The Immunomodulatory Capacity of an Epstein-Barr Virus Abortive Lytic Cycle: Potential Contribution to Viral Tumorigenesis


Abstract
:1. Introduction
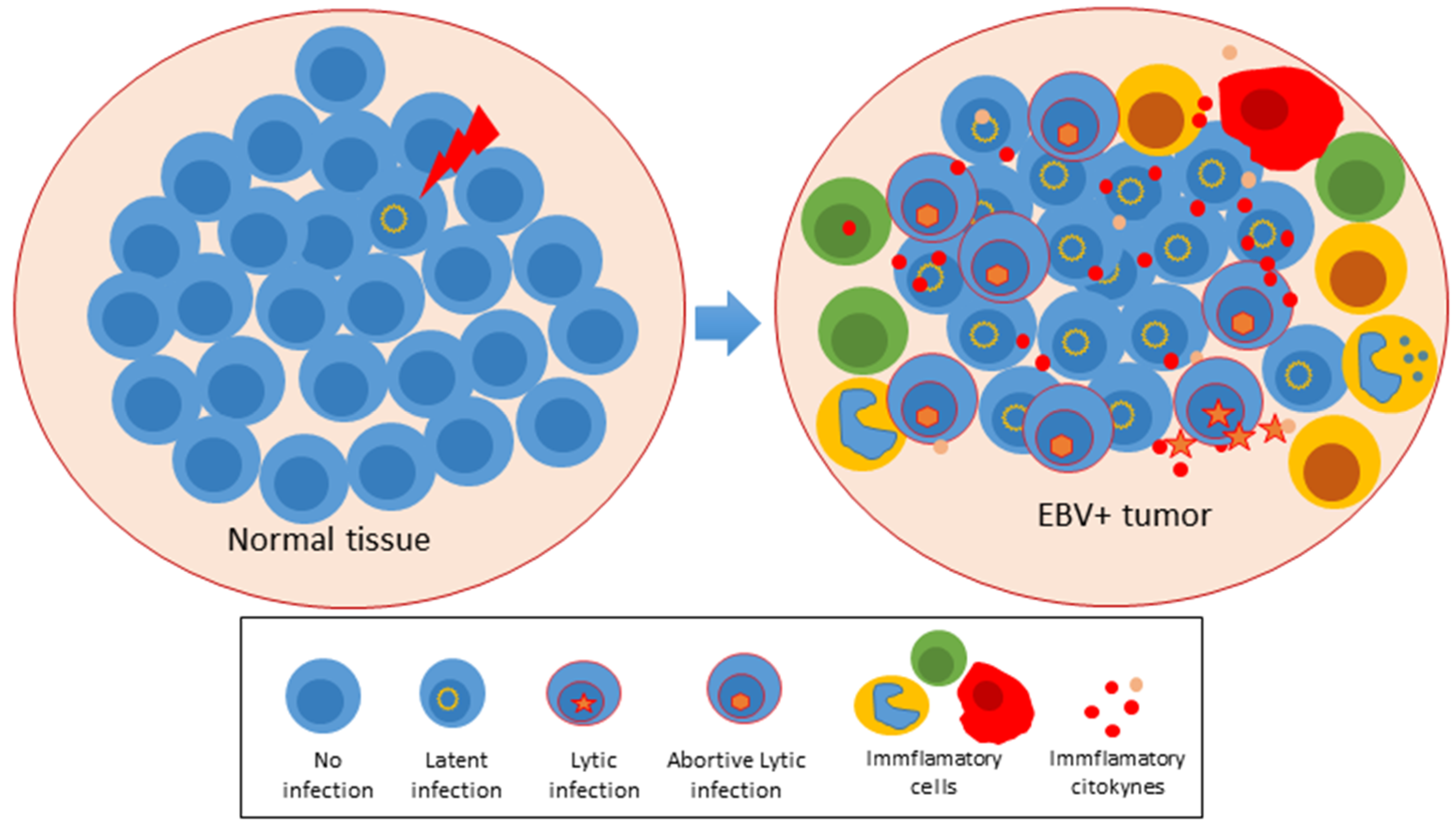
2. An Abortive Lytic Cycle in Pre-Latent Cells and Established Tumors
3. Evidence That the Abortive Lytic Cycle Contributes with the EBV-Induced Tumorigenesis
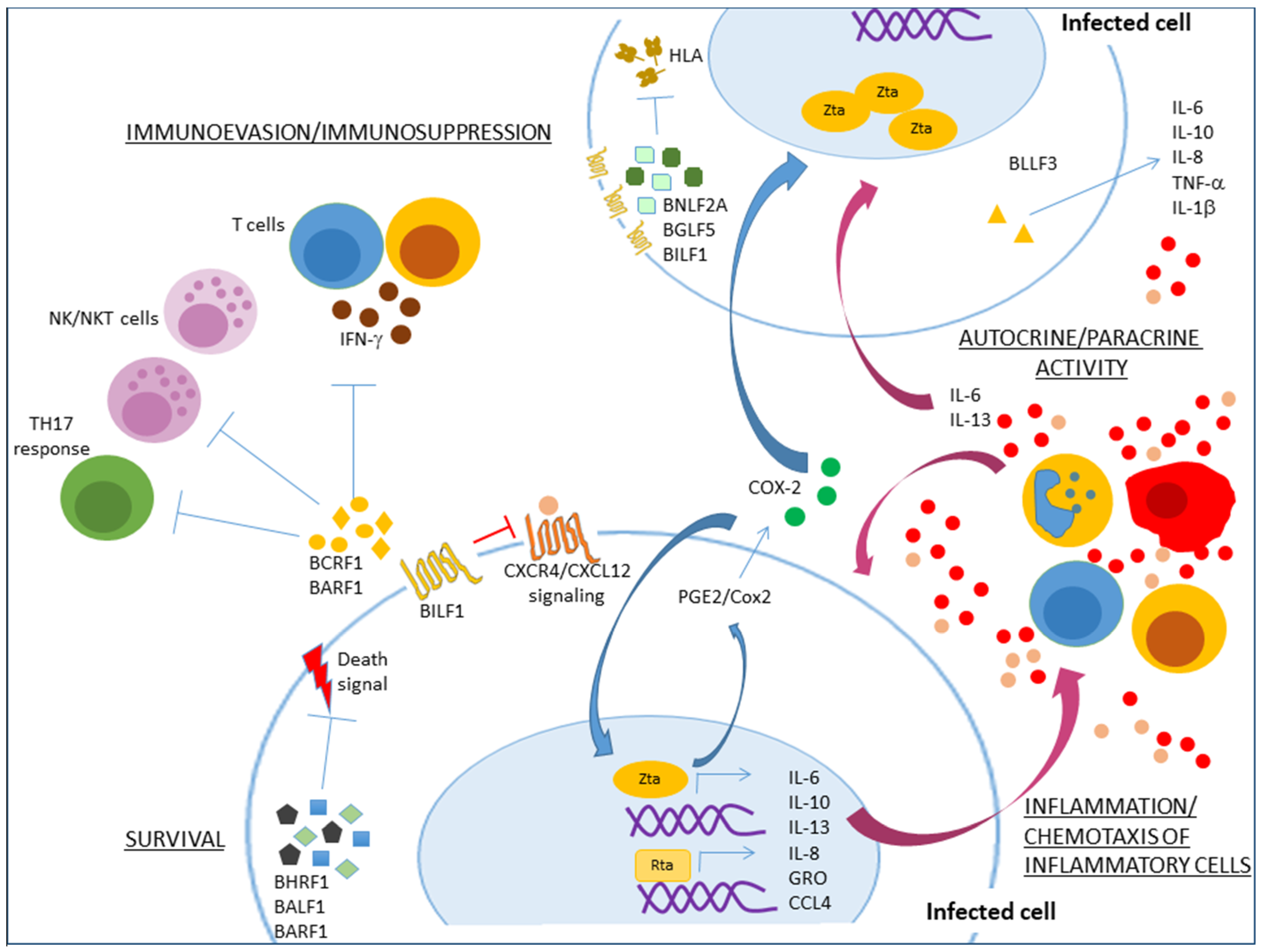
4. Lytic Cycle Proteins with Anti-Apoptotic and Immunomodulatory Functions
5. An Autocrine/Paracrine Role of γ-Herpesvirus Inflammatory Mediators in Tumor Initiation and Maintenance
6. An HCMV Oncomodulatory Role
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Bcl-2 | B cell lymphoma 2 |
| BCR | B cell antigen receptor |
| BL | Burkitt lymphoma |
| CMV | cytomegalovirus |
| COX-2 | cyclooxygenase-2 |
| CTL | cytotoxic T cell |
| DLBCL | diffuse large B cell lymphomas |
| dUTPases | deoxyuridine triphosphate nucleotide hydrolases |
| E | early |
| EBER | EBV-encoded small RNA |
| EBNA | Epstein-Barr Nuclear Protein |
| EBV | Epstein-Barr Virus |
| EBVaGC | EBV associated gastric cancers |
| GPCR | G protein-coupled receptor |
| GRO | growth-regulated oncogene |
| HCMV | human CMV |
| HDAC | histone deacetylase |
| HHV6 | human herpesvirus 6 |
| HHV7 | human herpesvirus 7 |
| HLA | human leucocyte antigen |
| HMB | hypersensitivity to mosquito bites |
| HL | Hodgkin lymphoma |
| HUVEC | human umbilical vein endothelial cells |
| HV | hydroa vacciniforme |
| IE | immediate early |
| IL | interleukin |
| IRF | interferon regulatory factor |
| IRIS | immune reconstitution inflammatory syndrome |
| ITAM | Immunoreceptor Tyrosine-Based Activation Motif |
| KCP | KSHV complement regulatory protein |
| KS | Kaposi sarcoma |
| KSHV | Kaposi Sarcoma herpesvirus |
| L | late |
| LCLs | lymphoblastoid cell lines |
| LP | leader protein |
| LPD | lymphoproliferative disease |
| MAPK | mitogen activated protein kinase |
| MCD | multicentric castleman disease |
| miRNA | micro RNA |
| NK | Natural Killer |
| ORF | Open Reading Frame |
| PBMC | peripheral blood mononuclear cell |
| PEL | primary effusion lymphoma |
| PGE2 | prostaglandin E2 |
| PKC | protein kinase C |
| PKR | protein kinase R |
| PLC | phospholipase C gamma |
| sBARF1 | soluble BARF1 |
| shRNAs | small hairpin RNAs |
| TAP1 | peptide transporter associated with antigen processing 1 |
| TGFβ | transforming growth factor-β |
| TLR | Toll-like Receptor |
| TPA | 12-O-tetradecanoylphorbol-13-acetate |
| TRAF | TNF receptor-associated factors |
| USP | ubiquitin-specific cysteine protease |
| VCA | viral capsid antigen |
References
- Morales-Sanchez, A.; Fuentes-Panana, E.M. Human viruses and cancer. Viruses 2014, 6, 4047–4079. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Munoz, M.E.; Fuentes-Panana, E.M. Beta and gamma human herpesviruses: Agonistic and antagonistic interactions with the host immune system. Front. Microbiol. 2017, 8, 2521. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Rowe, C.L.; Jardetzky, T.S.; Longnecker, R. The KGD motif of Epstein-Barr virus gH/gL is bifunctional, orchestrating infection of B cells and epithelial cells. mBio 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Chesnokova, L.S.; Hutt-Fletcher, L.M. Fusion of Epstein-Barr virus with epithelial cells can be triggered by alphavbeta5 in addition to alphavbeta6 and alphavbeta8, and integrin binding triggers a conformational change in glycoproteins gHgL. J. Virol. 2011, 85, 13214–13223. [Google Scholar] [CrossRef] [PubMed]
- Chesnokova, L.S.; Nishimura, S.L.; Hutt-Fletcher, L.M. Fusion of epithelial cells by Epstein-Barr virus proteins is triggered by binding of viral glycoproteins gHgL to integrins alphavbeta6 or alphavbeta8. Proc. Natl. Acad. Sci. USA 2009, 106, 20464–20469. [Google Scholar] [CrossRef] [PubMed]
- Shannon-Lowe, C.D.; Neuhierl, B.; Baldwin, G.; Rickinson, A.B.; Delecluse, H.J. Resting B cells as a transfer vehicle for epstein-barr virus infection of epithelial cells. Proc. Natl. Acad. Sci. USA 2006, 103, 7065–7070. [Google Scholar] [CrossRef] [PubMed]
- Tugizov, S.M.; Berline, J.W.; Palefsky, J.M. Epstein-Barr virus infection of polarized tongue and nasopharyngeal epithelial cells. Nat. Med. 2003, 9, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.B.; Zhang, H.; Zhang, J.P.; Li, Y.; Zhao, B.; Feng, G.K.; Du, Y.; Xiong, D.; Zhong, Q.; Liu, W.L.; et al. Neuropilin 1 is an entry factor that promotes EBV infection of nasopharyngeal epithelial cells. Nat. Commun. 2015, 6, 6240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Kenyon, W.J.; Li, Q.; Mullberg, J.; Hutt-Fletcher, L.M. Epstein-Barr virus uses different complexes of glycoproteins gH and gL to infect B lymphocytes and epithelial cells. J. Virol. 1998, 72, 5552–5558. [Google Scholar] [PubMed]
- Li, Q.X.; Young, L.S.; Niedobitek, G.; Dawson, C.W.; Birkenbach, M.; Wang, F.; Rickinson, A.B. Epstein-Barr virus infection and replication in a human epithelial cell system. Nature 1992, 356, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Tsao, S.W.; Tsang, C.M.; Pang, P.S.; Zhang, G.; Chen, H.; Lo, K.W. The biology of EBV infection in human epithelial cells. Semin. Cancer Biol. 2012, 22, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Thorley-Lawson, D.A. EBV persistence—Introducing the virus. Curr. Top. Microbiol. Immunol. 2015, 390, 151–209. [Google Scholar] [PubMed]
- Baumann, M.; Mischak, H.; Dammeier, S.; Kolch, W.; Gires, O.; Pich, D.; Zeidler, R.; Delecluse, H.J.; Hammerschmidt, W. Activation of the Epstein-Barr virus transcription factor BZLF1 by 12-O-tetradecanoylphorbol-13-acetate-induced phosphorylation. J. Virol. 1998, 72, 8105–8114. [Google Scholar] [PubMed]
- Nawandar, D.M.; Wang, A.; Makielski, K.; Lee, D.; Ma, S.; Barlow, E.; Reusch, J.; Jiang, R.; Wille, C.K.; Greenspan, D.; et al. Differentiation-dependent KLF4 expression promotes lytic Epstein-Barr virus infection in epithelial cells. PLoS Pathog. 2015, 11, e1005195. [Google Scholar] [CrossRef] [PubMed]
- Kraus, R.J.; Yu, X.; Cordes, B.A.; Sathiamoorthi, S.; Iempridee, T.; Nawandar, D.M.; Ma, S.; Romero-Masters, J.C.; McChesney, K.G.; Lin, Z.; et al. Hypoxia-inducible factor-1alpha plays roles in Epstein-Barr virus’s natural life cycle and tumorigenesis by inducing lytic infection through direct binding to the immediate-early BZLF1 gene promoter. PLoS Pathog. 2017, 13, e1006404. [Google Scholar] [CrossRef] [PubMed]
