Histone Deacetylase Inhibitors and Phenotypical Transformation of Cancer Cells

 ,
,  and
and
Abstract
:1. Introduction
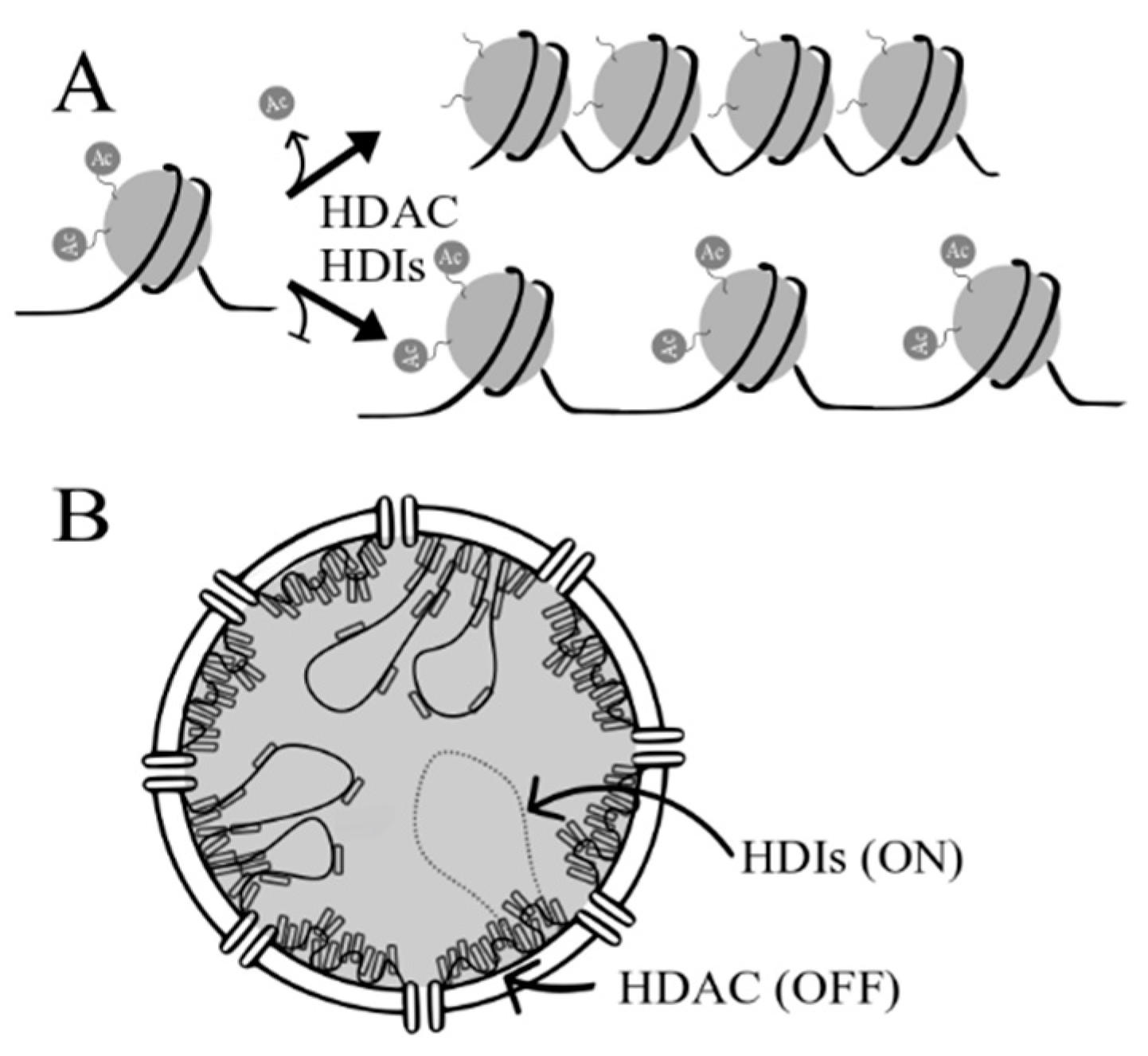
2. Histone Deacetylases (HDACs) and Histone Deacetylase Inhibitors (HDIs)
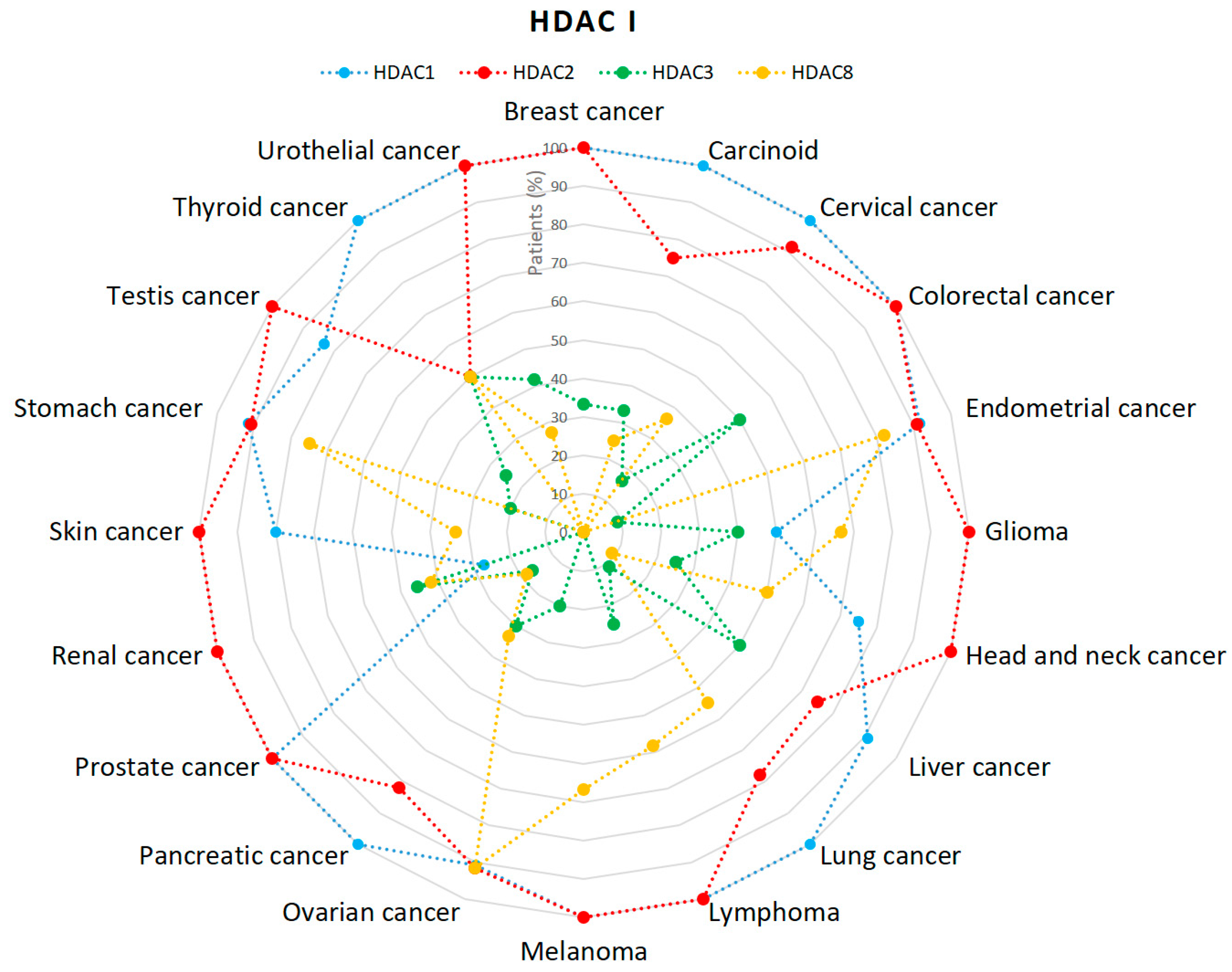
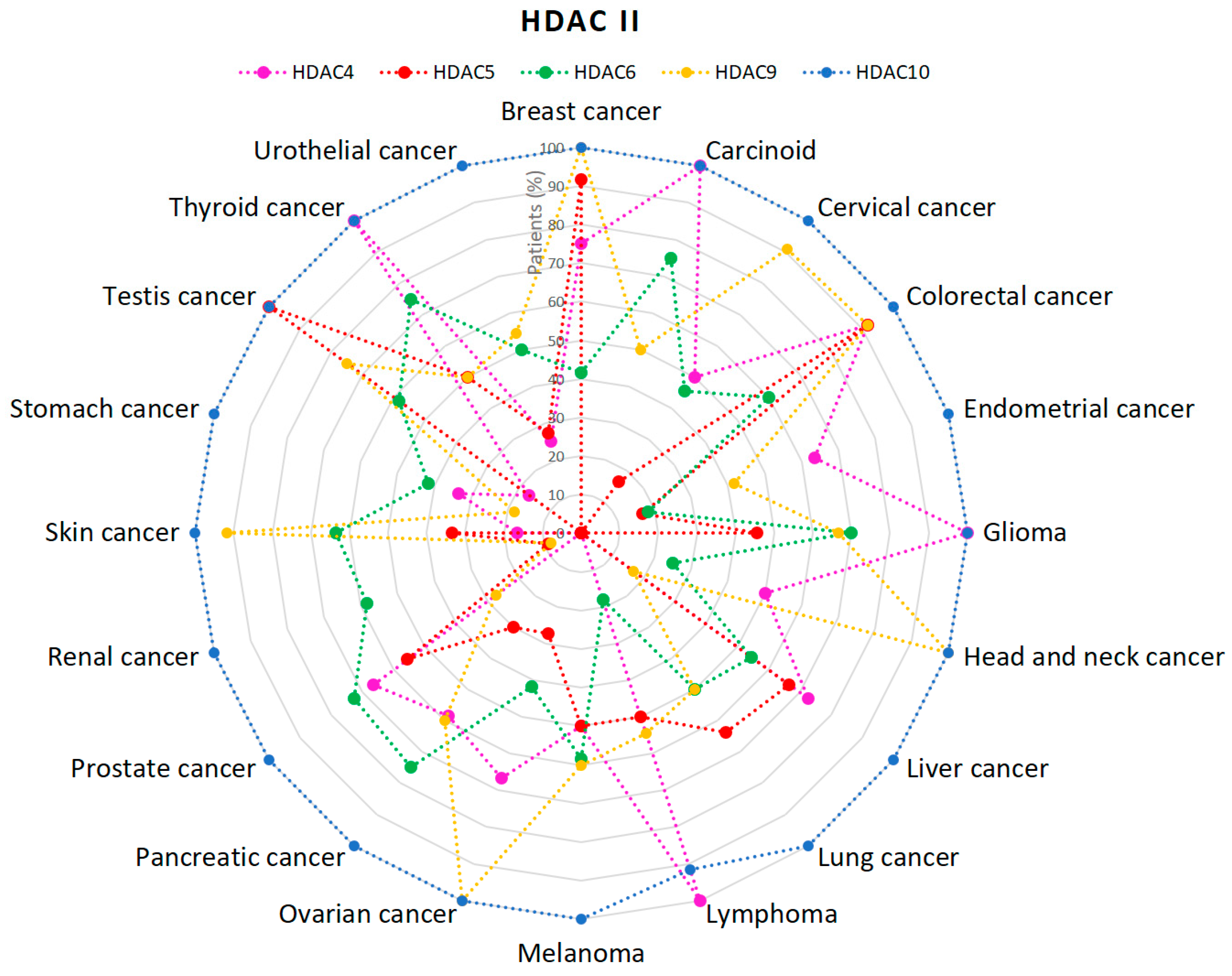
2.1. HDACs
2.2. HDIs
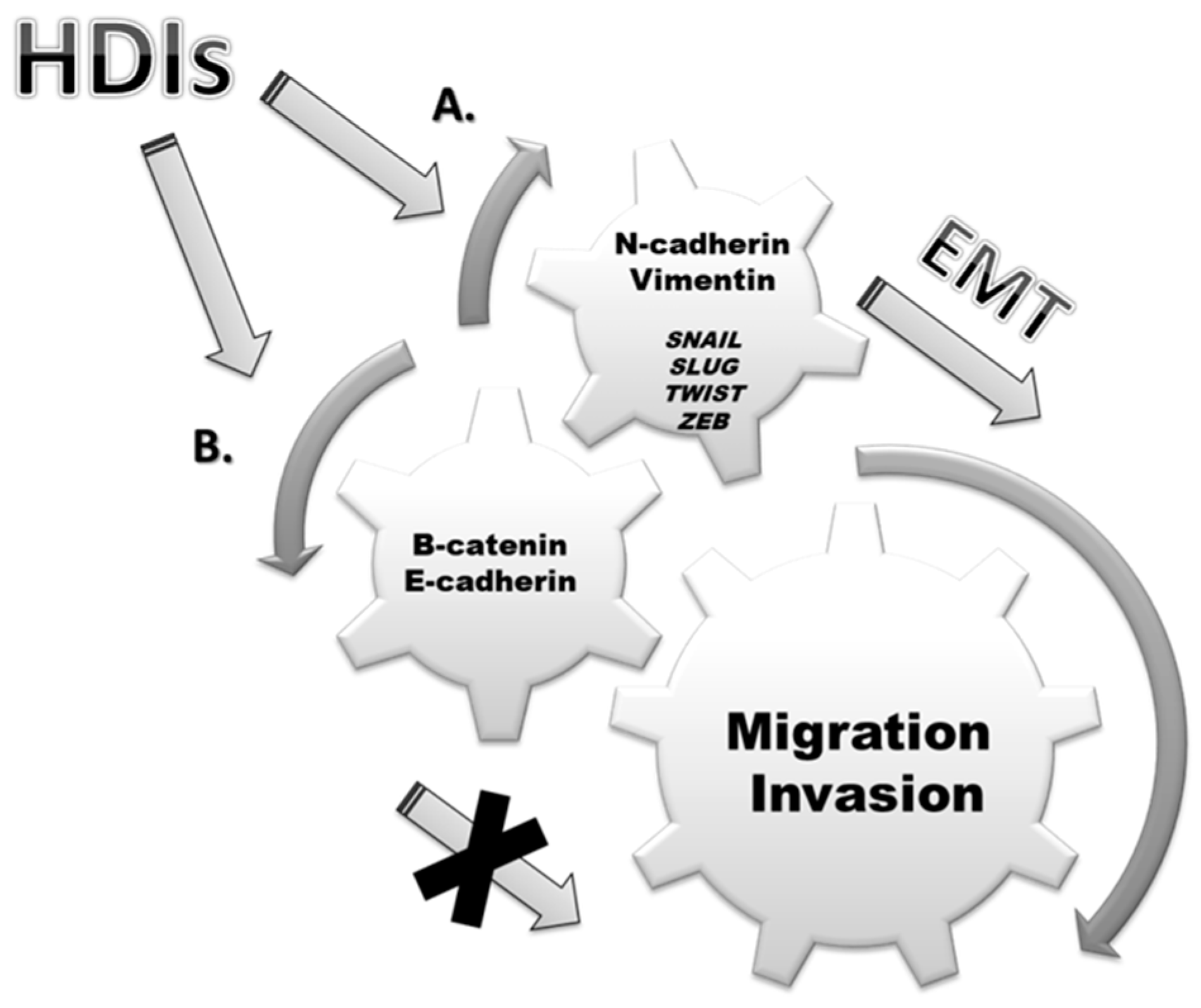
3. Epithelial-Mesenchymal Transition (EMT)
4. EMT and Cancers
4.1. Lung Cancer
4.2. Hepatocellular Carcinoma
4.3. Cholangiocarcinoma
4.4. Pancreatic Cancer
4.5. Colorectal Cancer
4.6. Renal Cancer
4.7. Urothelial Carcinoma
4.8. Prostate Cancer
4.9. Breast Cancer
4.10. Ovarian Cancer
4.11. Head and Neck Cancer
4.12. Malignant Glioma
5. Discussion
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Davis, F.M.; Stewart, T.A.; Thompson, E.W.; Monteith, G.R. Targeting EMT in cancer: Opportunities for pharmacological intervention. Trends Pharmacol. Sci. 2014, 35, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fardi, M.; Solali, S.; Farshdousti Hagh, M. Epigenetic mechanisms as a new approach in cancer treatment: An updated review. Genes Dis. 2018, 5, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Jiang, S.; Zhang, C. Recent advances in histone modification and histone modifying enzyme assays. Expert Rev. Mol. Diagn. 2018, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Grabarska, A.; Łuszczki, J.J.; Nowosadzka, E.; Gumbarewicz, E.; Jeleniewicz, W.; Dmoszyńska-Graniczka, M.; Kowalczuk, K.; Kupisz, K.; Polberg, K.; Stepulak, A. Histone Deacetylase Inhibitor SAHA as Potential Targeted Therapy Agent for Larynx Cancer Cells. J. Cancer 2017, 8, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Gumbarewicz, E.; Luszczki, J.J.; Wawruszak, A.; Dmoszynska-Graniczka, M.; Grabarska, A.J.; Jarząb, A.M.; Polberg, K.; Stepulak, A. Isobolographic analysis demonstrates additive effect of cisplatin and HDIs combined treatment augmenting their anti-cancer activity in lung cancer cell lines. Am. J. Cancer Res. 2016, 6, 2831–2845. [Google Scholar]
