
Is It Time to Test the Antiseizure Potential of Palmitoylethanolamide in Human Studies? A Systematic Review of Preclinical Evidence




Abstract
:1. Introduction
Objectives
2. Experimental Procedures
2.1. Inclusion and Exclusion Criteria
2.2. Search Strategy and Data Extraction
2.3. Risk of Bias
3. Results
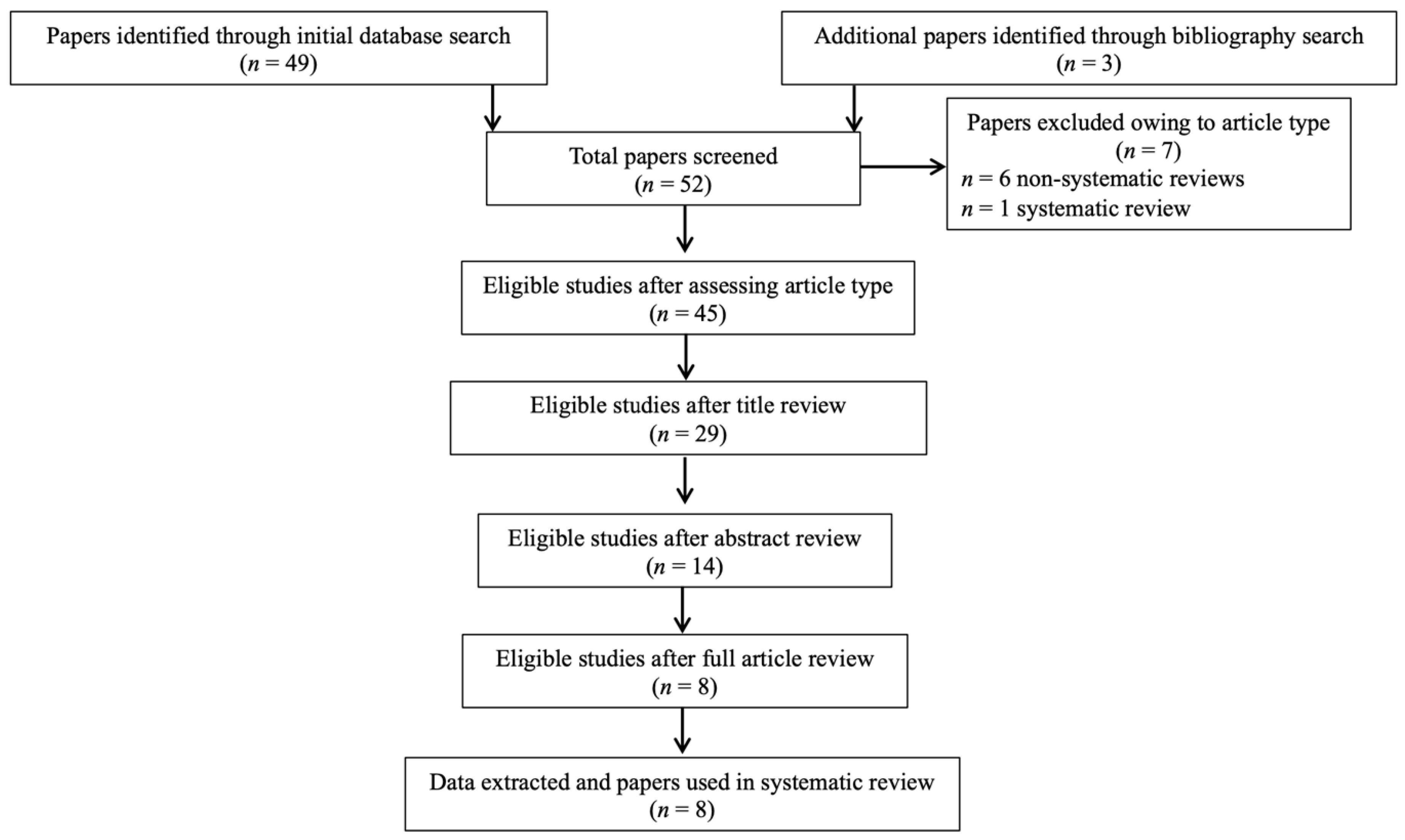
3.1. Study Selection
3.2. In Vivo Acute and Subchronic PEA Treatment Exposure and Comparison with Other Endocannabinoids (eCBs) and ASMs Exposure in Animal Models of Epilepsy and Acute Seizures
3.3. PEA Quantitative Brain and Peripheral Tissue Assessment and Comparison as a Function of Age in Animal Models of Epilepsy and Acute Seizures
3.4. Endocannabinoid (eCB) and Eicosanoid (eiC) Quantitative Brain and Blood Assessment in PEA-Treated Animal Models of Acute Seizures
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fisher, R.S.; Acevedo, C.; Arzimanoglou, A.; Bogacz, A.; Cross, J.H.; Elger, C.E.; Engel, J.; Forsgren, L.; French, J.A.; Glynn, M.; et al. ILAE official report: A practical clinical definition of epilepsy. Epilepsia 2014, 55, 475–482. [Google Scholar] [CrossRef] [Green Version]
- Falco-Walter, J. Epilepsy-Definition, Classification, Pathophysiology, and Epidemiology. Semin. Neurol. 2020, 40, 617–623. [Google Scholar] [CrossRef]
- Kaur, S.; Garg, R.; Aggarwal, S.; Chawla, S.P.S.; Pal, R. Adult onset seizures: Clinical, etiological, and radiological profile. J. Fam. Med. Prim. Care 2018, 7, 191–197. [Google Scholar] [CrossRef]
- Carpay, J.A.; Aldenkamp, A.P.; van Donselaar, C.A. Complaints associated with the use of antiepileptic drugs: Results from a community-based study. Seizure 2005, 14, 198–206. [Google Scholar] [CrossRef] [Green Version]
- Ochoa, J.G.; Kilgo, W.A. The Role of Benzodiazepines in the Treatment of Epilepsy. Curr. Treat. Options Neurol. 2016, 18, 18. [Google Scholar] [CrossRef]
- Marsicano, G.; Goodenough, S.; Monory, K.; Hermann, H.; Eder, M.; Cannich, A.; Azad, S.C.; Cascio, M.G.; Gutiérrez, S.O.; van der Stelt, M.; et al. CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science 2003, 302, 84–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monory, K.; Massa, F.; Egertová, M.; Eder, M.; Blaudzun, H.; Westenbroek, R.; Kelsch, W.; Jacob, W.; Marsch, R.; Ekker, M.; et al. The endocannabinoid system controls key epileptogenic circuits in the hippocampus. Neuron 2006, 51, 455–466. [Google Scholar] [CrossRef] [Green Version]
- Rankin, L.; Fowler, C.J. The Basal Pharmacology of Palmitoylethanolamide. Int. J. Mol. Sci. 2020, 21, 7942. [Google Scholar] [CrossRef] [PubMed]
- Lo Verme, J.; Fu, J.; Astarita, G.; La Rana, G.; Russo, R.; Calignano, A.; Piomelli, D. The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Mol. Pharmacol. 2005, 67, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Solorzano, C.; Zhu, C.; Battista, N.; Astarita, G.; Lodola, A.; Rivara, S.; Mor, M.; Russo, R.; Maccarrone, M.; Antonietti, F.; et al. Selective N-acylethanolamine-hydrolyzing acid amidase inhibition reveals a key role for endogenous palmitoylethanolamide in inflammation. Proc. Natl. Acad. Sci. USA 2009, 106, 20966–20971. [Google Scholar] [CrossRef] [Green Version]
- Jaggar, S.I.; Hasnie, F.S.; Sellaturay, S.; Rice, A.S. The anti-hyperalgesic actions of the cannabinoid anandamide and the putative CB2 receptor agonist palmitoylethanolamide in visceral and somatic inflammatory pain. Pain 1998, 76, 189–199. [Google Scholar] [CrossRef]
- Yu, H.L.; Deng, X.Q.; Li, Y.J.; Li, Y.C.; Quan, Z.S.; Sun, X.Y. N-palmitoylethanolamide, an endocannabinoid, exhibits antidepressant effects in the forced swim test and the tail suspension test in mice. Pharmacol. Rep. 2011, 63, 834–839. [Google Scholar] [CrossRef]
- Colizzi, M.; Bortoletto, R.; Costa, R.; Zoccante, L. Palmitoylethanolamide and Its Biobehavioral Correlates in Autism Spectrum Disorder: A Systematic Review of Human and Animal Evidence. Nutrients 2021, 13, 1346. [Google Scholar] [CrossRef]
