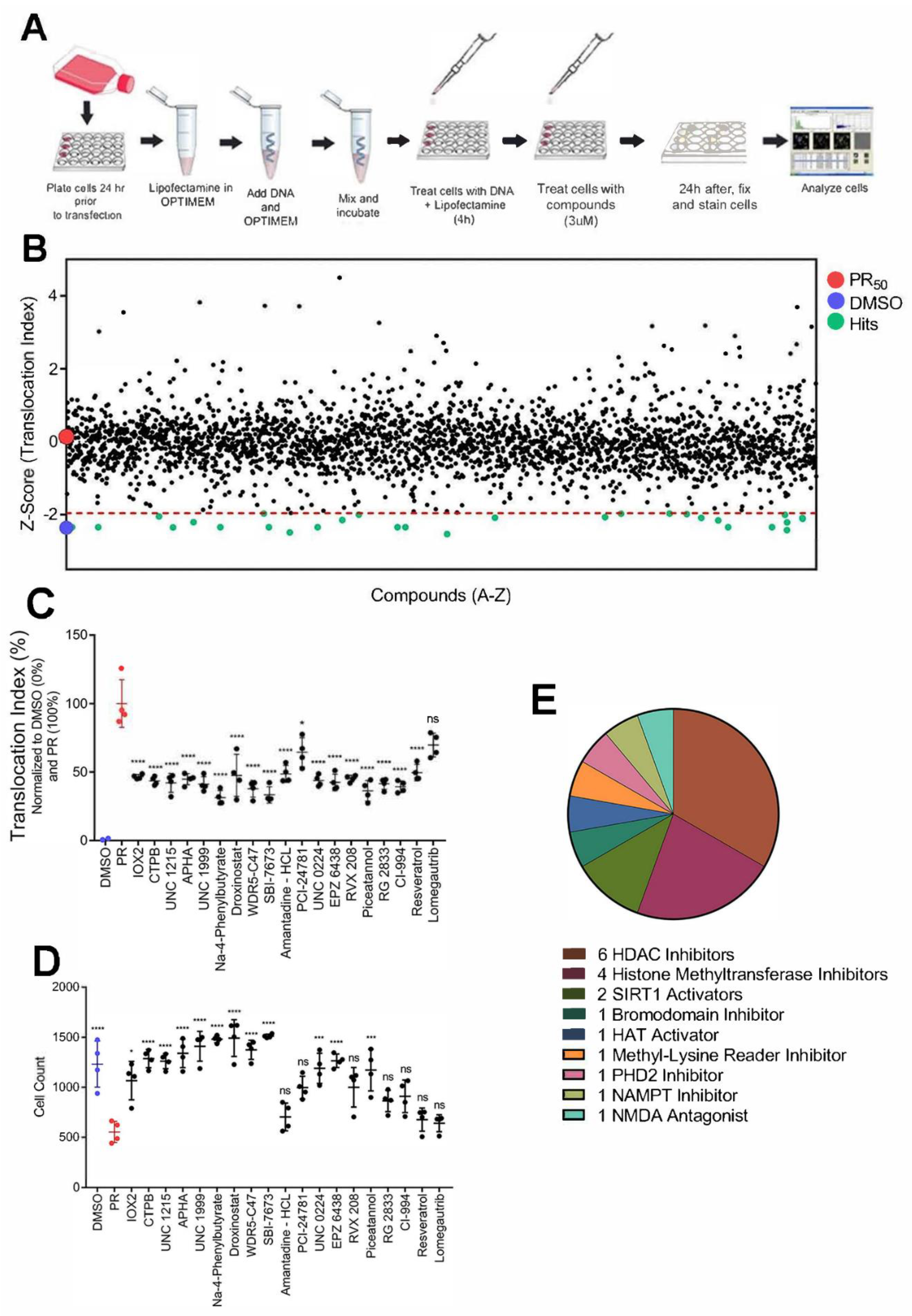
3.5. Small Molecules Targeting Epigenetic Modifiers Restore Disrupted Nucleocytoplasmic Transport in PR50 Expressing U-2 OS Cells
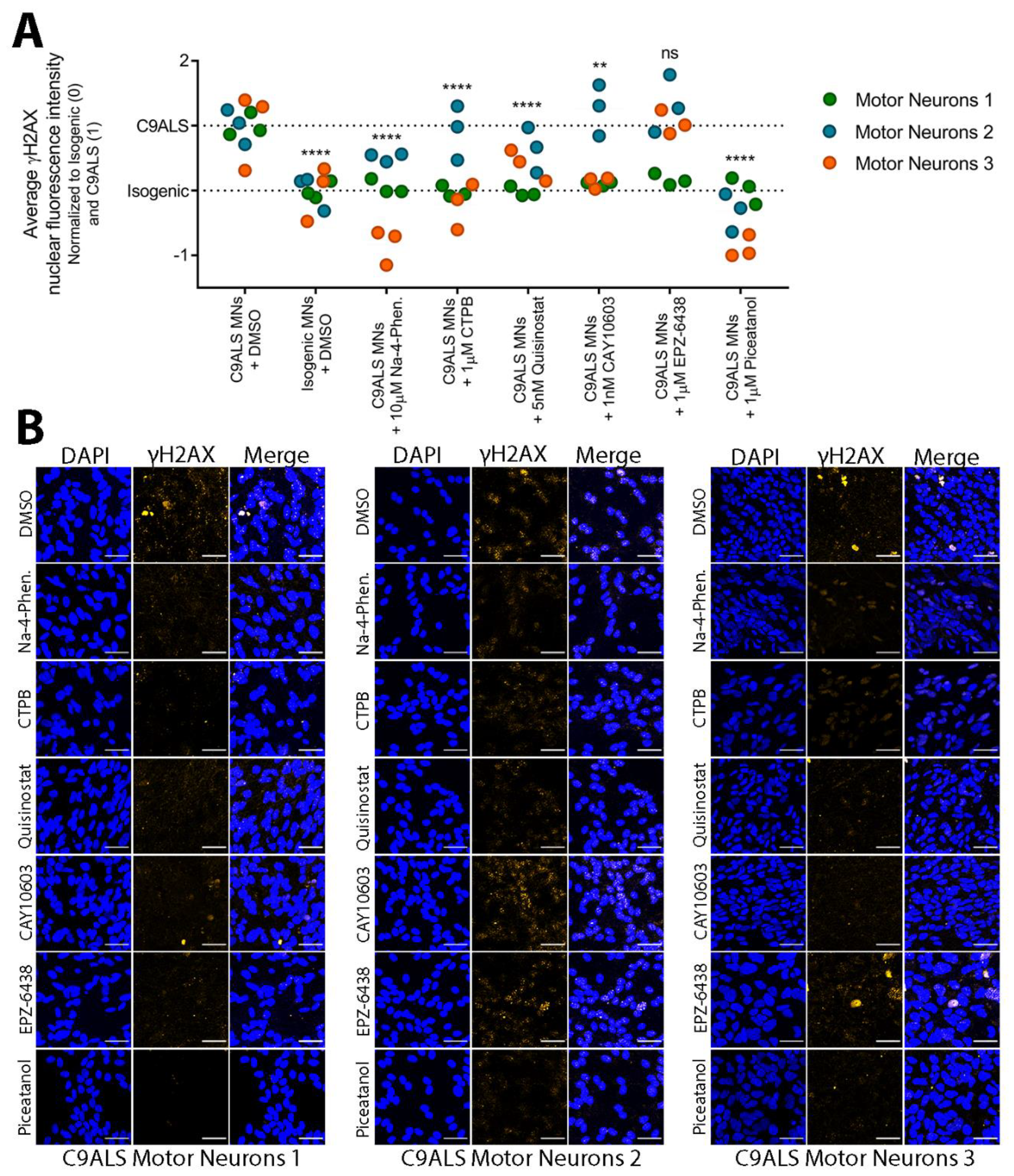
3.6. Small Molecules Targeting Epigenetic Modifiers Reduce γH2AX Immunoreactivity in C9ALS/FTD Patient iPSC-Derived Motor Neurons
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Ethics Approval and Consent to Participate
References
- Al-Chalabi, A.; Jones, A.; Troakes, C.; King, A.; Al-Sarraj, S.; van den Berg, L.H. The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol. 2012, 124, 339–352. [Google Scholar] [CrossRef]
- Doble, A. The pharmacology and mechanism of action of riluzole. Neurology 1996, 47, 233S–241S. [Google Scholar] [CrossRef]
- Abe, K.; Aoki, M.; Tsuji, S.; Itoyama, Y.; Sobue, G.; Togo, M.; Hamada, C.; Tanaka, M.; Akimoto, M.; Nakamura, K.; et al. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017, 16, 505–512. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Saánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, J.; Poulter, M.; Hensman, D.; Rohrer, J.D.; Mahoney, C.; Adamson, G.; Campbell, T.; Uphill, J.; Borg, A.; Fratta, P.; et al. Large C9orf72 Hexanucleotide Repeat Expansions Are Seen in Multiple Neurodegenerative Syndromes and Are More Frequent Than Expected in the UK Population. Am. J. Hum. Genet. 2013, 92, 345–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benussi, L.; Rossi, G.; Glionna, M.; Tonoli, E.; Piccoli, E.; Fostinelli, S.; Paterlini, A.; Flocco, R.; Albani, D.; Pantieri, R.; et al. C9ORF72 Hexanucleotide Repeat Number in Frontotemporal Lobar Degeneration: A Genotype-Phenotype Correlation Study. J. Alzheimer’s Dis. 2013, 38, 799–808. [Google Scholar] [CrossRef]
- Buchman, V.L.; Cooper-Knock, J.; Connor-Robson, N.; Higginbottom, A.; Kirby, J.; Razinskaya, O.D.; Ninkina, N.; Shaw, P. Simultaneous and independent detection of C9ORF72 alleles with low and high number of GGGGCC repeats using an optimised protocol of Southern blot hybridisation. Mol. Neurodegener. 2013, 8, 12. [Google Scholar] [CrossRef] [Green Version]
- Icardo, O.D.; García-Redondo, A.; Rojas-García, R.; Sanchez-Valle, R.; Noguera, A.; Gómez-Tortosa, E.; Pastor, P.; Hernández, I.; Esteban-Perez, J.; Suárez-Calvet, M.; et al. Characterization of the repeat expansion size in C9orf72 in amyotrophic lateral sclerosis and frontotemporal dementia. Hum. Mol. Genet. 2014, 23, 749–754. [Google Scholar] [CrossRef] [Green Version]
- Gijselinck, I.; On Behalf of the BELNEU CONSORTIUM; Van Mossevelde, S.; van der Zee, J.; Sieben, A.; Engelborghs, S.; De Bleecker, J.; Ivanoiu, A.; Deryck, O.; Edbauer, D.; et al. The C9orf72 repeat size correlates with onset age of disease, DNA methylation and transcriptional downregulation of the promoter. Mol. Psychiatry 2016, 21, 1112–1124. [Google Scholar] [CrossRef] [Green Version]
- Nordin, A.; Akimoto, C.; Wuolikainen, A.; Alstermark, H.; Jonsson, P.; Birve, A.; Marklund, S.L.; Graffmo, K.S.; Forsberg, K.; Brännström, T.; et al. Extensive size variability of the GGGGCC expansion in C9orf72 in both neuronal and non-neuronal tissues in 18 patients with ALS or FTD. Hum. Mol. Genet. 2015, 24, 3133–3142. [Google Scholar] [CrossRef]
- Suh, E.; Lee, E.B.; Neal, D.; Wood, E.M.; Toledo, J.; Rennert, L.; Irwin, D.; McMillan, C.; Krock, B.; Elman, L.B.; et al. Semi-automated quantification of C9orf72 expansion size reveals inverse correlation between hexanucleotide repeat number and disease duration in frontotemporal degeneration. Acta Neuropathol. 2015, 130, 363–372. [Google Scholar] [CrossRef]