- Wawruszak, A.; Luszczki, J.J.; Grabarska, A.; Gumbarewicz, E.; Dmoszynska-Graniczka, M.; Polberg, K.; Stepulak, A. Assessment of Interactions between Cisplatin and Two Histone Deacetylase Inhibitors in MCF7, T47D and MDA-MB-231 Human Breast Cancer Cell Lines—An Isobolographic Analysis. PLoS ONE 2015, 10, e0143013. [Google Scholar] [CrossRef]
- Falkenberg, K.J.; Johnstone, R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 2014, 13, 673–691. [Google Scholar] [CrossRef]
- Hölscher, A.S.; Schulz, W.A.; Pinkerneil, M.; Niegisch, G.; Hoffmann, M.J. Combined inhibition of BET proteins and class I HDACs synergistically induces apoptosis in urothelial carcinoma cell lines. Clin. Epigenetics 2018, 10, 1. [Google Scholar] [CrossRef] [Green Version]
- Yao, D.; Dai, C.; Peng, S. Mechanism of the mesenchymal-epithelial transition and its relationship with metastatic tumor formation. Mol. Cancer Res. 2011, 9, 1608–1620. [Google Scholar] [CrossRef]
- Lan, X.; Lu, G.; Yuan, C.; Mao, S.; Jiang, W.; Chen, Y.; Jin, X.; Xia, Q. Valproic acid (VPA) inhibits the epithelial–mesenchymal transition in prostate carcinoma via the dual suppression of SMAD4. J. Cancer Res. Clin. Oncol. 2016, 142, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Nalls, D.; Tang, S.-N.; Rodova, M.; Srivastava, R.K.; Shankar, S. Targeting epigenetic regulation of miR-34a for treatment of pancreatic cancer by inhibition of pancreatic cancer stem cells. PLoS ONE 2011, 6, e24099. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xu, J.; Wang, H.; Wu, L.; Yuan, W.; Du, J.; Cai, S. Trichostatin A, a histone deacetylase inhibitor, reverses epithelial–mesenchymal transition in colorectal cancer SW480 and prostate cancer PC3 cells. Biochem. Biophys. Res. Commun. 2015, 456, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.; Gau, Y.; Sabnis, G. Histone deacetylase inhibitor entinostat reverses epithelial to mesenchymal transition of breast cancer cells by reversing the repression of E-cadherin. Breast Cancer Res. Treat. 