- Babcock, G.J.; Hochberg, D.; Thorley-Lawson, A.D. The expression pattern of Epstein-Barr virus latent genes in vivo is dependent upon the differentiation stage of the infected B cell. Immunity 2000, 13, 497–506. [Google Scholar] [CrossRef]
- Wen, W.; Iwakiri, D.; Yamamoto, K.; Maruo, S.; Kanda, T.; Takada, K. Epstein-Barr virus BZLF1 gene, a switch from latency to lytic infection, is expressed as an immediate-early gene after primary infection of B lymphocytes. J. Virol. 2007, 81, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- Dickerson, S.J.; Xing, Y.; Robinson, A.R.; Seaman, W.T.; Gruffat, H.; Kenney, S.C. Methylation-dependent binding of the Epstein-Barr virus BZLF1 protein to viral promoters. PLoS Pathog. 2009, 5, e1000356. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, A.J. Epigenetic control of Epstein-Barr virus transcription—Relevance to viral life cycle? Front. Genet. 2013, 4, 161. [Google Scholar] [CrossRef] [PubMed]
- Kalla, M.; Schmeinck, A.; Bergbauer, M.; Pich, D.; Hammerschmidt, W. Ap-1 homolog BZLF1 of Epstein-Barr virus has two essential functions dependent on the epigenetic state of the viral genome. Proc. Natl. Acad. Sci. USA 2010, 107, 850–855. [Google Scholar] [CrossRef] [PubMed]
- Shannon-Lowe, C.; Adland, E.; Bell, A.I.; Delecluse, H.J.; Rickinson, A.B.; Rowe, M. Features distinguishing Epstein-Barr virus infections of epithelial cells and B cells: Viral genome expression, genome maintenance, and genome amplification. J. Virol. 2009, 83, 7749–7760. [Google Scholar] [CrossRef] [PubMed]
- Tsang, C.M.; Zhang, G.; Seto, E.; Takada, K.; Deng, W.; Yip, Y.L.; Man, C.; Hau, P.M.; Chen, H.; Cao, Y.; et al. Epstein-Barr virus infection in immortalized nasopharyngeal epithelial cells: Regulation of infection and phenotypic characterization. Int. J. Cancer 2010, 127, 1570–1583. [Google Scholar] [CrossRef] [PubMed]
- Kalla, M.; Hammerschmidt, W. Human B cells on their route to latent infection—Early but transient expression of lytic genes of Epstein-Barr virus. Eur. J. Cell Biol. 2012, 91, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Martel-Renoir, D.; Grunewald, V.; Touitou, R.; Schwaab, G.; Joab, I. Qualitative analysis of the expression of Epstein-Barr virus lytic genes in nasopharyngeal carcinoma biopsies. J. Gener. Virol. 1995, 76, 1401–1408. [Google Scholar] [CrossRef] [PubMed]
- Ramayanti, O.; Juwana, H.; Verkuijlen, S.A.; Adham, M.; Pegtel, M.D.; Greijer, A.E.; Middeldorp, J.M. Epstein-Barr virus mRNA profiles and viral DNA methylation status in nasopharyngeal brushings from nasopharyngeal carcinoma patients reflect tumor origin. Int. J. Cancer 2017, 140, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Borozan, I.; Zapatka, M.; Frappier, L.; Ferretti, V. Analysis of Epstein-Barr virus genomes and expression profiles in gastric adenocarcinoma. J. Virol. 2018, 92, e01239-17. [Google Scholar] [CrossRef] [PubMed]
- Vrzalikova, K.; Vockerodt, M.; Leonard, S.; Bell, A.; Wei, W.; Schrader, A.; Wright, K.L.; Kube, D.; Rowe, M.; Woodman, C.B.; et al. Down-regulation of BLIMP1α by the EBV oncogene, LMP-1, disrupts the plasma cell differentiation program and prevents viral replication in B cells: Implications for the pathogenesis of EBV-associated B-cell lymphomas. Blood 2011, 117, 5907–5917. [Google Scholar] [CrossRef] [PubMed]
- Al Tabaa, Y.; Tuaillon, E.; Bollore, K.; Foulongne, V.; Petitjean, G.; Seigneurin, J.M.; Duperray, C.; Desgranges, C.; Vendrell, J.P. Functional Epstein-Barr virus reservoir in plasma cells derived from infected peripheral blood memory B cells. Blood 2009, 113, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Al Tabaa, Y.; Tuaillon, E.; Jeziorski, E.; Ouedraogo, D.E.; Bollore, K.; Rubbo, P.A.; Foulongne, V.; Rodiere, M.; Vendrell, J.P. B-cell polyclonal activation and Epstein-Barr viral abortive lytic cycle are two key features in acute infectious mononucleosis. J. Clin. Virol. 2011, 52, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Miyake, T.; Yamamoto, T.; Hirai, Y.; Otsuka, M.; Hamada, T.; Tsuji, K.; Morizane, S.; Suzuki, D.; Aoyama, Y.; Iwatsuki, K. Survival rates and prognostic factors of Epstein-Barr virus-associated hydroa vacciniforme and hypersensitivity to mosquito bites. Br. J. Dermatol. 2015, 172, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.D.; Hegde, S.; Young, K.H.; Sullivan, R.; Rajesh, D.; Zhou, Y.; Jankowska-Gan, E.; Burlingham, W.J.; Sun, X.; Gulley, M.L.; et al. A new model of Epstein-Barr virus infection reveals an important role for early lytic viral protein expression in the development of lymphomas. J. Virol. 2011, 85, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Hong, G.K.; Gulley, M.L.; Feng, W.H.; Delecluse, H.J.; Holley-Guthrie, E.; Kenney, S.C. Epstein-Barr virus lytic infection contributes to lymphoproliferative disease in a scid mouse model. J. Virol. 2005, 79, 13993–14003. [Google Scholar] [CrossRef] [PubMed]
- Kvansakul, M.; Wei, A.H.; Fletcher, J.I.; Willis, S.N.; Chen, L.; Roberts, A.W.; Huang, D.C.; Colman, P.M. Structural basis for apoptosis inhibition by Epstein-Barr virus BHRF1. PLoS Pathog. 2010, 6, e1001236. [Google Scholar] [CrossRef] [PubMed]
- Marshall, W.L.; Yim, C.; Gustafson, E.; Graf, T.; Sage, D.R.; Hanify, K.; Williams, L.; Fingeroth, J.; Finberg, R.W. Epstein-Barr virus encodes a novel homolog of the Bcl-2 oncogene that inhibits apoptosis and associates with Bax and Bak. J. Virol. 1999, 73, 5181–5185. [Google Scholar] [PubMed]
- Altmann, M.; Hammerschmidt, W. Epstein-Barr virus provides a new paradigm: A requirement for the immediate inhibition of apoptosis. PLoS Biol. 2005, 3, e404. [Google Scholar] [CrossRef] [PubMed]
- Kelly, G.L.; Long, H.M.; Stylianou, J.; Thomas, W.A.; Leese, A.; Bell, A.I.; Bornkamm, G.W.; Mautner, J.; Rickinson, A.B.; Rowe, M. An Epstein-Barr virus anti-apoptotic protein constitutively expressed in transformed cells and implicated in burkitt lymphomagenesis: The Wp/BHRF1 link. PLoS Pathog. 2009, 5, e1000341. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.; Vistarop, A.G.; Huaman, F.; Narbaitz, M.; Metrebian, F.; De Matteo, E.; Preciado, M.V.; Chabay, P.A. Epstein-Barr virus lytic cycle involvement in diffuse large B cell lymphoma. Hematol. Oncol. 2017, 36, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Sun, P.; Zhang, Y.; Yan, L.; Luo, B. Expression of c-myc and PCNA in Epstein-Barr virus-associated gastric carcinoma. Exp. Ther. Med. 2013, 5, 1030–1034. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, A.; Brink, A.A.; Craanen, M.E.; Middeldorp, J.M.; Meijer, C.J.; van den Brule, A.J. Unique transcription pattern of Epstein-Barr virus (EBV) in EBV-carrying gastric adenocarcinomas: Expression of the transforming BARF1 gene. Cancer Res. 2000, 60, 2745–2748. [Google Scholar] [PubMed]