- van Blitterswijk, M.; DeJesus-Hernandez, M.; Niemantsverdriet, E.; Murray, M.; Heckman, M.G.; Diehl, N.N.; Brown, P.H.; Baker, M.C.; A Finch, N.; Bauer, P.; et al. Association between repeat sizes and clinical and pathological characteristics in carriers of C9ORF72 repeat expansions (Xpansize-72): A cross-sectional cohort study. Lancet Neurol. 2013, 12, 978–988. [Google Scholar] [CrossRef] [Green Version]
- Esanov, R.; Belle, K.C.; van Blitterswijk, M.; Belzil, V.V.; Rademakers, R.; Dickson, D.W.; Petrucelli, L.; Boylan, K.B.; Dykxhoorn, D.M.; Wuu, J.; et al. C9orf72 promoter hypermethylation is reduced while hydroxymethylation is acquired during reprogramming of ALS patient cells. Exp. Neurol. 2016, 277, 171–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esanov, R.; Cabrera, G.T.; Andrade, N.S.; Gendron, T.F.; Brown, R.H.; Benatar, M.; Wahlestedt, C.; Mueller, C.; Zeier, Z. A C9ORF72 BAC mouse model recapitulates key epigenetic perturbations of ALS/FTD. Mol. Neurodegener. 2017, 12, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ash, P.E.A.; Bieniek, K.F.; Gendron, T.F.; Caulfield, T.; Lin, W.-L.; DeJesus-Hernandez, M.; van Blitterswijk, M.M.; Jansen-West, K.; Paul, J.W., III; Rademakers, R.; et al. Unconventional Translation of C9ORF72 GGGGCC Expansion Generates Insoluble Polypeptides Specific to c9FTD/ALS. Neuron 2013, 77, 639–646. [Google Scholar] [CrossRef] [Green Version]
- Mori, K.; Weng, S.-M.; Arzberger, T.; May, S.; Rentzsch, K.; Kremmer, E.; Schmid, B.; Kretzschmar, H.A.; Cruts, M.; Van Broeckhoven, C.; et al. The C9orf72 GGGGCC Repeat Is Translated into Aggregating Dipeptide-Repeat Proteins in FTLD/ALS. Science 2013, 339, 1335–1338. [Google Scholar] [CrossRef]
- Wen, X.; Tan, W.; Westergard, T.; Krishnamurthy, K.; Markandaiah, S.S.; Shi, Y.; Lin, S.; Shneider, N.; Monaghan, J.; Pandey, U.B.; et al. Antisense Proline-Arginine RAN Dipeptides Linked to C9ORF72-ALS/FTD Form Toxic Nuclear Aggregates that Initiate In Vitro and In Vivo Neuronal Death. Neuron 2014, 84, 1213–1225. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-J.; Gendron, T.F.; Grima, J.C.; Sasaguri, H.; Jansen-West, K.; Xu, Y.-F.; Katzman, R.B.; Gass, J.; E Murray, M.; Shinohara, M.; et al. C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat. Neurosci. 2016, 19, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Nihei, Y.; German Consortium for Frontotemporal Lobar Degeneration; Mori, K.; Werner, G.; Arzberger, T.; Zhou, Q.; Khosravi, B.; Japtok, J.; Hermann, A.; Sommacal, A.; et al. Poly-glycine–alanine exacerbates C9orf72 repeat expansion-mediated DNA damage via sequestration of phosphorylated ATM and loss of nuclear hnRNPA3. Acta Neuropathol. 2020, 139, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Hayes, L.R.; Duan, L.; Bowen, K.; Kalab, P.; Rothstein, J.D. C9orf72 arginine-rich dipeptide repeat proteins disrupt karyopherin-mediated nuclear import. eLife 2020, 9, e51685. [Google Scholar] [CrossRef] [PubMed]
- White, M.; Mitrea, D.M.; Zhang, P.; Stanley, C.; Cassidy, D.; Nourse, A.; Phillips, A.H.; Tolbert, M.; Taylor, J.P.; Kriwacki, R.W. C9orf72 Poly(PR) Dipeptide Repeats Disturb Biomolecular Phase Separation and Disrupt Nucleolar Function. Mol. Cell 2019, 74, 713–728.e6. [Google Scholar] [CrossRef]