2014, 143, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Ji, M.; Lee, E.J.; Kim, K.B.; Kim, Y.; Sung, R.; Lee, S.-J.; Kim, D.S.; Park, S.M. HDAC inhibitors induce epithelial-mesenchymal transition in colon carcinoma cells. Oncol. Rep. 2015, 33, 2299–2308. [Google Scholar] [CrossRef] [PubMed]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination Therapy with Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef] [PubMed]
- Ronnekleiv-Kelly, S.M.; Sharma, A.; Ahuja, N. Epigenetic therapy and chemosensitization in solid malignancy. Cancer Treat. Rev. 2017, 55, 200–208. [Google Scholar] [CrossRef]
- Manal, M.; Chandrasekar, M.J.N.; Gomathi Priya, J.; Nanjan, M.J. Inhibitors of histone deacetylase as antitumor agents: A critical review. Bioorg. Chem. 2016, 67, 18–42. [Google Scholar] [CrossRef]
- Sun, X.-J.; Man, N.; Tan, Y.; Nimer, S.D.; Wang, L. The Role of Histone Acetyltransferases in Normal and Malignant Hematopoiesis. Front. Oncol. 2015, 5, 108. [Google Scholar] [CrossRef]
- Roche, J.; Bertrand, P. Inside HDACs with more selective HDAC inhibitors. Eur. J. Med. Chem. 2016, 121, 451–483. [Google Scholar] [CrossRef]
- Yang, X.-J.; Seto, E. HATs and HDACs: From structure, function and regulation to novel strategies for therapy and prevention. Oncogene 2007, 26, 5310–5318. [Google Scholar] [CrossRef] [PubMed]
- Chrun, E.S.; Modolo, F.; Daniel, F.I. Histone modifications: A review about the presence of this epigenetic phenomenon in carcinogenesis. Pathol. Res. Pract. 2017, 213, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- Barneda-Zahonero, B.; Parra, M. Histone deacetylases and cancer. Mol. Oncol. 2012, 6, 579–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Cai, C.; Tang, D.; Sun, S.; Li, H. Effect of histone deacetylase inhibitors trichostatin A and valproic acid on hair cell regeneration in zebrafish lateral line neuromasts. Front. Cell. Neurosci. 2014, 8, 382. [Google Scholar] [CrossRef] [PubMed]