- Seto, E.; Yang, L.; Middeldorp, J.; Sheen, T.S.; Chen, J.Y.; Fukayama, M.; Eizuru, Y.; Ooka, T.; Takada, K. Epstein-Barr virus (EBV)-encoded BARF1 gene is expressed in nasopharyngeal carcinoma and EBV-associated gastric carcinoma tissues in the absence of lytic gene expression. J. Med. Virol. 2005, 76, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Stevens, S.J.; Verkuijlen, S.A.; Hariwiyanto, B.; Paramita, D.K.; Fachiroh, J.; Adham, M.; Tan, I.B.; Haryana, S.M.; Middeldorp, J.M. Noninvasive diagnosis of nasopharyngeal carcinoma: Nasopharyngeal brushings reveal high Epstein-Barr virus DNA load and carcinoma-specific viral BARF1 mRNA. Int. J. Cancer 2006, 119, 608–614. [Google Scholar] [CrossRef] [PubMed]
- De Turenne-Tessier, M.; Jolicoeur, P.; Ooka, T. Expression of the protein encoded by Epstein-Barr virus (EBV) BARF1 open reading frame from a recombinant adenovirus system. Virus Res. 1997, 52, 73–85. [Google Scholar] [CrossRef]
- Paramita, D.K.; Fatmawati, C.; Juwana, H.; van Schaijk, F.G.; Fachiroh, J.; Haryana, S.M.; Middeldorp, J.M. Humoral immune responses to Epstein-Barr virus encoded tumor associated proteins and their putative extracellular domains in nasopharyngeal carcinoma patients and regional controls. J. Med. Virol. 2011, 83, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Hoebe, E.K.; Wille, C.; Hopmans, E.S.; Robinson, A.R.; Middeldorp, J.M.; Kenney, S.C.; Greijer, A.E. Epstein-Barr virus transcription activator r upregulates BARF1 expression by direct binding to its promoter, independent of methylation. J. Virol. 2012, 86, 11322–11332. [Google Scholar] [CrossRef] [PubMed]
- Fiorini, S.; Ooka, T. Secretion of Epstein-Barr virus-encoded BARF1 oncoprotein from latently infected B cells. Virol. J. 2008, 5, 70. [Google Scholar] [CrossRef] [PubMed]
- Elegheert, J.; Bracke, N.; Pouliot, P.; Gutsche, I.; Shkumatov, A.V.; Tarbouriech, N.; Verstraete, K.; Bekaert, A.; Burmeister, W.P.; Svergun, D.I.; et al. Allosteric competitive inactivation of hematopoietic CSF-1 signaling by the viral decoy receptor BARF1. Nat. Struct. Mol. Biol. 2012, 19, 938–947. [Google Scholar] [CrossRef] [PubMed]
- Tarbouriech, N.; Ruggiero, F.; de Turenne-Tessier, M.; Ooka, T.; Burmeister, W.P. Structure of the Epstein-Barr virus oncogene BARF1. J. Mol. Biol. 2006, 359, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Hoebe, E.K.; Le Large, T.Y.; Tarbouriech, N.; Oosterhoff, D.; De Gruijl, T.D.; Middeldorp, J.M.; Greijer, A.E. Epstein-Barr virus-encoded BARF1 protein is a decoy receptor for macrophage colony stimulating factor and interferes with macrophage differentiation and activation. Viral Immunol. 2012, 25, 461–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strockbine, L.D.; Cohen, J.I.; Farrah, T.; Lyman, S.D.; Wagener, F.; DuBose, R.F.; Armitage, R.J.; Spriggs, M.K. The Epstein-Barr virus BARF1 gene encodes a novel, soluble colony-stimulating factor-1 receptor. J. Virol. 1998, 72, 4015–4021. [Google Scholar] [PubMed]
- Cohen, J.I.; Lekstrom, K. Epstein-Barr virus BARF1 protein is dispensable for B-cell transformation and inhibits alpha interferon secretion from mononuclear cells. J. Virol. 1999, 73, 7627–7632. [Google Scholar]
- Sheng, W.; Decaussin, G.; Sumner, S.; Ooka, T. N-terminal domain of BARF1 gene encoded by Epstein-Barr virus is essential for malignant transformation of rodent fibroblasts and activation of Bcl-2. Oncogene 2001, 20, 1176–1185. [Google Scholar] [CrossRef] [PubMed]
- Wiech, T.; Nikolopoulos, E.; Lassman, S.; Heidt, T.; Schopflin, A.; Sarbia, M.; Werner, M.; Shimizu, Y.; Sakka, E.; Ooka, T.; et al. Cyclin D1 expression is induced by viral BARF1 and is overexpressed in EBV-associated gastric cancer. Virchows Arch. Int. J. Pathol. 2008, 452, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Tsao, S.W.; Ooka, T.; Nicholls, J.M.; Cheung, H.W.; Fu, S.; Wong, Y.C.; Wang, X. Anti-apoptotic role of BARF1 in gastric cancer cells. Cancer Lett. 2006, 238, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Cabras, G.; Sheng, W.; Zeng, Y.; Ooka, T. Synergism of BARF1 with ras induces malignant transformation in primary primate epithelial cells and human nasopharyngeal epithelial cells. Neoplasia 2009, 11, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.X.; Ooka, T. A transforming function of the BARF1 gene encoded by Epstein-Barr virus. EMBO J. 1989, 8, 2897–2903. [Google Scholar] [PubMed]
- Xu, Z.G.; Iwatsuki, K.; Oyama, N.; Ohtsuka, M.; Satoh, M.; Kikuchi, S.; Akiba, H.; Kaneko, F. The latency pattern of Epstein-Barr virus infection and viral IL-10 expression in cutaneous natural killer/T-cell lymphomas. Br. J. Cancer 2001, 84, 920–925. [Google Scholar] [CrossRef] [PubMed]
- Xue, S.A.; Labrecque, L.G.; Lu, Q.L.; Ong, S.K.; Lampert, I.A.; Kazembe, P.; Molyneux, E.; Broadhead, R.L.; Borgstein, E.; Griffin, B.E. Promiscuous expression of Epstein-Barr virus genes in burkitt’s lymphoma from the central african country malawi. Int. J. Cancer 2002, 99, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Kurilla, M.G.; Swaminathan, S.; Welsh, R.M.; Kieff, E.; Brutkiewicz, R.R. Effects of virally expressed interleukin-10 on vaccinia virus infection in mice. J. Virol. 1993, 67, 7623–7628. [Google Scholar] [PubMed]
- Miyazaki, I.; Cheung, R.K.; Dosch, H.M. Viral interleukin 10 is critical for the induction of B cell growth transformation by Epstein-Barr virus. J. Exp. Med. 1993, 178, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Jochum, S.; Moosmann, A.; Lang, S.; Hammerschmidt, W.; Zeidler, R. The EBV immunoevasins vIL-10 and BNLF2a protect newly infected B cells from immune recognition and elimination. PLoS Pathog. 2012, 8, e1002704. [Google Scholar] [CrossRef] [PubMed]
- Zeidler, R.; Eissner, G.; Meissner, P.; Uebel, S.; Tampe, R.; Lazis, S.; Hammerschmidt, W. Downregulation of tap1 in B lymphocytes by cellular and Epstein-Barr virus-encoded interleukin-10. Blood 1997, 90, 2390–2397. [Google Scholar] [PubMed]