- Zhang, Y.-J.; Gendron, T.F.; Ebbert, M.T.W.; O’Raw, A.; Yue, M.; Jansen-West, K.; Zhang, X.; Prudencio, M.; Chew, J.; Cook, C.N.; et al. Poly(GR) impairs protein translation and stress granule dynamics in C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis. Nat. Med. 2018, 24, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Gonzalez, R.; Lu, Y.; Gendron, T.F.; Karydas, A.; Tran, H.; Yang, D.; Petrucelli, L.; Miller, B.L.; Almeida, S.; Gao, F.-B. Poly(GR) in C9ORF72 -Related ALS/FTD Compromises Mitochondrial Function and Increases Oxidative Stress and DNA Damage in iPSC-Derived Motor Neurons. Neuron 2016, 92, 383–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamakawa, M.; Ito, D.; Honda, T.; Kubo, K.-I.; Noda, M.; Nakajima, K.; Suzuki, N. Characterization of the dipeptide repeat protein in the molecular pathogenesis of c9FTD/ALS. Hum. Mol. Genet. 2015, 24, 1630–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogacheva, M.; Egorova, A.; Slita, A.; Maretina, M.; Baranov, V.; Kiselev, A. Arginine-rich cross-linking peptides with different SV40 nuclear localization signal content as vectors for intranuclear DNA delivery. Bioorganic Med. Chem. Lett. 2017, 27, 4781–4785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magin, C.; Hesse, J.; Löwer, J.; Löwer, R. Corf, the Rev/Rex Homologue of HTDV/HERV-K, Encodes an Arginine-Rich Nuclear Localization Signal That Exerts a trans-Dominant Phenotype When Mutated. Virology 2000, 274, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Musinova, Y.; Kananykhina, E.Y.; Potashnikova, D.; Lisitsyna, O.; Sheval, E.V. A charge-dependent mechanism is responsible for the dynamic accumulation of proteins inside nucleoli. Biochim. Biophys. Acta 2015, 1853, 101–110. [Google Scholar] [CrossRef] [Green Version]
- Kwon, I.; Xiang, S.; Kato, M.; Wu, L.; Theodoropoulos, P.; Wang, T.; Kim, J.; Yun, J.; Xie, Y.; McKnight, S.L. Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science 2014, 345, 1139–1145. [Google Scholar] [CrossRef] [Green Version]
- Andrade, N.S.; Ramic, M.; Esanov, R.; Liu, W.; Rybin, M.J.; Gaidosh, G.; Abdallah, A.; Del’Olio, S.; Huff, T.C.; Chee, N.T.; et al. Dipeptide repeat proteins inhibit homology-directed DNA double strand break repair in C9ORF72 ALS/FTD. Mol. Neurodegener. 2020, 15, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Farg, M.A.; Konopka, A.; Soo, K.Y.; Ito, D.; Atkin, J.D. The DNA damage response (DDR) is induced by the C9orf72 repeat expansion in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2017, 26, 2882–2896. [Google Scholar] [CrossRef]
- Tao, Z.; Wang, H.; Xia, Q.; Li, K.; Li, K.; Jiang, X.; Xu, G.; Wang, G.; Ying, Z. Nucleolar stress and impaired stress granule formation contribute to C9orf72 RAN translation-induced cytotoxicity. Hum. Mol. Genet. 2015, 24, 2426–2441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freibaum, B.D.; Lu, Y.; Lopez-Gonzalez, R.; Kim, N.C.; Almeida, S.; Lee, K.-H.; Badders, N.; Valentine, M.; Miller, B.L.; Wong, P.C.; et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nat. Cell Biol. 2015, 525, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Donnelly, C.J.; Haeusler, A.R.; Grima, J.C.; Machamer, J.B.; Steinwald, P.; Daley, E.; Miller, S.J.; Cunningham, K.; Vidensky, S.; et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nat. Cell Biol. 2015, 525, 56–61. [Google Scholar] [CrossRef] [Green Version]