- Vickers, C.J.; Olsen, C.A.; Leman, L.J.; Ghadiri, M.R. Discovery of HDAC Inhibitors That Lack an Active Site Zn(2+)-Binding Functional Group. ACS Med. Chem. Lett. 2012, 3, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Dang, W. The controversial world of sirtuins. Drug Discov. Today Technol. 2014, 12, e9–e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef]
- Lahm, A.; Paolini, C.; Pallaoro, M.; Nardi, M.C.; Jones, P.; Neddermann, P.; Sambucini, S.; Bottomley, M.J.; Lo Surdo, P.; Carfí, A.; et al. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc. Natl. Acad. Sci. USA 2007, 104, 17335–17340. [Google Scholar] [CrossRef] [Green Version]
- Kaletsch, A.; Pinkerneil, M.; Hoffmann, M.J.; Jaguva Vasudevan, A.A.; Wang, C.; Hansen, F.K.; Wiek, C.; Hanenberg, H.; Gertzen, C.; Gohlke, H.; et al. Effects of novel HDAC inhibitors on urothelial carcinoma cells. Clin. Epigenetics 2018, 10, 100. [Google Scholar] [CrossRef]
- Fischle, W.; Dequiedt, F.; Hendzel, M.J.; Guenther, M.G.; Lazar, M.A.; Voelter, W.; Verdin, E. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol. Cell 2002, 9, 45–57. [Google Scholar] [CrossRef]
- Martin, M.; Kettmann, R.; Dequiedt, F. Class IIa histone deacetylases: Regulating the regulators. Oncogene 2007, 26, 5450–5467. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Bishayee, A.; Pandey, A. Targeting Histone Deacetylases with Natural and Synthetic Agents: An Emerging Anticancer Strategy. Nutrients 2018, 10, 731. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, H.; Huang, G. Histone acetylation and congenital heart diseases. Chinese J. Pediatr. 2013, 51, 552–554. [Google Scholar]
- Angiolilli, C.; Baeten, D.L.; Radstake, T.R.; Reedquist, K.A. The acetyl code in rheumatoid arthritis and other rheumatic diseases. Epigenomics 2017, 9, 447–461. [Google Scholar] [CrossRef] [PubMed]