- Beisser, P.S.; Verzijl, D.; Gruijthuijsen, Y.K.; Beuken, E.; Smit, M.J.; Leurs, R.; Bruggeman, C.A.; Vink, C. The Epstein-Barr virus BILF1 gene encodes a g protein-coupled receptor that inhibits phosphorylation of RNA-dependent protein kinase. J. Virol. 2005, 79, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Zuo, J.; Currin, A.; Griffin, B.D.; Shannon-Lowe, C.; Thomas, W.A.; Ressing, M.E.; Wiertz, E.J.; Rowe, M. The Epstein-Barr virus g-protein-coupled receptor contributes to immune evasion by targeting MHC class I molecules for degradation. PLoS Pathog. 2009, 5, e1000255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, J.; Quinn, L.L.; Tamblyn, J.; Thomas, W.A.; Feederle, R.; Delecluse, H.J.; Hislop, A.D.; Rowe, M. The Epstein-Barr virus-encoded BILF1 protein modulates immune recognition of endogenously processed antigen by targeting major histocompatibility complex class I molecules trafficking on both the exocytic and endocytic pathways. J. Virol. 2011, 85, 1604–1614. [Google Scholar] [CrossRef] [PubMed]
- Lyngaa, R.; Norregaard, K.; Kristensen, M.; Kubale, V.; Rosenkilde, M.M.; Kledal, T.N. Cell transformation mediated by the Epstein-Barr virus G protein-coupled receptor BILF1 is dependent on constitutive signaling. Oncogene 2010, 29, 4388–4398. [Google Scholar] [CrossRef] [PubMed]
- Spiess, K.; Fares, S.; Sparre-Ulrich, A.H.; Hilgenberg, E.; Jarvis, M.A.; Ehlers, B.; Rosenkilde, M.M. Identification and functional comparison of seven-transmembrane g-protein-coupled BILF1 receptors in recently discovered nonhuman primate lymphocryptoviruses. J. Virol. 2015, 89, 2253–2267. [Google Scholar] [CrossRef] [PubMed]
- Daum, R.S.; Siber, G.R.; Ballanco, G.A.; Sood, S.K. Serum anticapsular antibody response in the first week after immunization of adults and infants with the haemophilus influenzae type b-neisseria meningitidis outer membrane protein complex conjugate vaccine. J. Infect. Dis. 1991, 164, 1154–1159. [Google Scholar] [CrossRef] [PubMed]
- Hislop, A.D.; Ressing, M.E.; van Leeuwen, D.; Pudney, V.A.; Horst, D.; Koppers-Lalic, D.; Croft, N.P.; Neefjes, J.J.; Rickinson, A.B.; Wiertz, E.J. A CD8+ T cell immune evasion protein specific to Epstein-Barr virus and its close relatives in old world primates. J. Exp. Med. 2007, 204, 1863–1873. [Google Scholar] [CrossRef] [PubMed]
- Horst, D.; van Leeuwen, D.; Croft, N.P.; Garstka, M.A.; Hislop, A.D.; Kremmer, E.; Rickinson, A.B.; Wiertz, E.J.; Ressing, M.E. Specific targeting of the EBV lytic phase protein BNLF2a to the transporter associated with antigen processing results in impairment of hla class I-restricted antigen presentation. J. Immunol. 2009, 182, 2313–2324. [Google Scholar] [CrossRef] [PubMed]
- Strong, M.J.; Laskow, T.; Nakhoul, H.; Blanchard, E.; Liu, Y.; Wang, X.; Baddoo, M.; Lin, Z.; Yin, Q.; Flemington, E.K. Latent expression of the Epstein-Barr virus (EBV)-encoded major histocompatibility complex class I tap inhibitor, BNLF2a, in EBV-positive gastric carcinomas. J. Virol. 2015, 89, 10110–10114. [Google Scholar] [CrossRef] [PubMed]
- Strong, M.J.; Xu, G.; Coco, J.; Baribault, C.; Vinay, D.S.; Lacey, M.R.; Strong, A.L.; Lehman, T.A.; Seddon, M.B.; Lin, Z.; et al. Differences in gastric carcinoma microenvironment stratify according to EBV infection intensity: Implications for possible immune adjuvant therapy. PLoS Pathog. 2013, 9, e1003341. [Google Scholar] [CrossRef] [PubMed]
- Rowe, M.; Glaunsinger, B.; van Leeuwen, D.; Zuo, J.; Sweetman, D.; Ganem, D.; Middeldorp, J.; Wiertz, E.J.; Ressing, M.E. Host shutoff during productive Epstein-Barr virus infection is mediated by BGLF5 and may contribute to immune evasion. Proc. Natl. Acad. Sci. USA 2007, 104, 3366–3371. [Google Scholar] [CrossRef] [PubMed]
- Van Gent, M.; Griffin, B.D.; Berkhoff, E.G.; van Leeuwen, D.; Boer, I.G.; Buisson, M.; Hartgers, F.C.; Burmeister, W.P.; Wiertz, E.J.; Ressing, M.E. EBV lytic-phase protein BGLF5 contributes to tlr9 downregulation during productive infection. J. Immunol. 2011, 186, 1694–1702. [Google Scholar] [CrossRef] [PubMed]
- Zuo, J.; Thomas, W.; van Leeuwen, D.; Middeldorp, J.M.; Wiertz, E.J.; Ressing, M.E.; Rowe, M. The dnase of gammaherpesviruses impairs recognition by virus-specific CD8+ T cells through an additional host shutoff function. J. Virol. 2008, 82, 2385–2393. [Google Scholar] [CrossRef] [PubMed]
- Van Gent, M.; Gram, A.M.; Boer, I.G.; Geerdink, R.J.; Lindenbergh, M.F.; Lebbink, R.J.; Wiertz, E.J.; Ressing, M.E. Silencing the shutoff protein of Epstein-Barr virus in productively infected B cells points to (innate) targets for immune evasion. J. Gener. Virol. 2015, 96, 858–865. [Google Scholar] [CrossRef] [PubMed]
- Quinn, L.L.; Zuo, J.; Abbott, R.J.; Shannon-Lowe, C.; Tierney, R.J.; Hislop, A.D.; Rowe, M. Cooperation between Epstein-Barr virus immune evasion proteins spreads protection from CD8+ T cell recognition across all three phases of the lytic cycle. PLoS Pathog. 2014, 10, e1004322. [Google Scholar] [CrossRef] [PubMed]
- Feederle, R.; Bannert, H.; Lips, H.; Muller-Lantzsch, N.; Delecluse, H.J. The Epstein-Barr virus alkaline exonuclease BGLF5 serves pleiotropic functions in virus replication. J. Virol. 2009, 83, 4952–4962. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; Liu, M.Y.; Hsu, T.Y.; Cho, S.M.; Yang, C.S. Use of bacterially-expressed antigen for detection of antibodies to the EBV-specific deoxyribonuclease in sera from patients with nasopharyngeal carcinoma. J. Virol. Meth. 1993, 45, 49–66. [Google Scholar] [CrossRef]
- Sbih-Lammali, F.; Berger, F.; Busson, P.; Ooka, T. Expression of the dnase encoded by the BGLF5 gene of Epstein-Barr virus in nasopharyngeal carcinoma epithelial cells. Virology 1996, 222, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.V.; Cox, B.; Ariza, M.E. Herpesviruses dutpases: A new family of pathogen-associated molecular pattern (PAMP) proteins with implications for human disease. Pathogens 2016, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Ariza, M.E.; Glaser, R.; Kaumaya, P.T.; Jones, C.; Williams, M.V. The EBV-encoded dutpase activates NF-kappa B through the tlr2 and myd88-dependent signaling pathway. J. Immunol. 2009, 182, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Glaser, R.; Litsky, M.L.; Padgett, D.A.; Baiocchi, R.A.; Yang, E.V.; Chen, M.; Yeh, P.E.; Green-Church, K.B.; Caligiuri, M.A.; Williams, M.V. EBV-encoded dutpase induces immune dysregulation: Implications for the pathophysiology of EBV-associated disease. Virology 2006, 346, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Ariza, M.E.; Rivailler, P.; Glaser, R.; Chen, M.; Williams, M.V. Epstein-Barr virus encoded dutpase containing exosomes modulate innate and adaptive immune responses in human dendritic cells and peripheral blood mononuclear cells. PLoS ONE 2013, 8, e69827. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.V.; Webster Marketon, J.I.; Chen, M.; Lo, K.W.; Kim, S.J.; Glaser, R. Glucocorticoids activate epstein barr virus lytic replication through the upregulation of immediate early BZLF1 gene expression. Brain Behav. Immun. 2010, 24, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Halpin, P.; Williams, M.V.; Klimas, N.G.; Fletcher, M.A.; Barnes, Z.; Ariza, M.E. Myalgic encephalomyelitis/chronic fatigue syndrome and gulf war illness patients exhibit increased humoral responses to the herpesviruses-encoded dutpase: Implications in disease pathophysiology. J. Med. Virol. 2017, 89, 1636–1645. [Google Scholar] [CrossRef] [PubMed]