- Jovičić, A.; Mertens, J.; Boeynaems, S.; Bogaert, E.; Chai, N.; Yamada, S.B.; Paul, J.W., 3rd; Sun, S.; Herdy, J.R.; Bieri, G.; et al. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat. Neurosci. 2015, 18, 1226–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boeynaems, S.; Bogaert, E.; Michiels, E.; Gijselinck, I.; Sieben, A.; Jovičić, A.; De Baets, G.; Scheveneels, W.; Steyaert, J.; Cuijt, I.; et al. Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci. Rep. 2016, 6, 20877. [Google Scholar] [CrossRef]
- Chai, N.; Gitler, A.D. Yeast screen for modifiers of C9orf72 poly(glycine-arginine) dipeptide repeat toxicity. FEMS Yeast Res. 2018, 18, foy024. [Google Scholar] [CrossRef]
- Chou, C.-C.; Zhang, Y.; Umoh, M.E.; Vaughan, S.W.; Lorenzini, I.; Liu, F.; Sayegh, M.; Donlin-Asp, P.; Chen, Y.H.; Duong, D.; et al. TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat. Neurosci. 2018, 21, 228–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knauer, S.K.; Moodt, S.; Berg, T.; Liebel, U.; Pepperkok, R.; Stauber, R.H. Translocation Biosensors to Study Signal-Specific Nucleo-Cytoplasmic Transport, Protease Activity and Protein-Protein Interactions. Traffic 2005, 6, 594–606. [Google Scholar] [CrossRef]
- Henderson, B.R.; Percipallea, P. Interactions between HIV rev and nuclear import and export factors: The rev nuclear localisation signal mediates specific binding to human importin-β. J. Mol. Biol. 1997, 274, 693–707. [Google Scholar] [CrossRef]
- Henderson, B.R.; Eleftheriou, A. A Comparison of the Activity, Sequence Specificity, and CRM1-Dependence of Different Nuclear Export Signals. Exp. Cell Res. 2000, 256, 213–224. [Google Scholar] [CrossRef]
- Zhang, J.-H.; Chung, T.D.Y.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef]
- Sorokin, A.V.; Kim, E.R.; Ovchinnikov, L.P. Nucleocytoplasmic transport of proteins. Biochemistry 2007, 72, 1439–1457. [Google Scholar] [CrossRef] [PubMed]
- Marfori, M.; Mynott, A.; Ellis, J.; Mehdi, A.M.; Saunders, N.; Curmi, P.; Forwood, J.; Bodén, M.; Kobe, B. Molecular basis for specificity of nuclear import and prediction of nuclear localization. Biochim. Biophys. Acta 2011, 1813, 1562–1577. [Google Scholar] [CrossRef] [PubMed]
- Fontes, M.; Teh, T.; Kobe, B. Structural basis of recognition of monopartite and bipartite nuclear localization sequences by mammalian importin-α. J. Mol. Biol. 2000, 297, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Lange, A.; McLane, L.M.; Mills, R.E.; Devine, S.E.; Corbett, A.H. Expanding the Definition of the Classical Bipartite Nuclear Localization Signal. Traffic 2010, 11, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.J.; Cansizoglu, A.E.; Süel, K.E.; Louis, T.H.; Zhang, Z.; Chook, Y.M. Rules for Nuclear Localization Sequence Recognition by Karyopherin β2. Cell 2006, 126, 543–558. [Google Scholar] [CrossRef] [Green Version]
- Lai, M.-C.; Lin, R.-I.; Huang, S.-Y.; Tsai, C.-W.; Tarn, W.-Y. A Human Importin-β Family Protein, Transportin-SR2, Interacts with the Phosphorylated RS Domain of SR Proteins. J. Biol. Chem. 2000, 275, 7950–7957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pradeepa, M.M.; Manjunatha, S.; Sathish, V.; Agrawal, S.; Rao, M.R.S. Involvement of Importin-4 in the Transport of Transition Protein 2 into the Spermatid Nucleus. Mol. Cell. Biol. 2008, 28, 4331–4341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ploski, J.E.; Shamsher, M.K.; Radu, A. Paired-Type Homeodomain Transcription Factors Are Imported into the Nucleus by Karyopherin Mol. Cell. Biol. 2004, 24, 4824–4834. [Google Scholar] [CrossRef] [Green Version]
- Tabata, H.; Koinui, A.; Ogura, A.; Nishihara, D.; Yamamoto, H. A novel nuclear localization signal spans the linker of the two DNA-binding subdomains in the conserved paired domain of Pax6. Genes Genet. Syst. 2018, 93, 75–81. [Google Scholar] [CrossRef]