- Bonnaud, E.M.; Suberbielle, E.; Malnou, C.E. Histone acetylation in neuronal (dys)function. Biomol. Concepts 2016, 7, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Bayat, S.; Shekari Khaniani, M.; Choupani, J.; Alivand, M.R.; Mansoori Derakhshan, S. HDACis (class I), cancer stem cell, and phytochemicals: Cancer therapy and prevention implications. Biomed. Pharmacother. 2018, 97, 1445–1453. [Google Scholar] [CrossRef] [PubMed]
- Giaginis, C.; Damaskos, C.; Koutsounas, I.; Zizi-Serbetzoglou, A.; Tsoukalas, N.; Patsouris, E.; Kouraklis, G.; Theocharis, S. Histone deacetylase (HDAC)-1, -2, -4 and -6 expression in human pancreatic adenocarcinoma: Associations with clinicopathological parameters, tumor proliferative capacity and patients’ survival. BMC Gastroenterol. 2015, 15, 148. [Google Scholar] [CrossRef] [PubMed]
- Shan, W.; Jiang, Y.; Yu, H.; Huang, Q.; Liu, L.; Guo, X.; Li, L.; Mi, Q.; Zhang, K.; Yang, Z. HDAC2 overexpression correlates with aggressive clinicopathological features and DNA-damage response pathway of breast cancer. Am. J. Cancer Res. 2017, 7, 1213–1226. [Google Scholar]
- Ramakrishnan, S.; Ku, S.; Ciamporcero, E.; Miles, K.M.; Attwood, K.; Chintala, S.; Shen, L.; Ellis, L.; Sotomayor, P.; Swetzig, W.; et al. HDAC 1 and 6 modulate cell invasion and migration in clear cell renal cell carcinoma. BMC Cancer 2016, 16, 617. [Google Scholar] [CrossRef]
- Damaskos, C.; Valsami, S.; Kontos, M.; Spartalis, E.; Kalampokas, T.; Kalampokas, E.; Athanasiou, A.; Moris, D.; Daskalopoulou, A.; Davakis, S.; et al. Histone Deacetylase Inhibitors: An Attractive Therapeutic Strategy Against Breast Cancer. Anticancer Res. 2017, 37, 35–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Shang, Y.-P.; Chen, H.; Li, J. Histone deacetylases function as novel potential therapeutic targets for cancer. Hepatol. Res. 2017, 47, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Expression of HDAC1 in Cancer—Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000116478-HDAC1/pathology (accessed on 10 January 2019).
- Expression of HDAC2 in Cancer—Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000196591-HDAC2/pathology (accessed on 10 January 2019).