- West, S.; King, V.; Carey, T.S.; Lohr, K.N.; McKoy, N.; Sutton, S.F.; Lux, L. Systems to rate the strength of scientific evidence. Evid. Rep./Technol. Assess. (Summ.) 2002, 1–11. Available online: https://europepmc.org/article/nbk/nbk33881 (accessed on 4 December 2021).
- Lambert, D.M.; Vandevoorde, S.; Diependaele, G.; Govaerts, S.J.; Robert, A.R. Anticonvulsant activity of N-palmitoylethanolamide, a putative endocannabinoid, in mice. Epilepsia 2001, 42, 321–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Citraro, R.; Russo, E.; Leo, A.; Russo, R.; Avagliano, C.; Navarra, M.; Calignano, A.; De Sarro, G. Pharmacokinetic-pharmacodynamic influence of N-palmitoylethanolamine, arachidonyl-2’-chloroethylamide and WIN 55,212-2 on the anticonvulsant activity of antiepileptic drugs against audiogenic seizures in DBA/2 mice. Eur. J. Pharmacol. 2016, 791, 523–534. [Google Scholar] [CrossRef] [Green Version]
- Post, J.M.; Loch, S.; Lerner, R.; Remmers, F.; Lomazzo, E.; Lutz, B.; Bindila, L. Antiepileptogenic Effect of Subchronic Palmitoylethanolamide Treatment in a Mouse Model of Acute Epilepsy. Front. Mol. Neurosci. 2018, 11, 67. [Google Scholar] [CrossRef] [Green Version]
- Sheerin, A.H.; Zhang, X.; Saucier, D.M.; Corcoran, M.E. Selective antiepileptic effects of N-palmitoylethanolamide, a putative endocannabinoid. Epilepsia 2004, 45, 1184–1188. [Google Scholar] [CrossRef]
- Citraro, R.; Russo, E.; Scicchitano, F.; van Rijn, C.M.; Cosco, D.; Avagliano, C.; Russo, R.; D’Agostino, G.; Petrosino, S.; Guida, F.; et al. Antiepileptic action of N-palmitoylethanolamine through CB1 and PPAR-α receptor activation in a genetic model of absence epilepsy. Neuropharmacology 2013, 69, 115–126. [Google Scholar] [CrossRef]
- Aghaei, I.; Rostampour, M.; Shabani, M.; Naderi, N.; Motamedi, F.; Babaei, P.; Khakpour-Taleghani, B. Palmitoylethanolamide attenuates PTZ-induced seizures through CB1 and CB2 receptors. Epilepsy Res. 2015, 117, 23–28. [Google Scholar] [CrossRef]
- Lerner, R.; Post, J.; Loch, S.; Lutz, B.; Bindila, L. Targeting brain and peripheral plasticity of the lipidome in acute kainic acid-induced epileptic seizures in mice via quantitative mass spectrometry. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Fezza, F.; Marrone, M.C.; Avvisati, R.; Di Tommaso, M.; Lanuti, M.; Rapino, C.; Mercuri, N.B.; Maccarrone, M.; Marinelli, S. Distinct modulation of the endocannabinoid system upon kainic acid-induced in vivo seizures and in vitro epileptiform bursting. Mol. Cell. Neurosci. 2014, 62, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Farrelly, A.M.; Vlachou, S.; Grintzalis, K. Efficacy of Phytocannabinoids in Epilepsy Treatment: Novel Approaches and Recent Advances. Int. J. Environ. Res. Public Health 2021, 18, 3993. [Google Scholar] [CrossRef]
- Espinosa-Jovel, C. Cannabinoids in epilepsy: Clinical efficacy and pharmacological considerations. Neurologia 2021, (in press). [Google Scholar] [CrossRef] [PubMed]
- Colizzi, M.; Ruggeri, M.; Bhattacharyya, S. Unraveling the Intoxicating and Therapeutic Effects of Cannabis Ingredients on Psychosis and Cognition. Front. Psychol. 2020, 11, 833. [Google Scholar] [CrossRef]
- Laprairie, R.B.; Bagher, A.M.; Kelly, M.E.; Denovan-Wright, E.M. Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. Br. J. Pharmacol. 2015, 172, 4790–4805. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, S.E. An update on PPAR activation by cannabinoids. Br. J. Pharmacol. 2016, 173, 1899–1910. [Google Scholar] [CrossRef] [Green Version]
- Filipiuc, L.E.; Ababei, D.C.; Alexa-Stratulat, T.; Pricope, C.V.; Bild, V.; Stefanescu, R.; Stanciu, G.D.; Tamba, B.I. Major Phytocannabinoids and Their Related Compounds: Should We Only Search for Drugs That Act on Cannabinoid Receptors? Pharmaceutics 2021, 13, 1823. [Google Scholar] [CrossRef]
- Maier, N.; Morris, G.; Schuchmann, S.; Korotkova, T.; Ponomarenko, A.; Böhm, C.; Wozny, C.; Schmitz, D. Cannabinoids disrupt hippocampal sharp wave-ripples via inhibition of glutamate release. Hippocampus 2012, 22, 1350–1362. [Google Scholar] [CrossRef] [PubMed]