- Dingwall, C.; Robbins, J.; Dilworth, S.M.; Roberts, B.; Richardson, W.D. The nucleoplasmin nuclear location sequence is larger and more complex than that of SV-40 large T antigen. J. Cell Biol. 1988, 107, 841–849. [Google Scholar] [CrossRef]
- Soniat, M.; Chook, Y.M. Karyopherin-β2 Recognition of a PY-NLS Variant that Lacks the Proline-Tyrosine Motif. Structure 2016, 24, 1802–1809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ossareh-Nazari, B.; Bachelerie, F.; Dargemont, C. Evidence for a Role of CRM1 in Signal-Mediated Nuclear Protein Export. Science 1997, 278, 141–144. [Google Scholar] [CrossRef]
- Stade, K.; Ford, C.S.; Guthrie, C.; Weis, K. Exportin 1 (Crm1p) Is an Essential Nuclear Export Factor. Cell 1997, 90, 1041–1050. [Google Scholar] [CrossRef] [Green Version]
- Neville, M.; Stutz, F.; Lee, L.; I Davis, L.; Rosbash, M. The importin-beta family member Crm1p bridges the interaction between Rev and the nuclear pore complex during nuclear export. Curr. Biol. 1997, 7, 767–775. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Zhang, H.; Wu, X.; He, Z.; Wang, L.; Yin, S.; Tian, B.; Li, G.; Cheng, H. ALYREF mainly binds to the 5′ and the 3′ regions of the mRNA in vivo. Nucleic Acids Res. 2017, 45, 9640–9653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Wang, K.; Du, X.; Wang, J.; Chen, S.; Wang, Y.; Shi, M.; Zhang, L.; Wu, X.; Zheng, D.; et al. ALYREF links 3′-end processing to nuclear export of non-polyadenylated mRNA s. EMBO J. 2019, 38, 99910. [Google Scholar] [CrossRef]
- Andrés-Benito, P.; Gelpi, E.; Povedano, M.; Ausín, K.; Fernández-Irigoyen, J.; Santamaría, E.; Ferrer, I. Combined Transcriptomics and Proteomics in Frontal Cortex Area 8 in Frontotemporal Lobar Degeneration Linked to C9ORF72 Expansion. J. Alzheimer’s Dis. 2019, 68, 1287–1307. [Google Scholar] [CrossRef] [Green Version]
- DeJesus-Hernandez, M.; Finch, N.A.; Wang, X.; Gendron, T.F.; Bieniek, K.; Heckman, M.G.; Vasilevich, A.; Murray, M.; Rousseau, L.; Weesner, R.; et al. In-depth clinico-pathological examination of RNA foci in a large cohort of C9ORF72 expansion carriers. Acta Neuropathol. 2017, 134, 255–269. [Google Scholar] [CrossRef]
- Yin, S.; Lopez-Gonzalez, R.; Kunz, R.C.; Gangopadhyay, J.; Borufka, C.; Gygi, S.P.; Gao, F.-B.; Reed, R. Evidence that C9ORF72 Dipeptide Repeat Proteins Associate with U2 snRNP to Cause Mis-splicing in ALS/FTD Patients. Cell Rep. 2017, 19, 2244–2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper-Knock, J.; Walsh, M.J.; Higginbottom, A.; Highley, R.; Dickman, M.; Edbauer, D.; Ince, P.G.; Wharton, S.B.; Wilson, S.; Kirby, J.; et al. Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions. Brain 2014, 137, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Kudo, N.; Matsumori, N.; Taoka, H.; Fujiwara, D.; Schreiner, E.P.; Wolff, B.; Yoshida, M.; Horinouchi, S. Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc. Natl. Acad. Sci. USA 1999, 96, 9112–9117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanella, F.; Rosado, A.; Blanco, F.; Henderson, B.R.; Carnero, A.; Link, W. An HTS Approach to Screen for Antagonists of the Nuclear Export Machinery Using High Content Cell-Based Assays. ASSAY Drug Dev. Technol. 