- Lerner, A.M.; Ariza, M.E.; Williams, M.; Jason, L.; Beqaj, S.; Fitzgerald, J.T.; Lemeshow, S.; Glaser, R. Antibody to Epstein-Barr virus deoxyuridine triphosphate nucleotidohydrolase and deoxyribonucleotide polymerase in a chronic fatigue syndrome subset. PLoS ONE 2012, 7, e47891. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, J.; Kremmer, E.; Greenspan, J.S.; Grasser, F.A.; Niedobitek, G. Expression of viral and human dutpase in Epstein-Barr virus-associated diseases. J. Med. Virol. 2002, 68, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, J.M.; Sommer, P.; Kremmer, E.; Ong, K.S.; Fung, K.; Lee, J.M.; Ng, M.H.; Grasser, F.A. A new lytic antibody, 7d6, detects Epstein-Barr virus dutpase in nonkeratinizing undifferentiated nasopharyngeal carcinomas. Lab. Investig. 1998, 78, 1031–1032. [Google Scholar] [PubMed]
- Schlieker, C.; Korbel, G.A.; Kattenhorn, L.M.; Ploegh, H.L. A deubiquitinating activity is conserved in the large tegument protein of the herpesviridae. J. Virol. 2005, 79, 15582–15585. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Cahir-McFarland, E.; Zhao, B.; Kieff, E. Virus and cell RNAs expressed during epstein-barr virus replication. J. Virol. 2006, 80, 2548–2565. [Google Scholar] [CrossRef] [PubMed]
- Gastaldello, S.; Hildebrand, S.; Faridani, O.; Callegari, S.; Palmkvist, M.; Di Guglielmo, C.; Masucci, M.G. A deneddylase encoded by Epstein-Barr virus promotes viral DNA replication by regulating the activity of cullin-ring ligases. Nat. Cell Biol. 2010, 12, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Van Gent, M.; Braem, S.G.; de Jong, A.; Delagic, N.; Peeters, J.G.; Boer, I.G.; Moynagh, P.N.; Kremmer, E.; Wiertz, E.J.; Ovaa, H.; et al. Epstein-Barr virus large tegument protein BPLF1 contributes to innate immune evasion through interference with toll-like receptor signaling. PLoS Pathog. 2014, 10, e1003960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, S.; Murata, T.; Kanda, T.; Isomura, H.; Narita, Y.; Sugimoto, A.; Kawashima, D.; Tsurumi, T. Epstein-Barr virus deubiquitinase downregulates TRAF6-mediated Nf-kappab signaling during productive replication. J. Virol. 2013, 87, 4060–4070. [Google Scholar] [CrossRef] [PubMed]
- Coghill, A.E.; Pfeiffer, R.M.; Proietti, C.; Hsu, W.L.; Chien, Y.C.; Lekieffre, L.; Krause, L.; Teng, A.; Pablo, J.; Yu, K.J.; et al. Identification of a novel, EBV-based antibody risk stratification signature for early detection of nasopharyngeal carcinoma in Taiwan. Clin. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Whitehurst, C.B.; Li, G.; Montgomery, S.A.; Montgomery, N.D.; Su, L.; Pagano, J.S. Knockout of Epstein-Barr virus BPLF1 retards b-cell transformation and lymphoma formation in humanized mice. mBio 2015, 6, e01574-15. [Google Scholar] [CrossRef] [PubMed]
- Bergbauer, M.; Kalla, M.; Schmeinck, A.; Gobel, C.; Rothbauer, U.; Eck, S.; Benet-Pages, A.; Strom, T.M.; Hammerschmidt, W. Cpg-methylation regulates a class of Epstein-Barr virus promoters. PLoS Pathog. 2010, 6, e1001114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Qian, L.; Chen, C.; Shi, M.; Yu, M.; Hu, M.; Song, L.; Shen, B.; Guo, N. Down-regulation of mhc class II expression through inhibition of ciita transcription by lytic transactivator zta during Epstein-Barr virus reactivation. J. Immunol. 2009, 182, 1799–1809. [Google Scholar] [CrossRef] [PubMed]
- Zuo, J.; Thomas, W.A.; Haigh, T.A.; Fitzsimmons, L.; Long, H.M.; Hislop, A.D.; Taylor, G.S.; Rowe, M. Epstein-Barr virus evades CD4+ T cell responses in lytic cycle through BZLF1-mediated downregulation of CD74 and the cooperation of VBCL-2. PLoS Pathog. 2011, 7, e1002455. [Google Scholar] [CrossRef] [PubMed]
- Morrison, T.E.; Mauser, A.; Wong, A.; Ting, J.P.; Kenney, S.C. Inhibition of IFN-gamma signaling by an Epstein-Barr virus immediate-early protein. Immunity 2001, 15, 787–799. [Google Scholar] [CrossRef]
- Morrison, T.E.; Mauser, A.; Klingelhutz, A.; Kenney, S.C. Epstein-Barr virus immediate-early protein BZLF1 inhibits tumor necrosis factor alpha-induced signaling and apoptosis by downregulating tumor necrosis factor receptor 1. J. Virol. 2004, 78, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Poppers, J.; Mulvey, M.; Perez, C.; Khoo, D.; Mohr, I. Identification of a lytic-cycle Epstein-Barr virus gene product that can regulate PKR activation. J. Virol. 2003, 77, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.; Wu, S.Y.; Chang, S.S.; Su, I.J.; Tsai, C.H.; Lai, S.J.; Shiau, A.L.; Takada, K.; Chang, Y. Epstein-Barr virus lytic transactivator zta enhances chemotactic activity through induction of interleukin-8 in nasopharyngeal carcinoma cells. J. Virol. 2008, 82, 3679–3688. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.J.; Seaman, W.T.; Feng, W.H.; Barlow, E.; Dickerson, S.; Delecluse, H.J.; Kenney, S.C. Roles of lytic viral infection and IL-6 in early versus late passage lymphoblastoid cell lines and EBV-associated lymphoproliferative disease. Int. J. Cancer 2007, 121, 1274–1281. [Google Scholar] [CrossRef] [PubMed]
- Haddad, E.; Paczesny, S.; Leblond, V.; Seigneurin, J.M.; Stern, M.; Achkar, A.; Bauwens, M.; Delwail, V.; Debray, D.; Duvoux, C.; et al. Treatment of b-lymphoproliferative disorder with a monoclonal anti-interleukin-6 antibody in 12 patients: A multicenter phase 1–2 clinical trial. Blood 2001, 97, 1590–1597. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Yeh, T.H.; Lai, H.C.; Wu, S.Y.; Su, I.J.; Takada, K.; Chang, Y. Epstein-Barr virus zta-induced immunomodulators from nasopharyngeal carcinoma cells upregulate interleukin-10 production from monocytes. J. Virol. 2011, 85, 7333–7342. [Google Scholar] [CrossRef] [PubMed]