- Polissidis, A.; Galanopoulos, A.; Naxakis, G.; Papahatjis, D.; Papadopoulou-Daifoti, Z.; Antoniou, K. The cannabinoid CB1 receptor biphasically modulates motor activity and regulates dopamine and glutamate release region dependently. Int. J. Neuropsychopharmacol. 2013, 16, 393–403. [Google Scholar] [CrossRef] [Green Version]
- Ruehle, S.; Remmers, F.; Romo-Parra, H.; Massa, F.; Wickert, M.; Wörtge, S.; Häring, M.; Kaiser, N.; Marsicano, G.; Pape, H.C.; et al. Cannabinoid CB1 receptor in dorsal telencephalic glutamatergic neurons: Distinctive sufficiency for hippocampus-dependent and amygdala-dependent synaptic and behavioral functions. J. Neurosci. 2013, 33, 10264–10277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Blázquez, P.; Rodríguez-Muñoz, M.; Garzón, J. The cannabinoid receptor 1 associates with NMDA receptors to produce glutamatergic hypofunction: Implications in psychosis and schizophrenia. Front. Pharmacol. 2014, 4, 169. [Google Scholar] [CrossRef] [Green Version]
- Albayram, O.; Alferink, J.; Pitsch, J.; Piyanova, A.; Neitzert, K.; Poppensieker, K.; Mauer, D.; Michel, K.; Legler, A.; Becker, A.; et al. Role of CB1 cannabinoid receptors on GABAergic neurons in brain aging. Proc. Natl. Acad. Sci. USA 2011, 108, 11256–11261. [Google Scholar] [CrossRef] [Green Version]
- Antonucci, F.; Alpár, A.; Kacza, J.; Caleo, M.; Verderio, C.; Giani, A.; Martens, H.; Chaudhry, F.A.; Allegra, M.; Grosche, J.; et al. Cracking down on inhibition: Selective removal of GABAergic interneurons from hippocampal networks. J. Neurosci. 2012, 32, 1989–2001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair, R.E.; Deshpande, L.S.; Sombati, S.; Elphick, M.R.; Martin, B.R.; DeLorenzo, R.J. Prolonged exposure to WIN55,212-2 causes downregulation of the CB1 receptor and the development of tolerance to its anticonvulsant effects in the hippocampal neuronal culture model of acquired epilepsy. Neuropharmacology 2009, 57, 208–218. [Google Scholar] [CrossRef] [Green Version]
- Karlócai, M.R.; Tóth, K.; Watanabe, M.; Ledent, C.; Juhász, G.; Freund, T.F.; Maglóczky, Z. Redistribution of CB1 cannabinoid receptors in the acute and chronic phases of pilocarpine-induced epilepsy. PLoS ONE 2011, 6, e27196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skaper, S.D.; Facci, L.; Giusti, P. Mast cells, glia and neuroinflammation: Partners in crime? Immunology 2014, 141, 314–327. [Google Scholar] [CrossRef]
- Skaper, S.D.; Facci, L. Mast cell-glia axis in neuroinflammation and therapeutic potential of the anandamide congener palmitoylethanolamide. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012, 367, 3312–3325. [Google Scholar] [CrossRef]
- Bracken, M.B. Why animal studies are often poor predictors of human reactions to exposure. J. R. Soc. Med. 2009, 102, 120–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Study (Country) | Aim of Study | PEA Type of Study | Population | N | Outcome Measure (Test Name or Description) | Seizure Model | Summary Results |
|---|---|---|---|---|---|---|---|
| Lambert et al. (2001) (Belgium) | 1. To assess PEA effects in tonic-clonic seizures in mice; 2. To compare PEA effects to other ECBs/palmitoyl derivatives and ASMs | In-vivo exposure in animals | 1. MES test, 16 groups: (a) VHI (30 min/4 h); (b) PEA (100 mg/kg) (30 min/4 h); (c) PEA (50 mg/kg) (30 min/4 h); (d) AEA (30 min/4 h); (e) PA (30 min/4 h); (f) PAA (30 min/4 h); (g) HXD (30 min/4 h); (h) 16HPA (30 min/4 h); 2. Time course calculation, 8 groups: (a) 30 min; (b) 1 h; (c) 1 h 30 min; (d) 2 h; (e) 3 h; (f) 4 h; (g) 5 h; (h) 7 h; 3. ED50 calculation, 8 groups: (PEA range 0.5–50 mg/kg); 4. CIS test, 18 groups: (a) PTZ (VHI, PEA, PHT); (b) MPA (VHI, PEA, PHT); (c) BC (VHI, PEA, PHT); (d) STR (VHI, PEA, PHT); (e) PIC (VHI, PEA, PHT); (f) NMA (VHI, PEA, PHT); 5. Rotarod test group | 1. MES test: 15–16 per group; 2. Time course calculation: 7–8 per group; 3. ED50 calculation: 6–10 per group; 4. CIS test: 15–16 per group; 5. Rotarod test: 6–10 per group | 1. Antiseizure activity (MES test, CIS test); 2. Neurologic impairment (rotarod performance test) | 1. Maximal electroshock seizure; 2. Chemically-induced seizure | ED50 was comparable between PEA and PHT; PEA was effective only against tonic seizures and showed a high protective index |