2007, 5, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.Y.; Mori, E.; Nizami, Z.F.; Lin, Y.; Kato, M.; Xiang, S.; Wu, L.C.; Ding, M.; Yu, Y.; Gall, J.G.; et al. Toxic PRn poly-dipeptides encoded by the C9orf72 repeat expansion block nuclear import and export. Proc. Natl. Acad. Sci. USA 2017, 114, E1111–E1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizielinska, S.; Grönke, S.; Niccoli, T.; Ridler, C.E.; Clayton, E.L.; Devoy, A.; Moens, T.; Norona, F.E.; Woollacott, I.O.C.; Pietrzyk, J.; et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 2014, 345, 1192–1194. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.-J.; Jeng, U.-S.; Chiang, Y.-L.; Hwang, I.-S.; Chen, Y.-R. The Glycine-Alanine Dipeptide Repeat from C9orf72 Hexanucleotide Expansions Forms Toxic Amyloids Possessing Cell-to-Cell Transmission Properties. J. Biol. Chem. 2016, 291, 4903–4911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- May, S.; Hornburg, D.; Schludi, M.H.; Arzberger, T.; Rentzsch, K.; Schwenk, B.M.; Grässer, F.A.; Mori, K.; Kremmer, E.; Banzhaf-Strathmann, J.; et al. C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 seques-tration. Acta Neuropathol. 2014, 128, 485–503. [Google Scholar] [CrossRef] [Green Version]
- Khosravi, B.; Hartmann, H.; May, S.; Möhl, C.; Ederle, H.; Michaelsen, M.; Schludi, M.H.; Dormann, D.; Edbauer, D. Cytoplasmic poly-GA aggregates impair nuclear import of TDP-43 in C9orf72 ALS/FTLD. Hum. Mol. Genet. 2016, 26, 790–800. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Kim, H.J.; Wang, H.; Monaghan, J.; Freyermuth, F.; Sung, J.C.; O’Donovan, K.; Fare, C.; Diaz, Z.; Singh, N.; et al. Nuclear-Import Receptors Reverse Aberrant Phase Transitions of RNA-Binding Proteins with Prion-like Domains. Cell 2018, 173, 677–692. [Google Scholar] [CrossRef] [Green Version]
- Hutten, S.; Usluer, S.; Bourgeois, B.; Simonetti, F.; Odeh, H.M.; Fare, C.M.; Czuppa, M.; Hruska-Plochan, M.; Hofweber, M.; Polymenidou, M.; et al. Nuclear Import Receptors Directly Bind to Arginine-Rich Dipeptide Repeat Proteins and Suppress Their Pathological Interactions. Cell Rep. 2020, 33, 108538. [Google Scholar] [CrossRef]
- Fazal, R.; Boeynaems, S.; Swijsen, A.; De Decker, M.; Fumagalli, L.; Moisse, M.; Vanneste, J.; Guo, W.; Boon, R.; Vercruysse, T.; et al. HDAC6 inhibition restores TDP-43 pathology and axonal transport defects in human motor neurons with TARDBP mutations. EMBO J. 2021, 40, e106177. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Arai, T.; Nonaka, T.; Kametani, F.; Yoshida, M.; Hashizume, Y.; Beach, T.G.; Buratti, E.; Baralle, F.; Morita, M.; et al. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol. 2008, 64, 60–70. [Google Scholar] [CrossRef] [Green Version]
- Bilican, B.; Serio, A.; Barmada, S.J.; Nishimura, A.; Sullivan, G.; Carrasco, M.; Phatnani, H.P.; Puddifoot, C.A.; Story, D.; Fletcher, J.; et al. Mutant induced pluripotent stem cell lines recapitulate aspects of TDP-43 proteinopathies and reveal cell-specific vulnerability. Proc. Natl. Acad. Sci. USA 2012, 109, 5803–5808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geser, F.; Winton, M.J.; Kwong, L.K.; Xu, Y.; Xie, S.X.; Igaz, L.M.; Garruto, R.M.; Perl, D.P.; Galasko, D.; Lee, V.M.-Y.; et al. Pathological TDP-43 in parkinsonism–dementia complex and amyotrophic lateral sclerosis of Guam. Acta Neuropathol. 2007, 115, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Gendron, T.F.; Bieniek, K.; Zhang, Y.-J.; Jansen-West, K.; Ash, P.E.A.; Caulfield, T.; Daughrity, L.; Dunmore, J.H.; Castanedes-Casey, M.; Chew, J.; et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 2013, 126, 829–844. [Google Scholar] [CrossRef] [Green Version]