- Expression of HDAC3 in Cancer—Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000171720-HDAC3/pathology (accessed on 10 January 2019).
- Expression of HDAC8 in Cancer—Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000147099-HDAC8/pathology (accessed on 10 January 2019).
- Expression of HDAC4 in Cancer—Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000068024-HDAC4/pathology (accessed on 10 January 2019).
- Expression of HDAC5 in Cancer—Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000108840-HDAC5/pathology (accessed on 10 January 2019).
- Expression of HDAC9 in Cancer—Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000048052-HDAC9/pathology (accessed on 10 January 2019).
- Lei, Y.; Liu, L.; Zhang, S.; Guo, S.; Li, X.; Wang, J.; Su, B.; Fang, Y.; Chen, X.; Ke, H.; et al. Hdac7 promotes lung tumorigenesis by inhibiting Stat3 activation. Mol. Cancer 2017, 16, 170. [Google Scholar] [CrossRef] [PubMed]
- Expression of HDAC10 in Cancer—Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000100429-HDAC10/pathology (accessed on 10 January 2019).
- Expression of HDAC6 in Cancer—Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000094631-HDAC6/pathology (accessed on 10 January 2019).
- Expression of SIRT3 in Cancer—Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000142082-SIRT3/pathology (accessed on 10 January 2019).
- Expression of SIRT5 in Cancer—Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000124523-SIRT5/pathology (accessed on 10 January 2019).
- Expression of SIRT6 in Cancer—Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000077463-SIRT6/pathology (accessed on 10 January 2019).
- Expression of SIRT7 in Cancer—Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000187531-SIRT7/pathology (accessed on 10 January 2019).
- Expression of SIRT2 in Cancer—Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000068903-SIRT2/pathology (accessed on 10 January 2019).