| Sheerin et al. (2004) (Canada) | To assess PEA effects in tonic-clonic seizures in rats | In-vivo exposure in animals | 1. KS groups: 10 VHI (DMSO); 10 PEA (1); 8 PEA (10); 4 PEA (100); 2. CIS groups: 5 VHI; 5 PEA | 42 | Antiseizure activity (KS test; CIS test) | 1. Kindled amygdaloid seizure; 2. PTZ-induced seizure | PEA increased latency to clonus at 1 mg/kg, was effective against tonic seizures, and increased the latency between convulsive episodes |
| Citraro et al. (2013) (Italy) | 1. To assess PEA effects and PPAR-α role in absence seizures in rats; 2. To quantify PEA and other ECBs/AEs brain levels in epileptic and control rats | 1. In-vivo exposure in animals; 2. Quantitative brain assessment in animals | 1. 6 VHI (icv); 2. 6 VHI (ip); 3. 24 PEA (icv); 4. 24 PEA (ip); 5. 24 AEA (icv); 6. 18 SR (icv); 7. 12 GW (icv); 8. 12 SR (icv) + PEA(ip); 9. 6 GW(icv) + PEA (icv); 10. 6 SR (icv) + AEA (ip); 11. 6 GW (icv) + AEA (icv) | 144 | 1. Antiepileptic activity (EEG recording); 2. Brain ECBs/AEs levels (LC/APCI/MS) | Genetic model of absence epilepsy | PEA showed anti-absence properties depending on PPAR-α and indirect CB1 receptors activation |
| Fezza et al. (2014) (Italy) | To quantify PEA and other ECBs/AEs brain levels in epileptic and control young and adult rats | Quantitative brain assessment in animals | 1. In-vivo saline-injection: (a) 5 P14; 11 P56–70; 2. In-vivo KA-injection: (a) 5 P14; (b) 12 P56–70; 3. In-vitro KA bath perfusion: (a) 27 P14; (b) 36 P56–70 | 96 | 1. Brain ECB system activity (TLC, LC-ESI-MS); 2. Epileptic activity post ECB system manipulation (burst duration, burst amplitude, PSs number) | KA-induced seizure | PEA levels were higher in the hippocampus of younger KA-treated rats while decreasing in adults |
| Aghaei et al. (2015) (Iran) | To assess PEA effects in tonic-clonic seizures in rats | In-vivo exposure in animals | 1. PTZ; 2. VHI (PTZ + saline or DMSO); 3. PEA + PTZ; 4. AM630 + PTZ; 5. AM630 + PEA + PTZ; 6. AM251 + PTZ; 7. AM251 + PEA + PTZ; 8. AM251 + AM630 + PTZ; 9. AM251 + AM630 + PEA + PTZ | 220 | Antiseizure activity (CIS test) | PTZ-induced seizure | PEA reduced PTZ-induced seizures, and CB1/CB2 blockage reduced its effectiveness |
| Citraro et al. (2016) (Italy) | To assess PEA effects in tonic-clonic seizures in mice | In-vivo exposure in animals | 1. VHI; 2. PEA, 3. GW + PEA, 4. NIDA + PEA; 5. ACEA, 6. GW + ACEA, 7. NIDA + ACEA; 8. WIN, 9. GW + WIN, 10. NIDA + WIN; 11. (ASMs) + VHI/PEA/WIN/ACEA/NIDA/GW | X | 1. Antiepileptic activity (audiogenic seizures test); 2. Neurologic impairment (rotarod performance test); 3. Brain/plasma ASMs levels | Genetic model of reflex audiogenic epilepsy | PEA showed antiepileptic properties and potentiated the effect of several ASMs |
| Lerner et al. (2017) (Germany) | To quantify PEA, other ECBs/AEs and PLs/eiCs brain and peripheral tissue levels in epileptic and control mice | Brain and other tissues assessment in animals | 1. 9 KA; 2. 9 saline | 18 | Lipid profiling (LC/MRM quantitative and qualitative assessment of PLs/ECBs/AEs/eiCs) | KA-induced seizure | PEA levels were lower in the striatum, cerebellum, lung, and plasma of epileptic animals compared to controls |
| Post et al. (2018) (Germany) | 1. To assess acute and subchronic PEA effects in tonic-clonic seizures in mice; 2. To assess PEA effects on ECBs/eiCs brain and plasma levels; 3. To assess PEA effects on neurodegeneration | 1. In-vivo exposure in animals; 2. Quantitative brain and blood assessment in animals | 1. 48 KA; 2. 24 PEA2/KA; 3. 48 PEA/KA; 4. 24 CTRL1; 5. 24 CTRL2; 6. 24 PEA2; 7. 24 PEA + URB597/KA; 8. 24 URB597/KA; 9. 24 PEA + URB937/KA; 10. 24 URB937/KA (n = 6 per time point each group) | 288 | 1. Antiepileptogenic activity (behavioral assessment in KA-induced seizures, LC/MRM quantification of ECBs/eiCs); 2. Hippocampus neurodegenerative process (immunohistochemistry) | KA-induced seizure | PEA subchronic administration reduced seizure intensity, enhanced neuroprotection, and modulated ECBs/eiCs brain and plasma levels |