- Melamed, Z.; López-Erauskin, J.; Baughn, M.; Zhang, O.; Drenner, K.; Sun, Y.; Freyermuth, F.; McMahon, M.A.; Beccari, M.S.; Artates, J.W.; et al. Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat. Neurosci. 2019, 22, 180–190. [Google Scholar] [CrossRef]
- Prudencio, M.; Humphrey, J.; Pickles, S.; Brown, A.-L.; Hill, S.E.; Kachergus, J.M.; Shi, J.; Heckman, M.G.; Spiegel, M.R.; Cook, C.; et al. Truncated stathmin-2 is a marker of TDP-43 pathology in frontotemporal dementia. J. Clin. Investig. 2020, 130, 6080–6092. [Google Scholar] [CrossRef]
- Kalderon, D.; Roberts, B.L.; Richardson, W.D.; Smith, A.E. A short amino acid sequence able to specify nuclear location. Cell 1984, 39, 499–509. [Google Scholar] [CrossRef]
- Zhang, Y.-J.; Jansen-West, K.; Xu, Y.-F.; Gendron, T.F.; Bieniek, K.; Lin, W.-L.; Sasaguri, H.; Caulfield, T.; Hubbard, J.; Daughrity, L.; et al. Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol. 2014, 128, 505–524. [Google Scholar] [CrossRef] [Green Version]
- Archbold, H.C.; Jackson, K.L.; Arora, A.; Weskamp, K.; Tank, E.M.-H.; Li, X.; Miguez, R.; Dayton, R.D.; Tamir, S.; Klein, R.L.; et al. TDP43 nuclear export and neurodegeneration in models of amyotrophic lateral sclerosis and frontotemporal dementia. Sci. Rep. 2018, 8, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Bruneteau, G.; Simonet, T.; Bauché, S.; Mandjee, N.; Malfatti, E.; Girard, E.; Tanguy, M.-L.; Béhin, A.; Khiami, F.; Sariali, E.; et al. Muscle histone deacetylase 4 upregulation in amyotrophic lateral sclerosis: Potential role in reinnervation ability and disease progression. Brain 2013, 136, 2359–2368. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Goodrich, K.J.; Conlon, E.G.; Gao, J.; Erbse, A.; Manley, J.L.; Cech, T.R. C9orf72 and triplet repeat disorder RNAs: G-quadruplex formation, binding to PRC2 and implications for disease mechanisms. RNA 2019, 25, 935–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belzil, V.V.; Bauer, P.O.; Prudencio, M.; Gendron, T.F.; Stetler, C.T.; Yan, I.K.; Pregent, L.; Daughrity, L.; Baker, M.C.; Rademakers, R.; et al. Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol. 2013, 126, 895–905. [Google Scholar] [CrossRef] [Green Version]
- Liu, E.Y.; Russ, J.; Wu, K.; Neal, D.; Suh, E.; McNally, A.G.; Irwin, D.; Van Deerlin, V.M.; Lee, E.B. C9orf72 hypermethylation protects against repeat expansion-associated pathology in ALS/FTD. Acta Neuropathol. 2014, 128, 525–541. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Chatterjee, S.; Bousiges, O.; Selvi, B.R.; Swaminathan, A.; Cassel, R.; Blanc, F.; Kundu, T.K.; Boutillier, A.-L. Acetyltransferases (HATs) as Targets for Neurological Therapeutics. Neurotherapeutics 2013, 10, 568–588. [Google Scholar] [CrossRef] [PubMed]
- Corman, A.; Jung, B.; Häggblad, M.; Bräutigam, L.; Lafarga, V.; Lidemalm, L.; Hühn, D.; Carreras-Puigvert, J.; Fernandez-Capetillo, O. A Chemical Screen Identifies Compounds Limiting the Toxicity of C9ORF72 Dipeptide Repeats. Cell Chem. Biol. 2019, 26, 235–243.e5. [Google Scholar] [CrossRef]
- Ryu, H.; Smith, K.; Camelo, S.I.; Carreras, I.; Lee, J.; Iglesias, A.H.; Dangond, F.; Cormier, K.A.; Cudkowicz, M.E.; Brown, R.H.; et al. Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J. Neurochem. 2005, 93, 1087–1098. [Google Scholar] [CrossRef]
- Paganoni, S.; Macklin, E.A.; Hendrix, S.; Berry, J.D.; Elliott, M.A.; Maiser, S.; Karam, C.; Caress, J.B.; Owegi, M.A.; Quick, A.; et al. Trial of Sodium Phenylbutyrate–Taurursodiol for Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2020, 383, 919–930. [Google Scholar] [CrossRef]