- Thun, M.J.; Henley, S.J.; Patrono, C. Nonsteroidal anti-inflammatory drugs as anticancer agents: Mechanistic, pharmacologic, and clinical issues. J. Natl. Cancer Inst. 2002, 94, 252–266. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, J.; Gaur, N.; Khera, L.; Kaul, R.; Robertson, E.S. Cox-2 induces lytic reactivation of EBV through PGE2 by modulating the EP receptor signaling pathway. Virology 2015, 484, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Katsumura, K.R.; Maruo, S.; Takada, K. EBV lytic infection enhances transformation of B-lymphocytes infected with EBV in the presence of t-lymphocytes. J. Med. Virol. 2012, 84, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Sall, A.; Caserta, S.; Jolicoeur, P.; Franqueville, L.; de Turenne-Tessier, M.; Ooka, T. Mitogenic activity of Epstein-Barr virus-encoded BARF1 protein. Oncogene 2004, 23, 4938–4944. [Google Scholar] [CrossRef] [PubMed]
- Houali, K.; Wang, X.; Shimizu, Y.; Djennaoui, D.; Nicholls, J.; Fiorini, S.; Bouguermouh, A.; Ooka, T. A new diagnostic marker for secreted Epstein-Barr virus encoded LMP1 and BARF1 oncoproteins in the serum and saliva of patients with nasopharyngeal carcinoma. Clin. Cancer Res. 2007, 13, 4993–5000. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Ooka, T.; Middeldorp, J.; Takada, K. Reconstitution of nasopharyngeal carcinoma-type EBV infection induces tumorigenicity. Cancer Res. 2008, 68, 1030–1036. [Google Scholar] [CrossRef] [PubMed]
- Stack, G.; Stacey, M.A.; Humphreys, I.R. Herpesvirus exploitation of host immune inhibitory pathways. Viruses 2012, 4, 1182–1201. [Google Scholar] [CrossRef]
- Moore, K.W.; Vieira, P.; Fiorentino, D.F.; Trounstine, M.L.; Khan, T.A.; Mosmann, T.R. Homology of cytokine synthesis inhibitory factor (IL-10) to the Epstein-Barr virus gene BCRFI. Science 1990, 248, 1230–1234. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, S.; Hesselton, R.; Sullivan, J.; Kieff, E. Epstein-Barr virus recombinants with specifically mutated BCRF1 genes. J. Virol. 1993, 67, 7406–7413. [Google Scholar] [PubMed]
- Vischer, H.F.; Nijmeijer, S.; Smit, M.J.; Leurs, R. Viral hijacking of human receptors through heterodimerization. Biochem. Biophys. Res. Commun. 2008, 377, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Nijmeijer, S.; Leurs, R.; Smit, M.J.; Vischer, H.F. The epstein-barr virus-encoded g protein-coupled receptor BILF1 hetero-oligomerizes with human cxcr4, scavenges galphai proteins, and constitutively impairs CXCR4 functioning. J. Biol. Chem. 2010, 285, 29632–29641. [Google Scholar] [CrossRef] [PubMed]
- Giffin, L.; Damania, B. Kshv: Pathways to tumorigenesis and persistent infection. Adv. Virus Res. 2014, 88, 111–159. [Google Scholar] [PubMed]
- Potouridou, I.; Katsambas, A.; Pantazi, V.; Armenaka, M.; Vareltzidis, A.; Stratigos, J. Koebner phenomenon in classic Kaposi’s sarcoma. Acta Derm. Venereol. 1997, 77, 481. [Google Scholar] [PubMed]
- Siegel, M.O.; Ghafouri, S.; Ajmera, R.; Simon, G.L. Immune reconstitution inflammatory syndrome, human herpesvirus 8 viremia, and HIV-associated multicentric castleman disease. Int. J. Infect. Dis. 2016, 48, 49–51. [Google Scholar] [CrossRef] [PubMed]
- Walker, N.F.; Scriven, J.; Meintjes, G.; Wilkinson, R.J. Immune reconstitution inflammatory syndrome in hiv-infected patients. HIV Aids 2015, 7, 49–64. [Google Scholar]
- Monini, P.; Colombini, S.; Sturzl, M.; Goletti, D.; Cafaro, A.; Sgadari, C.; Butto, S.; Franco, M.; Leone, P.; Fais, S.; et al. Reactivation and persistence of human herpesvirus-8 infection in B cells and monocytes by Th-1 cytokines increased in kaposi’s sarcoma. Blood 1999, 93, 4044–4058. [Google Scholar] [PubMed]
- Dittmer, D.P.; Damania, B. Kaposi sarcoma-associated herpesvirus: Immunobiology, oncogenesis, and therapy. J. Clin. Investig. 2016, 126, 3165–3175. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, H.H.; Naranatt, P.P.; Smith, M.S.; Zeng, L.; Bloomer, C.; Chandran, B. Concurrent expression of latent and a limited number of lytic genes with immune modulation and antiapoptotic function by Kaposi’s sarcoma-associated herpesvirus early during infection of primary endothelial and fibroblast cells and subsequent decline of lytic gene expression. J. Virol. 2004, 78, 3601–3620. [Google Scholar] [PubMed]
- Mutlu, A.D.; Cavallin, L.E.; Vincent, L.; Chiozzini, C.; Eroles, P.; Duran, E.M.; Asgari, Z.; Hooper, A.T.; La Perle, K.M.; Hilsher, C.; et al. In vivo-restricted and reversible malignancy induced by human herpesvirus-8 kshv: A cell and animal model of virally induced Kaposi’s sarcoma. Cancer Cell 2007, 11, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Grundhoff, A.; Ganem, D. Inefficient establishment of kshv latency suggests an additional role for continued lytic replication in Kaposi sarcoma pathogenesis. J. Clin. Investig. 2004, 113, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Bais, C.; Van Geelen, A.; Eroles, P.; Mutlu, A.; Chiozzini, C.; Dias, S.; Silverstein, R.L.; Rafii, S.; Mesri, E.A. Kaposi’s sarcoma associated herpesvirus g protein-coupled receptor immortalizes human endothelial cells by activation of the VEGF receptor-2/KDR. Cancer Cell 2003, 3, 131–143. [Google Scholar] [CrossRef]
- Montaner, S.; Sodhi, A.; Ramsdell, A.K.; Martin, D.; Hu, J.; Sawai, E.T.; Gutkind, J.S. The Kaposi’s sarcoma-associated herpesvirus g protein-coupled receptor as a therapeutic target for the treatment of Kaposi’s sarcoma. Cancer Res. 2006, 66, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Vischer, H.F.; Siderius, M.; Leurs, R.; Smit, M.J. Herpesvirus-encoded gpcrs: Neglected players in inflammatory and proliferative diseases? Nat. Rev. Drug Discov. 2014, 13, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.; Ganem, D. A unique herpesviral transcriptional program in kshv-infected lymphatic endothelial cells leads to mTORC1 activation and rapamycin sensitivity. Cell Host Microbe 2013, 13, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.X.; King, S.M.; Smith, E.J.; Levy, D.E.; Yuan, Y. A Kaposi’s sarcoma-associated herpesviral protein inhibits virus-mediated induction of type I interferon by blocking IRF-7 phosphorylation and nuclear accumulation. Proc. Natl. Acad. Sci. USA 2002, 99, 5573–5578. [Google Scholar] [CrossRef] [PubMed]