| Study (Country) | Results | ||||||
| Lambert et al. (2001) (Belgium) | 1. MES test: (a) % mice exhibiting seizures after 30 min: PEA (100–50 mg/kg), AEA, PAA < VHI; PA, HXD, 16HPA vs. VHI, NS; (b) Time of peak effect, ED50: PEA vs. PHT, NS; 2. CIS test: (a) % mice exhibiting clonic seizures: PEA vs. VHI, NS after each compound; PEA > PHT after PTZ, BC; (b) % mice exhibiting tonic seizures: PEA < VHI after PTZ, MPA, BC; PEA vs. VHI, NS after STR, PIC, NMA; PEA vs. PHT, NS after PTZ, MPA, BC; PEA > PHT, after PIC; 3. Rotarod test: (a) TD50: PEA > Ameltolide, PHT, CBZ, PB; PEA < VPA; (b) PI (TD50/ED50): PEA > each ASM | ||||||
| Sheerin et al. (2004) (Canada) | 1. KS test: (a) latency to clonus: PEA(1), ↑; VHI, PEA(10), PEA(100), NS; (b) Duration of clonus: VHI, PEA(1), PEA(10), PEA(100), NS; (c) AD duration: VHI, PEA(1), PEA(10), PEA(100), NS; 2. CIS test: (a) tonic seizures: PEA < VHI; (b) clonic convulsions: PEA vs. VHI, NS; (c) 1st convulsion latency, PEA vs. VHI, NS; (d) 1st convulsion duration, PEA vs. VHI, NS; (e) 2nd convulsion latency, PEA > VHI; (f) 2nd convulsion duration, PEA vs. VHI, NS | ||||||
| Citraro et al. (2013) (Italy) | 1. SWDs number and duration: PEA (1, 3, and 10 µg/2 µL (icv) and 20, 40, and 60 mg/kg (ip)) < VHI; PEA (0.5 µg/2 µL (icv) and 10 mg/kg (ip)) vs. VHI, NS; AEA < VHI (1, 3, and 10 µg/2 µL) with dose-dependent effect; AEA vs. VHI (0.5 µg/2 µL), NS; SR > VHI; GW vs. VHI, NS; SR (icv) + PEA (ip) vs. VHI, NS; GW (icv) + PEA (icv) vs. VHI, NS; SR (icv) + AEA (ip) vs. VHI, NS; GW (icv) + AEA (icv) < VHI; 2. AEA levels: ↓ with age in WAG/Rij in amygdala and cortex; ↓ with age in Wistar in thalamus; 2-AG levels: ↑ with age in ACI in amygdala; ↑ with age in Wistar in cortex; PEA levels: ↓ with age in ACI in thalamus; amygdala PEA levels: 6-month WAG-Rij < 6-month Wistar, ACI; cortex PEA levels: 6-month WAG-Rij > 6-month Wistar, ACI; thalamus PEA-levels: 2/6-month WAG-Rij < 2/6-month Wistar, ACI; amygdala AEA and 2-AG levels: 6-month WAG/Rij < 6-month ACI; 6-month WAG/Rij vs. 6-month Wistar, NS. | ||||||
| Fezza et al. (2014) (Italy) | 1. Hippocampi ECB system analysis post saline: PEA and AEA levels, FAAH and NAPE-PLD activity, CBR binding affinity: P14 < P56–70; MAGL and DAGL activity, 2-AG levels: P14 vs. P56–70, NS; 2. Seizure onset rapidity post KA: P14 > P56–70; female P56–70 vs. male P56–70, NS; 3. Hippocampi ECB system analysis post KA: FAAH and MAGL activity, CBR binding affinity: KA/P14 vs. saline/P14, KA/P56–70 vs. saline/P56–70, NS; PEA and AEA levels, NAPE-PLD activity: KA/P14 > saline/P14, KA/P56–70 < saline/P56–70; 2-AG levels, DAGL activity: KA/P14 < saline/P14, KA/P56–70 > saline/P56–70; 4. Burst duration: KA > saline; P14 > P56–70; P14: KA vs. URB597 + KA, NS; P14: KA > JZL + KA; P56–70: KA > URB597 + KA; P56–70: KA vs. JZL + KA, NS; P56–70: KA > KA + WIN; P56–70: KA + WIN < KA + WIN + SR; P56–70: KA < SR; KA < KA + SR; 5. Burst amplitude: KA > saline; P14 > P56–70; P14: KA > URB597 + KA; P14: KA > KA + WIN; P56–70: KA + WIN < KA + WIN + SR; P56–70: KA vs. SR, NS. 6. PSs number: P56–70: KA > KA + WIN; P56–70: KA vs. SR, NS; P56–70: KA < KA + SR. | ||||||
| Aghaei et al. (2015) (Iran) | 1. LP: PEA + PTZ > VHI; AM251 + PTZ (1.25, 2.5, 5 µg/kg) vs. VHI, NS; AM251 + PTZ (10 µg/kg) > VHI; AM630 + PTZ (2.5, 5 µg/kg) vs. VHI, NS; AM630 + PTZ (10, 20, 40 µg/kg) < VHI; PEA + PTZ > AM251 + PEA + PTZ > VHI; PEA + PTZ > AM630 + PEA + PTZ > VHI; AM251 + AM630 + PTZ < VHI; PEA + PTZ > AM251 + AM630 + PEA + PTZ > VHI; 2. S5D: PEA + PTZ < VHI; AM251(1.25, 2.5 µg/kg) + PTZ > VHI; AM251 (5, 10 µg/kg) + PTZ < VHI; AM630 (2.5 µg/kg) + PTZ vs.VHI, NS; AM630 (5, 10, 20, 40 µg/kg) + PTZ > VHI; PEA + PTZ < AM251 + PEA + PTZ < VHI; AM251 + AM630 + PTZ > VHI; PEA + PTZ < AM251 + AM630 + PEA + PTZ < VHI; PEA + PTZ < AM630 + PEA + PTZ < VHI; 3. S5L(-1): PEA + PTZ < VHI; AM251(5, 10 µg/kg) + PTZ < VHI; AM251 (2.5 µg/kg) + PTZ > VHI; AM630 (2.5 µg/kg) + PTZ vs.VHI, NS; AM630 (5, 10, 20, 40 µg/kg) + PTZ > VHI; PEA + PTZ < AM251 + PEA + PTZ < VHI; AM251 (2.5 µg/kg) + AM630 (20 µg/kg) + PTZ > VHI; VHI > AM251 + AM630 + PEA + PTZ > PEA + PTZ; PEA + PTZ < AM630 + PEA + PTZ < VHI; 4. SS: PEA(2.5, 5, 10, 25 µg/kg) + PTZ < VHI; PEA (1 µg/kg) + PTZ vs. VHI, NS; AM251 (5, 10 µg/kg) + PTZ < VHI; AM251 (1.25, 2.5 µg/kg) + PTZ vs. VHI, NS; AM630 + PTZ vs. VHI, NS | ||||||