- Guo, W.; Naujock, M.; Fumagalli, L.; Vandoorne, T.; Baatsen, P.; Boon, R.; Ordovás, L.; Patel, A.; Welters, M.; Vanwelden, T.; et al. HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat. Commun. 2017, 8, 1–15. [Google Scholar] [CrossRef]
- Zeier, Z.; Esanov, R.; Belle, K.C.; Volmar, C.-H.; Johnstone, A.L.; Halley, P.; DeRosa, B.A.; Khoury, N.; van Blitterswijk, M.; Rademakers, R.; et al. Bromodomain inhibitors regulate the C9ORF72 locus in ALS. Exp. Neurol. 2015, 271, 241–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woerner, A.C.; Frottin, F.; Hornburg, D.; Feng, L.R.; Meissner, F.; Patra, M.; Tatzelt, J.; Mann, M.; Winklhofer, K.F.; Hartl, F.U.; et al. Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science 2016, 351, 173–176. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biosensor | NLS Type | NLS Sequence | Importin Recognized | NES Type | NES Sequence | Exportin Recognized |
|---|---|---|---|---|---|---|
| NCT-C | cNLS | PKKKRKV | KNPA1-KPNB1 | cNES | KEVDQLRLERLQIDEQL | XPO1 |
| NCT-01 | cNLS R5A mutant | PKKKAKV | KNPA1-KPNB1 | cNES | KEVDQLRLERLQIDEQL | XPO1 |
| NCT-02 | cNLS K6A mutant | PKKKRAVE | KNPA1-KPNB1 | cNES | KEVDQLRLERLQIDEQL | XPO1 |
| NCT-03 | cNLS (bipartite) | KRPAATKKAGQAKKKK | KNPA1-KPNB1 | cNES | KEVDQLRLERLQIDEQL | XPO1 |
| NCT-04 | Non-classical NLS | GKISKHWTG | KNPA1-KPNB1 | cNES | KEVDQLRLERLQIDEQL | XPO1 |
| NCT-05 | IB1-NLS | RRKKKEYVK | KNPB1 | cNES | KEVDQLRLERLQIDEQL | XPO1 |
| NCT-06 | IB1-NLS | RKKRRQRRR | KNPB1 | cNES | KEVDQLRLERLQIDEQL | XPO1 |
| NCT-07 | PY-NLS (basic, M9) | FGNYNNQSSNFGPMKGGNFGGRSSGPY | KNPB2 | cNES | KEVDQLRLERLQIDEQL | XPO1 |
| NCT-08 | PY-NLS (hydrophobic) | YGDYSNQQSGYGKVSRRGGHQNSYKPY | KNPB2 | cNES | KEVDQLRLERLQIDEQL | XPO1 |
| NCT-09 | PY-NLS (atypical) | GPGKMDSRGEHRQDRR-ERPY | KNPB2 | cNES | KEVDQLRLERLQIDEQL | XPO1 |
| NCT-10 | I4-NLS | GKVSKRKAV | IPO4 | cNES | KEVDQLRLERLQIDEQL | XPO1 |
| NCT-11 | I5-NLS | HTPQRVLPLKKPPMKSLRKKGSGKILTPAKKSFL | IPO5 | cNES | KEVDQLRLERLQIDEQL | XPO1 |
| NCT-13 | RS-NLS (RD mimic) | RDPSYG(RD)8NDRDRDYSPRRDRGSPRYSPRHDRDRDRT | TNPO3 | cNES | KEVDQLRLERLQIDEQL | XPO1 |
| NCT-14 | Homeodomain-NLS | RKLQRNRTSFTQEQIEALEKEFERTHYPDVFARERLAAKIDLPEARIQVWFSNRRAKWRREE | IPO13 | cNES | KEVDQLRLERLQIDEQL | XPO1 |
| NCT-15 | cNLS (monopartite) | PKKKRKV | KNPA1-KPNB1 | cNES | KEVDQLRLERLQIDEQL | XPO1 |
| NCT-A | cNLS | PKKKRKV | KNPA1-KPNB1 | NXF1-NXT1 mRNA | APPAQPPSQPQQHYSEGELEEDEDSDDA | ALYREF Adapter |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramic, M.; Andrade, N.S.; Rybin, M.J.; Esanov, R.; Wahlestedt, C.; Benatar, M.; Zeier, Z. Epigenetic Small Molecules Rescue Nucleocytoplasmic Transport and DNA Damage Phenotypes in C9ORF72 ALS/FTD. Brain Sci. 2021, 11, 1543. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci11111543
Ramic M, Andrade NS, Rybin MJ, Esanov R, Wahlestedt C, Benatar M, Zeier Z. Epigenetic Small Molecules Rescue Nucleocytoplasmic Transport and DNA Damage Phenotypes in C9ORF72 ALS/FTD. Brain Sciences. 2021; 11(11):1543. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci11111543
Chicago/Turabian StyleRamic, Melina, Nadja S. Andrade, Matthew J. Rybin, Rustam Esanov, Claes Wahlestedt, Michael Benatar, and Zane Zeier. 2021. "Epigenetic Small Molecules Rescue Nucleocytoplasmic Transport and DNA Damage Phenotypes in C9ORF72 ALS/FTD" Brain Sciences 11, no. 11: 1543. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci11111543