- Deubzer, H.E.; Schier, M.C.; Oehme, I.; Lodrini, M.; Haendler, B.; Sommer, A.; Witt, O. HDAC11 is a novel drug target in carcinomas. Int. J. Cancer 2013, 132, 2200–2208. [Google Scholar] [CrossRef] [PubMed]
- Expression of SIRT1 in Cancer—Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000096717-SIRT1/pathology (accessed on 10 January 2019).
- Expression of SIRT4 in Cancer—Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000089163-SIRT4/pathology (accessed on 10 January 2019).
- Niegisch, G.; Knievel, J.; Koch, A.; Hader, C.; Fischer, U.; Albers, P.; Schulz, W.A. Changes in histone deacetylase (HDAC) expression patterns and activity of HDAC inhibitors in urothelial cancers. Urol. Oncol. Semin. Orig. Investig. 2013, 31, 1770–1779. [Google Scholar] [CrossRef] [PubMed]
- Pinkerneil, M.; Hoffmann, M.J.; Deenen, R.; Kohrer, K.; Arent, T.; Schulz, W.A.; Niegisch, G. Inhibition of Class I Histone Deacetylases 1 and 2 Promotes Urothelial Carcinoma Cell Death by Various Mechanisms. Mol. Cancer Ther. 2016, 15, 299–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, M.; Hoffmann, M.J.; Koch, A.; Ulrich, S.M.; Schulz, W.A.; Niegisch, G. Histone deacetylase 8 is deregulated in urothelial cancer but not a target for efficient treatment. J. Exp. Clin. Cancer Res. 2014, 33, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chervona, Y.; Costa, M. Histone modifications and cancer: Biomarkers of prognosis? Am. J. Cancer Res. 2012, 2, 589–597. [Google Scholar]
- Gobinet, J.; Carascossa, S.; Cavaillès, V.; Vignon, F.; Nicolas, J.-C.; Jalaguier, S. SHP represses transcriptional activity via recruitment of histone deacetylases. Biochemistry 2005, 44, 6312–6320. [Google Scholar] [CrossRef] [PubMed]
- Narita, T.; Weinert, B.T.; Choudhary, C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2018. [Google Scholar] [CrossRef]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Galvin, K.M.; See, R.H.; Eckner, R.; Livingston, D.; Moran, E.; Shi, Y. Relief of YY1 transcriptional repression by adenovirus E1A is mediated by E1A-associated protein p300. Genes Dev. 1995, 9, 1188–1198. [Google Scholar] [CrossRef] [PubMed]
- Lührs, H.; Hock, R.; Schauber, J.; Weihrauch, M.; Harrer, M.; Melcher, R.; Scheppach, W.; Bustin, M.; Menzel, T. Modulation of HMG-N2 binding to chromatin by butyrate-induced acetylation in human colon adenocarcinoma cells. Int. J. Cancer 2002, 97, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Edberg, D.D.; Bruce, J.E.; Siems, W.F.; Reeves, R. In Vivo Posttranslational Modifications of the High Mobility Group A1a Proteins in Breast Cancer Cells of Differing Metastatic Potential. Biochemistry 2004, 43, 11500–11515. [Google Scholar] [CrossRef] [PubMed]
- Gaughan, L.; Logan, I.R.; Cook, S.; Neal, D.E.; Robson, C.N. Tip60 and Histone Deacetylase 1 Regulate Androgen Receptor Activity through Changes to the Acetylation Status of the Receptor. J. Biol. Chem. 2002, 277, 25904–25913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyes, J.; Byfield, P.; Nakatani, Y.; Ogryzko, V. Regulation of activity of the transcription factor GATA-1 by acetylation. Nature 1998, 396, 594–598. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Bieker, J.J. Acetylation and modulation of erythroid Krüppel-like factor (EKLF) activity by interaction with histone acetyltransferases. Proc. Natl. Acad. Sci. USA 1998, 95, 9855–9860. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Mu, Y.; Greene, W.C. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. EMBO J. 2002, 21, 6539–6548. [Google Scholar] [CrossRef] [Green Version]
- Bali, P.; Pranpat, M.; Bradner, J.; Balasis, M.; Fiskus, W.; Guo, F.; Rocha, K.; Kumaraswamy, S.; Boyapalle, S.; Atadja, P.; et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: A novel basis for antileukemia activity of histone deacetylase inhibitors. J. Biol. Chem. 2005, 280, 26729–26734. [Google Scholar] [CrossRef]
- Jeong, J.W.; Bae, M.K.; Ahn, M.Y.; Kim, S.H.; Sohn, T.K.; Bae, M.H.; Yoo, M.A.; Song, E.J.; Lee, K.J.; Kim, K.W. Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell 2002, 111, 709–720. [Google Scholar] [CrossRef]