| Citraro et al. (2016) (Italy) | 1. (a) ED50, wild running phase: PEA (60 min) < GW (30 min) + PEA (60 min); ACEA (60 min) < NIDA (45 min) + ACEA (60 min); WIN (20 min) < NIDA (45 min) + WIN (20 min); differences among other concurrent groups, NS; (CBZ, DZP, FBM, GPT, LTG, OXC, PB, TPM, VPA) + PEA < (same ASMs) + VHI; (LEV, PHT) + PEA vs. (same ASMs) + VHI, NS; (CBZ, DZP, FBM, GPT, LTG, TPM, VPA) + WIN/ACEA < (same ASMs) + VHI; (LEV, PHT, OXC, PB) + WIN/ACEA vs. (same ASMs) + VHI, NS; ASMs + NIDA/GW vs. ASMs + VHI, NS; (b) ED50, clonic phase: PEA (90 min) < NIDA (45 min) + PEA (90 min); PEA (60 min) < GW (30 min) + PEA (60 min); PEA (90 min) < GW (30 min) + PEA (90 min); WIN (20 min) < NIDA (45 min) + WIN (20 min); ACEA (60 min) < NIDA (45 min) + ACEA (60 min); differences among other concurrent groups, NS; (CBZ, DZP, FBM, GPT, LTG, OXC, PB, TPM, VPA) + PEA < (same ASMs) + VHI; (LEV, PHT) + PEA vs. (same ASMs) + VHI, NS; (CBZ, DZP, FBM, GPT, LTG, PB, TPM, VPA) + WIN < (same ASMs) + VHI; (LEV, PHT, OXC) + WIN vs. (same ASMs) + VHI, NS; (CBZ, DZP, FBM, GPT, PB, VPA) + ACEA < (same ASMs) + VHI; (LEV, PHT, OXC, LTG, TPM) + ACEA vs. (same ASMs) + VHI, NS; ASMs + NIDA/GW vs. ASMs + VHI, NS; (c) ED50, tonic phase: PEA (60 min) < NIDA (45 min) + PEA (60 min); PEA (90 min) < NIDA (45 min) + PEA (90 min); PEA (60 min) < GW (30 min) + PEA (60 min); PEA (90 min) < GW (30 min) + PEA (90 min); WIN (20 min) < NIDA (45 min) + WIN (20 min); ACEA (60 min) < NIDA (45 min) + ACEA (60 min); differences among other concurrent groups, NS; (CBZ, DZP, FBM, GPT, LTG, OXC, PB, TPM, VPA, PHT) + PEA < (same ASMs) + VHI; LEV + PEA vs. LEV + VHI, NS; (CBZ, DZP, FBM, GPT, LTG, PB, TPM, VPA) + WIN < (same ASMs) + VHI; (LEV, PHT, OXC) + WIN vs. (same ASMs) + VHI, NS; (CBZ, DZP, FBM, GPT, PB, VPA, TPM) + ACEA < (same ASMs) + VHI; (LEV, PHT, OXC, LTG) + ACEA vs. (same ASMs) + VHI, NS; ASMs + NIDA/GW vs. ASMs + VHI, NS. 2. TD50, (DZP, FBM, GPT, LTG, OXC, PB, TPM, VPA, PHT) + PEA < (same ASMs) + VHI; (LEV, CBZ) + PEA vs. (same ASMs) + VHI, NS; (DZP, FBM, GPT, LTG, OXC, PB, TPM, VPA) + WIN < (same ASMs) + VHI; (LEV, CBZ, PHT) + WIN vs. (same ASMs) + VHI, NS; (DZP, FBM, GPT, LTG, PB, PHT, TPM, VPA) + ACEA < (same ASMs) + VHI; (LEV, CBZ, OXC) + ACEA vs. (same ASMs) + VHI, NS; ASMs + NIDA/GW vs. ASMs + VHI, NS. 3. Brain/plasma ASMs levels: ASMs + PEA/WIN/ACEA/NIDA vs. ASMs + VHI, NS. | ||||||
| Lerner et al. (2017) (Germany) | (a) cCTX levels: PA 16:0_18:1 and SM d18:1/18:0, KA > saline; PE 16:0_18:1, PE 20:2_20:4, 12(S)-HETE and 15(S)-HETE, KA < saline; other PLs, ECBs, AEs and eiCs, KA vs. saline, NS; (b) CER levels: OEA, PEA, LPC 20:4 and PC 18:2_20:4, KA < saline; other PLs, ECBs, AEs and eiCs, KA vs. saline, NS; (c) THL levels: PE 20:0_20:4 and C16:0, KA > saline; PA 16:0_18:1 and PS 16:0_18:1, KA < saline; other PLS, ECBs, AEs and eiCs, KA vs. saline, NS; (d) HYP levels: PE 16:0_18:1, PG 18:1_20:4, PE 18:0_20:4, PE 20:2_20:4, LPC 18:0, PC 16:0_18:1, PG 16:0_18:1, PI 16:0_18:1, PC 18:2_20:4, PC 18:0_20:4, PG 16:1_20:4, PE 18:2_20:4, PE 20:0_20:4, PS 16:0_18:1, SM d18:1/18:0, LPA 16:0, LPA 20:4 and C18:1, KA < saline; PGD2, KA > saline; other PLS, ECBs, AEs and eiCs, KA vs. saline, NS; (e) HC levels: PGD2 and PGF2α, KA > saline; other PLS, ECBs, AEs and eiCs, KA vs. saline, NS; (f) STR levels: PEA, PC 16:0_18:1 and LPA 20:4, KA < saline; PGF2α, KA > saline; other PLS, ECBs, AEs and eiCs, KA vs. saline, NS; (g) Heart levels: SM d18:1/18:0, PG 16:0_18:1, PS 16:0_18:1, PI 16:0_18:1, C18:1, C16:0 and C20:4, KA > saline; PC 18:0_20:4 and PC 18.2_20:4, KA < saline; other PLS, ECBs, AEs and eiCs, KA vs. saline, NS; (h) Lung levels: PEA and LPA 20:4, KA > saline; PC 16:0_18:1, LPA 16:0 and PG 16:0_18, KA < saline; other PLS, ECBs, AEs and eiCs, KA vs. saline, NS; (i) Plasma levels: AEA, OEA, PEA, AA, PG 16:0_18:1, PS 16:0_18:1, SM d18:1/18:0, PE 16:0_18:1 and PG 18:1_20:4, KA < saline; 2-AG and PGD2, KA > saline; other PLS, ECBs, AEs and eiCs, KA vs. saline, NS. | ||||||