- Du, M.Q.; Liu, H.; Diss, T.C.; Ye, H.; Hamoudi, R.A.; Dupin, N.; Meignin, V.; Oksenhendler, E.; Boshoff, C.; Isaacson, P.G. Kaposi sarcoma-associated herpesvirus infects monotypic (IgM lambda) but polyclonal naive B cells in castleman disease and associated lymphoproliferative disorders. Blood 2001, 97, 2130–2136. [Google Scholar] [CrossRef] [PubMed]
- Asahi-Ozaki, Y.; Sato, Y.; Kanno, T.; Sata, T.; Katano, H. Quantitative analysis of kaposi sarcoma-associated herpesvirus (KSHV) in kshv-associated diseases. J. Infect. Dis. 2006, 193, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Naresh, K.N.; Rice, A.J.; Bower, M. Lymph nodes involved by multicentric castleman disease among hiv-positive individuals are often involved by Kaposi sarcoma. Am. J. Surg. Pathol. 2008, 32, 1006–1012. [Google Scholar] [CrossRef] [PubMed]
- Polizzotto, M.N.; Uldrick, T.S.; Hu, D.; Yarchoan, R. Clinical manifestations of kaposi sarcoma herpesvirus lytic activation: Multicentric castleman disease (KSHV-MCD) and the kshv inflammatory cytokine syndrome. Front. Microbiol. 2012, 3, 73. [Google Scholar] [CrossRef] [PubMed]
- Brandt, S.J.; Bodine, D.M.; Dunbar, C.E.; Nienhuis, A.W. Dysregulated interleukin 6 expression produces a syndrome resembling castleman’s disease in mice. J. Clin. Investig. 1990, 86, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Mocarski, E.S., Jr. Immunomodulation by cytomegaloviruses: Manipulative strategies beyond evasion. Trends Microbiol. 2002, 10, 332–339. [Google Scholar] [CrossRef]
- Basta, S.; Bennink, J.R. A survival game of hide and seek: Cytomegaloviruses and MHC class I antigen presentation pathways. Viral Immunol. 2003, 16, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Beck, S.; Barrell, B.G. Human cytomegalovirus encodes a glycoprotein homologous to MHC class I antigens. Nature 1988, 331, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Bjorkman, P.J. Structure of ul18, a peptide-binding viral MHC mimic, bound to a host inhibitory receptor. Proc. Natl. Acad. Sci. USA 2008, 105, 10095–10100. [Google Scholar] [CrossRef] [PubMed]
- Tomasec, P.; Braud, V.M.; Rickards, C.; Powell, M.B.; McSharry, B.P.; Gadola, S.; Cerundolo, V.; Borysiewicz, L.K.; McMichael, A.J.; Wilkinson, G.W. Surface expression of HLA-e, an inhibitor of natural killer cells, enhanced by human cytomegalovirus GPUL40. Science 2000, 287, 1031. [Google Scholar] [CrossRef] [PubMed]
- Avdic, S.; McSharry, B.P.; Steain, M.; Poole, E.; Sinclair, J.; Abendroth, A.; Slobedman, B. Human cytomegalovirus-encoded human interleukin-10 (IL-10) homolog amplifies its immunomodulatory potential by upregulating human IL-10 in monocytes. J. Virol. 2016, 90, 3819–3827. [Google Scholar] [CrossRef] [PubMed]
- Dziurzynski, K.; Wei, J.; Qiao, W.; Hatiboglu, M.A.; Kong, L.Y.; Wu, A.; Wang, Y.; Cahill, D.; Levine, N.; Prabhu, S.; et al. Glioma-associated cytomegalovirus mediates subversion of the monocyte lineage to a tumor propagating phenotype. Clin. Cancer Res. 2011, 17, 4642–4649. [Google Scholar] [CrossRef] [PubMed]
- Cinatl, J., Jr.; Cinatl, J.; Vogel, J.U.; Rabenau, H.; Kornhuber, B.; Doerr, H.W. Modulatory effects of human cytomegalovirus infection on malignant properties of cancer cells. Intervirology 1996, 39, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Cinatl, J.; Scholz, M.; Kotchetkov, R.; Vogel, J.U.; Doerr, H.W. Molecular mechanisms of the modulatory effects of HCMV infection in tumor cell biology. Trends Mol. Med. 2004, 10, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Cobbs, C. Response to “human cytomegalovirus infection in tumor cells of the nervous system is not detectable with standardized pathologico-virological diagnostics”. Neuro Oncol. 2014, 16, 1435–1436. [Google Scholar] [CrossRef] [PubMed]
- Holdhoff, M.; Guner, G.; Rodriguez, F.J.; Hicks, J.L.; Zheng, Q.; Forman, M.S.; Ye, X.; Grossman, S.A.; Meeker, A.K.; Heaphy, C.M.; et al. Absence of cytomegalovirus in glioblastoma and other high-grade gliomas by real-time PCR, immunohistochemistry, and in situ hybridization. Clin. Cancer Res. 2017, 23, 3150–3157. [Google Scholar] [CrossRef] [PubMed]
- Strong, M.J.; Blanchard, E.T.; Lin, Z.; Morris, C.A.; Baddoo, M.; Taylor, C.M.; Ware, M.L.; Flemington, E.K. A comprehensive next generation sequencing-based virome assessment in brain tissue suggests no major virus-tumor association. Acta Neuropathol. Commun. 2016, 4, 71. [Google Scholar] [CrossRef] [PubMed]
- Soroceanu, L.; Matlaf, L.; Bezrookove, V.; Harkins, L.; Martinez, R.; Greene, M.; Soteropoulos, P.; Cobbs, C.S. Human cytomegalovirus US28 found in glioblastoma promotes an invasive and angiogenic phenotype. Cancer Res. 2011, 71, 6643–6653. [Google Scholar] [CrossRef] [PubMed]
- Martini, M.; Pallini, R.; Luongo, G.; Cenci, T.; Lucantoni, C.; Larocca, L.M. Prognostic relevance of SOCS3 hypermethylation in patients with glioblastoma multiforme. Int. J. Cancer 2008, 123, 2955–2960. [Google Scholar] [CrossRef] [PubMed]
- Slinger, E.; Maussang, D.; Schreiber, A.; Siderius, M.; Rahbar, A.; Fraile-Ramos, A.; Lira, S.A.; Soderberg-Naucler, C.; Smit, M.J. HCMV-encoded chemokine receptor US28 mediates proliferative signaling through the IL-6-STAT3 axis. Sci. Signal. 2010, 3, ra58. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lathia, J.D.; Wu, Q.; Wang, J.; Li, Z.; Heddleston, J.M.; Eyler, C.E.; Elderbroom, J.; Gallagher, J.; Schuschu, J.; et al. Targeting interleukin 6 signaling suppresses glioma stem cell survival and tumor growth. Stem Cells 2009, 27, 2393–2404. [Google Scholar] [CrossRef] [PubMed]
- Price, R.L.; Song, J.; Bingmer, K.; Kim, T.H.; Yi, J.Y.; Nowicki, M.O.; Mo, X.; Hollon, T.; Murnan, E.; Alvarez-Breckenridge, C.; et al. Cytomegalovirus contributes to glioblastoma in the context of tumor suppressor mutations. Cancer Res. 2013, 73, 3441–3450. [Google Scholar] [CrossRef] [PubMed]
- Maussang, D.; Verzijl, D.; van Walsum, M.; Leurs, R.; Holl, J.; Pleskoff, O.; Michel, D.; van Dongen, G.A.; Smit, M.J. Human cytomegalovirus-encoded chemokine receptor us28 promotes tumorigenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 13068–13073. [Google Scholar] [CrossRef] [PubMed]