| Post et al. (2018) (Germany) | 1. (a) Mean Behavioral Score: 20 min post KA: PEA/KA < KA; PEA2/KA vs. KA, NS; 40, 60, 90, 120 min post KA: PEA/KA, PEA2/KA < KA; 150, 180 min post KA: PEA2/KA < KA; PEA/KA vs. KA, NS; 180 min post KA: URB597/KA vs. PEA/KA vs. PEA + URB597/KA, NS; 10, 20 min post KA: PEA/KA < URB937/KA, PEA + URB937/KA; 40 min post KA: PEA/KA < PEA + URB937/KA; 60, 90 min post KA: PEA/KA, URB937/KA < PEA + URB937/KA; 120 min post: PEA/KA < PEA + URB937/KA; URB937/KA vs. PEA + URB937/KA, NS; (b) Hippocampus ECBs/eiCs levels (min post KA): AEA (20 min): KA > CTRL, PEA2/KA; AEA (60, 120, 180 min): PEA2/KA vs. KA vs. CTRL, NS; 2-AG (20, 60, 120, 180 min): PEA2/KA vs. KA vs. CTRL, NS; PEA (20 min), PEA2/KA > both; PEA (60 min): PEA2/KA > CTRL; PEA2/KA vs. KA, NS; PEA (120, 180 min): PEA2/KA vs. KA vs. CTRL, NS; AA (20 min): KA > both; AA (60, 120, 180 min): PEA2/KA vs. KA vs. CTRL, NS; PGE2 (20 min): KA > CTRL, PEA2/KA; PGE2 (60, 120, 180 min): PEA2/KA vs. KA vs. CTRL, NS; PGD2 (20, 60 min): KA > both; PGD2 (120, 180 min); PEA2/KA vs. KA vs. CTRL, NS; AEA (20, 60, 180 min): PEA/KA < URB597/KA, PEA + URB597/KA; AEA (120 min): PEA/KA < URB597/KA; PEA/KA vs. PEA + URB597/KA, NS; 2-AG (20, 60, 120, 180 min): PEA/KA vs. URB597/KA vs. PEA + URB597/KA, NS; PEA (20 min): PEA/KA < URB597/KA < PEA + URB597/KA; PEA (60, 120, 180 min); PEA/KA < both; AA (20 min): PEA/KA vs. URB597/KA vs. PEA + URB597/KA, NS; AA (60 min): PEA/KA < PEA + URB597/KA; AA(120 min): PEA/KA < URB597/KA; AA(180 min): PEA/KA < URB597/KA, PEA + URB597/KA; PGE2 (20, 60 min): PEA/KA vs. URB597/KA vs. PEA + URB597/KA, NS; PGE2 (120 min): PEA/KA < URB597/KA; PGE2 (180 min): PEA/KA < both; PGD2 (20 min): PEA/KA > PEA + URB597/KA; PGD2 (60 min): PEA/KA vs. URB597/KA vs. PEA + URB597/KA, NS; PGD2 (120 min): PEA/KA < URB597/KA; PGD2 (180 min): PEA/KA < both; AEA, 2-AG, AA and PGD2 (20, 60, 120, 180 min): PEA/KA vs. URB937/KA vs. PEA + URB937/KA, NS; PEA (20, 60, 120, 180 min): PEA/KA < URB937/KA, PEA + URB937/KA; PEA (20, 60, 120 min): URB937/KA < PEA + URB937/KA; PGE2 (20, 60, 120 min): PEA/KA vs. both, NS. PGE2 (180 min): PEA/KA < both; (c) Plasma ECBs/eiCs levels (min post KA): AEA (20, 60 min): CTRL > both; AEA (180 min); KA > both; AEA (120 min): PEA2/KA vs. KA vs. CTRL, NS; 2-AG (60 min): KA > CTRL, PEA2/KA; 2-AG (180 min): PEA2/KA < CTRL; 2-AG (20, 120 min): PEA2/KA vs. KA vs. CTRL, NS; PEA (20, 60, 120, 180 min): PEA2/KA > both; AA (20, 60 min); CTRL > both; AA (180 min): KA > both; AA (120 min): PEA2/KA vs. KA vs. CTRL, NS; PGE2 (60 min): KA > CTRL; PGE2 (120, 180 min): KA > both; PGE2 (20 min): PEA2/KA vs. KA vs. CTRL, NS; PGD2 (120, 180 min): CTRL < both; PGD2 (20, 60 min): PEA2/KA vs. KA vs. CTRL, NS; AEA (20, 60, 120, 180 min): PEA/KA < URB597/KA, PEA + URB597/KA; 2-AG (20, 60, 120, 180 min): PEA/KA > both; PEA (20, 60, 120, 180 min): PEA + URB597/KA > both; AA (20 min): PEA/KA < PEA + URB597/KA; AA (60, 120 min): PEA/KA < both; AA (180 min): PEA/KA vs. both, NS; AEA (20, 60, 120, 180 min): PEA/KA < URB937/KA, PEA + URB937/KA; 2-AG (20, 180 min): PEA/KA > both; 2-AG (120 min): PEA/KA > PEA + URB937/KA; 2-AG (60 min): PEA/KA vs. both, NS; PEA (20, 120 min): PEA/KA < PEA+ URB937/KA; PEA (60, 180 min): PEA/KA < both; AA and PGD2 (20, 60, 120, 180 min): PEA/KA vs. both, NS; PGE2 (20, 60 min): PEA/KA vs. both, NS; PGE2 (120, 180 min): PEA/KA < PEA + URB937/KA. 2. (a) NeuN staining: KA, ↓; PEA2/KA vs. saline, NS; (b) FJC staining (5 days post KA): PEA2/KA < KA; (c) Silver staining: KA, ↑↑; PEA2/KA ↑; saline, ─ | ||||||
| Study (Country) | Study Design | Defined Study Population | Age | Gender | PEA Measure | Adequate PEA Evaluation | Control Group | Statistical Analyses | Funding or Sponsorship |
|---|---|---|---|---|---|---|---|---|---|
| Lambert et al. (2001) (Belgium) | √ Analytic, observational, interventional | √ OF1 mice | X | √ Male | √ 1. MES test: 50, 100 mg/kg (ip); Time course calculation: 25 mg/kg (ip); ED50 calculation: 0.5–50 mg/kg (ip); CIS test: 25 mg/kg (ip) 2. Rotarod test: up to 250 mg/kg (ip) | √ 1. (a) MES test: double assessment (30 min, 4 h); (b) Time course calculation, ED50, CIS test: single administration. 2. Rotarod test: multiple administrations | √ 1. MES test: VHI (30 min/4 h); CIS test: VHI, PHT 2. Ameltolide, PHT, VPA, PB, CBZ | √ Fisher’s exact test | X |
| Sheerin et al. (2004) (Canada) | √ Analytic, observational, interventional | √ Long–Evans rats | X | √ Male | √ KS test: 1, 10, 100 mg/kg (ip); CIS test: 40 mg/kg (ip) | √ Single administration, 2 h before each kindling session | √ KS test: [VHI, PEA(1), PEA(10), PEA(100)] baseline; CIS test: VHI | √ ANOVA; Fisher’s exact test; t-test | √ |
| Citraro et al. (2013) (Italy) | √ Analytic, observational, interventional | √ WAG/Rij, Wistar, ACI rats | √ 1 month; 6–7 months | √ Male | √ 1. 0.5, 1, 3, and 10 µg/2 µL (icv); 10, 20, 40, and 60 mg/kg (ip); 20 and 40 mg/kg (ip post SR); 3 µg/2 µL (icv post GW) 2. Brain tissue levels | √ 1. (a) Single administration after 1 h baseline EEG recording; (b) single administration after 1 h baseline EEG recording and 30 min after SR or GW administration; 2. Double assessment (2 and 6 months) | √ 1. VHI (icv/ip) 2. Wistar, ACI rats | √ ANOVA; Tukey’s post-hoc test | √/X |
| Fezza et al. (2014) (Italy) | √ Analytic, observational | √ Wistar rats | √ P14 and P56–70 | √ Male and female | √ Brain tissue levels | √ Single assessment | √ Saline | √ ANOVA; t-test; Mann–Whitney U test | √ |
| Aghaei et al. (2015) (Iran) | √ Analytic, observational, interventional | √ Wistar rats | √ 8–10 weeks | √ Male | √ 1, 2.5, 5, 10, 25 µg/kg (icv) | √ Single administration | √ VHI | √ ANOVA; Mann–Whitney U test; Kruskal–Wallis test; Tukey’s test | √ |
| Citraro et al. (2016) (Italy) | √ Analytic, observational, interventional | √ DBA/2 mice | √ 22–26 days or 48–56 days | √ Male | √ 5–40 mg/kg (ip) | √ Single administration 30, 60, 90, or 120 min before auditory stimulation | √ VHI, ASMs + VHI | √ ANOVA; Fisher’s exact test; Dunnett’s test; χ2-test; t-test | √ |
| Lerner et al. (2017) (Germany) | √ Analytic, observational | √ C57BL/6N mice | √ 80–100 days | √ Male | √ Brain tissue, peripheral tissue, plasma levels | √ Single assessment after 1 h KA-injection | √ Saline | √ ANOVA; Shapiro–Wilk test; Kolmogorow–Smirnow test; t-test | √ |
| Post et al. (2018) (Germany) | √ Analytic, observational, interventional | √ C57BL/6N mice | √ 8–10 weeks | √ Male | √ 1. 40 mg/kg (ip); 2. Brain tissue levels; plasma levels | √ 1. (a) Single administration (acute treatment, 30 min prior to KA); (b) double administration (subchronic treatment, 7 h and 30 min prior to KA); 2. Multiple assessment | √ 1. KA, CTRL1, CTRL2; 2. saline | √ ANOVA; Greenhouse–Gasser correction, Bonferroni’s post-hoc analysis for multiple comparisons | √ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bortoletto, R.; Balestrieri, M.; Bhattacharyya, S.; Colizzi, M. Is It Time to Test the Antiseizure Potential of Palmitoylethanolamide in Human Studies? A Systematic Review of Preclinical Evidence. Brain Sci. 2022, 12, 101. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci12010101
Bortoletto R, Balestrieri M, Bhattacharyya S, Colizzi M. Is It Time to Test the Antiseizure Potential of Palmitoylethanolamide in Human Studies? A Systematic Review of Preclinical Evidence. Brain Sciences. 2022; 12(1):101. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci12010101
Chicago/Turabian StyleBortoletto, Riccardo, Matteo Balestrieri, Sagnik Bhattacharyya, and Marco Colizzi. 2022. "Is It Time to Test the Antiseizure Potential of Palmitoylethanolamide in Human Studies? A Systematic Review of Preclinical Evidence" Brain Sciences 12, no. 1: 101. https://0-doi-org.brum.beds.ac.uk/10.3390/brainsci12010101