- Bongers, G.; Maussang, D.; Muniz, L.R.; Noriega, V.M.; Fraile-Ramos, A.; Barker, N.; Marchesi, F.; Thirunarayanan, N.; Vischer, H.F.; Qin, L.; et al. The cytomegalovirus-encoded chemokine receptor US28 promotes intestinal neoplasia in transgenic mice. J. Clin. Investig. 2010, 120, 3969–3978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, X.; Li, X.J.; Liu, X.J.; Yuan, H.; Li, J.F.; Duan, Y.L.; Ye, H.Q.; Fu, Y.R.; Qiao, G.H.; Wu, C.C.; et al. Later passages of neural progenitor cells from neonatal brain are more permissive for human cytomegalovirus infection. J. Virol. 2013, 87, 10968–10979. [Google Scholar] [CrossRef] [PubMed]
- Fiallos, E.; Judkins, J.; Matlaf, L.; Prichard, M.; Dittmer, D.; Cobbs, C.; Soroceanu, L. Human cytomegalovirus gene expression in long-term infected glioma stem cells. PLoS ONE 2014, 9, e116178. [Google Scholar] [CrossRef] [PubMed]
- Soroceanu, L.; Matlaf, L.; Khan, S.; Akhavan, A.; Singer, E.; Bezrookove, V.; Decker, S.; Ghanny, S.; Hadaczek, P.; Bengtsson, H.; et al. Cytomegalovirus immediate-early proteins promote stemness properties in glioblastoma. Cancer Res. 2015, 75, 3065–3076. [Google Scholar] [CrossRef] [PubMed]
- Dumortier, J.; Streblow, D.N.; Moses, A.V.; Jacobs, J.M.; Kreklywich, C.N.; Camp, D.; Smith, R.D.; Orloff, S.L.; Nelson, J.A. Human cytomegalovirus secretome contains factors that induce angiogenesis and wound healing. J. Virol. 2008, 82, 6524–6535. [Google Scholar] [CrossRef] [PubMed]
- Baryawno, N.; Rahbar, A.; Wolmer-Solberg, N.; Taher, C.; Odeberg, J.; Darabi, A.; Khan, Z.; Sveinbjornsson, B.; FuskevAg, O.M.; Segerstrom, L.; et al. Detection of human cytomegalovirus in medulloblastomas reveals a potential therapeutic target. J. Clin. Investig. 2011, 121, 4043–4055. [Google Scholar] [CrossRef] [PubMed]
- Harkins, L.; Volk, A.L.; Samanta, M.; Mikolaenko, I.; Britt, W.J.; Bland, K.I.; Cobbs, C.S. Specific localisation of human cytomegalovirus nucleic acids and proteins in human colorectal cancer. Lancet 2002, 360, 1557–1563. [Google Scholar] [CrossRef]
- Maussang, D.; Langemeijer, E.; Fitzsimons, C.P.; Stigter-van Walsum, M.; Dijkman, R.; Borg, M.K.; Slinger, E.; Schreiber, A.; Michel, D.; Tensen, C.P.; et al. The human cytomegalovirus-encoded chemokine receptor us28 promotes angiogenesis and tumor formation via cyclooxygenase-2. Cancer Res. 2009, 69, 2861–2869. [Google Scholar] [CrossRef] [PubMed]
- Rahbar, A.; Orrego, A.; Peredo, I.; Dzabic, M.; Wolmer-Solberg, N.; Straat, K.; Stragliotto, G.; Soderberg-Naucler, C. Human cytomegalovirus infection levels in glioblastoma multiforme are of prognostic value for survival. J. Clin. Virol. 2013, 57, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Soderberg-Naucler, C.; Rahbar, A.; Stragliotto, G. Survival in patients with glioblastoma receiving valganciclovir. N. Engl. J. Med. 2013, 369, 985–986. [Google Scholar] [CrossRef] [PubMed]
- Stragliotto, G.; Rahbar, A.; Solberg, N.W.; Lilja, A.; Taher, C.; Orrego, A.; Bjurman, B.; Tammik, C.; Skarman, P.; Peredo, I.; et al. Effects of valganciclovir as an add-on therapy in patients with cytomegalovirus-positive glioblastoma: A randomized, double-blind, hypothesis-generating study. Int. J. Cancer 2013, 133, 1204–1213. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.M.; Glaser, R.; Malarkey, W.B.; Beversdorf, D.Q.; Peng, J.; Kiecolt-Glaser, J.K. Inflammation and reactivation of latent herpesviruses in older adults. Brain Behav. Immun. 2012, 26, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Whitehurst, C.B.; Pagano, J.S. The RAD6/18 ubiquitin complex interacts with the Epstein-Barr virus deubiquitinating enzyme, BPLF1, and contributes to virus infectivity. J. Virol. 2014, 88, 6411–6422. [Google Scholar] [CrossRef] [PubMed]
- Whitehurst, C.B.; Vaziri, C.; Shackelford, J.; Pagano, J.S. Epstein-Barr virus BPLF1 deubiquitinates pcna and attenuates polymerase eta recruitment to DNA damage sites. J. Virol. 2012, 86, 8097–8106. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Liu, M.T.; Chang, Y.T.; Fang, C.Y.; Chou, S.P.; Liao, H.W.; Kuo, K.L.; Hsu, S.L.; Chen, Y.R.; Wang, P.W.; et al. Epstein-Barr virus dnase (BGLF5) induces genomic instability in human epithelial cells. Nucleic Acids Res. 2010, 38, 1932–1949. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.H.; Lee, C.P.; Su, M.T.; Wang, J.T.; Chen, J.Y.; Lin, S.F.; Tsai, C.H.; Hsieh, M.J.; Takada, K.; Chen, M.R. Epstein-Barr virus BGLF4 kinase retards cellular s-phase progression and induces chromosomal abnormality. PLoS ONE 2012, 7, e39217. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Deng, W.; Hau, P.M.; Liu, J.; Lau, V.M.; Cheung, A.L.; Huen, M.S.; Tsao, S.W. Epstein-Barr virus BZLF1 protein impairs accumulation of host DNA damage proteins at damage sites in response to DNA damage. Lab. Investig. 2015, 95, 937–950. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| EBV | KSHV | HCMV | Function |
|---|---|---|---|
| BHRF1 | KSHV BCL-2 | IE1 | Anti-apoptotic |
| BALF1 | Anti-apoptotic | ||
| BARF1 | K14 | e127 | Anti-apoptotic, Paracrine/autocrine role in inflammation and immunomodulation, oncogene |
| BCRF1 (ebvIL-10) | cmvIL-10 | Immunomodulatory, immunosuppressive | |
| BILF1 | ORF74 (kshvGPCR) | US27, US28, UL33 & UL78 | Paracrine/autocrine role in inflammation and immunomodulation, immunoevasin |
| BGLF5 | Immunomodulatory, immunosuppressive, immunoevasin | ||
| BNLF2A | K3 & K5 | US2, US3, US6 & US11 | Immunomodulatory, immunoevasin |
| BLLF3 | ORF54 | UL72 | dUTPase, Paracrine/autocrine role in inflammation and immunomodulation |
| BPLF1 | ORF64 | UL48 | Deneddylase, ubiquitin-specific cysteine protease, immunomodulatory |
| BZLF1 (Zta) | AP1 orthologue, paracrine/autocrine role in inflammation and immunomodulation | ||
| BILF1 | Immunoevasin | ||
| BRLF1 (Rta) | Rta/ORF50 | Paracrine/autocrine role in inflammation and immunomodulation |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morales-Sánchez, A.; Fuentes-Panana, E.M. The Immunomodulatory Capacity of an Epstein-Barr Virus Abortive Lytic Cycle: Potential Contribution to Viral Tumorigenesis. Cancers 2018, 10, 98. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers10040098
Morales-Sánchez A, Fuentes-Panana EM. The Immunomodulatory Capacity of an Epstein-Barr Virus Abortive Lytic Cycle: Potential Contribution to Viral Tumorigenesis. Cancers. 2018; 10(4):98. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers10040098
Chicago/Turabian StyleMorales-Sánchez, Abigail, and Ezequiel M. Fuentes-Panana. 2018. "The Immunomodulatory Capacity of an Epstein-Barr Virus Abortive Lytic Cycle: Potential Contribution to Viral Tumorigenesis" Cancers 10, no. 4: 98. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers10040098