- Simonsson, M.; Heldin, C.-H.; Ericsson, J.; Grönroos, E. The balance between acetylation and deacetylation controls Smad7 stability. J. Biol. Chem. 2005, 280, 21797–21803. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Zhou, W.; Han, X.; Wang, Z.; Li, B.; Jeffries, S.; Tao, W.; Robbins, D.J.; Capobianco, A.J. Acetylation of Mastermind-like 1 by p300 Drives the Recruitment of NACK to Initiate Notch-Dependent Transcription. Cancer Res. 2017, 77, 4228–4237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, D.; Mo, Y.; Li, M.-T.; Zou, S.-W.; Cheng, Z.-L.; Sun, Y.-P.; Xiong, Y.; Guan, K.-L.; Lei, Q.-Y. NOTCH-induced aldehyde dehydrogenase 1A1 deacetylation promotes breast cancer stem cells. J. Clin. Investig. 2014, 124, 5453–5465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinkerneil, M.; Hoffmann, M.J.; Kohlhof, H.; Schulz, W.A.; Niegisch, G. Evaluation of the Therapeutic Potential of the Novel Isotype Specific HDAC Inhibitor 4SC-202 in Urothelial Carcinoma Cell Lines. Target. Oncol. 2016, 11, 783–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, X.; Xiao, X.; Li, N.; Li, Y. Histone deacetylases inhibitors (HDACis) as novel therapeutic application in various clinical diseases. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2017, 72, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Tasoulas, J.; Giaginis, C.; Patsouris, E.; Manolis, E.; Theocharis, S. Histone deacetylase inhibitors in oral squamous cell carcinoma treatment. Expert Opin. Investig. Drugs 2015, 24, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Tandon, N.; Ramakrishnan, V. Clinical use and applications of histone deacetylase inhibitors in multiple myeloma. Clin. Pharmacol. Adv. Appl. 2016, 8, 35. [Google Scholar] [CrossRef]
- Kusaczuk, M.; Krętowski, R.; Bartoszewicz, M.; Cechowska-Pasko, M. Phenylbutyrate-a pan-HDAC inhibitor-suppresses proliferation of glioblastoma LN-229 cell line. Tumour Biol. 2016, 37, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Park, M.J.; Sohrabji, F. The histone deacetylase inhibitor, sodium butyrate, exhibits neuroprotective effects for ischemic stroke in middle-aged female rats. J. Neuroinflamm. 2016, 13, 300. [Google Scholar] [CrossRef]
- Leng, Y.; Wang, J.; Wang, Z.; Liao, H.-M.; Wei, M.; Leeds, P.; Chuang, D.-M. Valproic Acid and Other HDAC Inhibitors Upregulate FGF21 Gene Expression and Promote Process Elongation in Glia by Inhibiting HDAC2 and 3. Int. J. Neuropsychopharmacol. 2016, 19. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Feng, X.; Han Han, H.; Guo, S.; Wang, G. Synergistic effects of combined treatment with histone deacetylase inhibitor suberoylanilide hydroxamic acid and TRAIL on human breast cancer cells. Sci. Rep. 2016, 6, 28004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foss, F.; Advani, R.; Duvic, M.; Hymes, K.B.; Intragumtornchai, T.; Lekhakula, A.; Shpilberg, O.; Lerner, A.; Belt, R.J.; Jacobsen, E.D.; et al. A Phase II trial of Belinostat (PXD101) in patients with relapsed or refractory peripheral or cutaneous T-cell lymphoma. Br. J. Haematol. 2015, 168, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Wightman, F.; Lu, H.K.; Solomon, A.E.; Saleh, S.; Harman, A.N.; Cunningham, A.L.; Gray, L.; Churchill, M.; Cameron, P.U.; Dear, A.E.; et al. Entinostat is a histone deacetylase inhibitor selective for class 1 histone deacetylases and activates HIV production from latently infected primary T cells. AIDS 2013, 27, 2853–2862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Sun, M.; Zhou, S.; Guo, B. Class I HDAC inhibitor mocetinostat induces apoptosis by activation of miR-31 expression and suppression of E2F6. Cell Death Discov. 2016, 2, 16036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrich, A.; Nabhan, C. Use of class I histone deacetylase inhibitor romidepsin in combination regimens. Leuk. Lymphoma 2016, 57, 1755–1765. [Google Scholar] [CrossRef] [PubMed]
- Igotti Abramova, M.V.; Pojidaeva, A.K.; Filippova, E.A.; Gnedina, O.O.; Svetlikova, S.B.; Pospelov, V.A. HDAC inhibitors induce apoptosis but not cellular senescence in Gadd45α-deficient E1A+Ras cells. Int. J. Biochem. Cell Biol. 2014, 51, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.; Diao, H.; Dong, N.; Su, X.; Wang, B.; Mo, Q.; Yu, H.; Wang, X.; Chen, C. Histone deacetylase inhibitor induces cell apoptosis and cycle arrest in lung cancer cells via mitochondrial injury and p53 up-acetylation. Cell Biol. Toxicol. 2016, 32, 469–482. [Google Scholar] [CrossRef] [Green Version]
- Stepulak, A.; Stryjecka-Zimmer, M.; Kupisz, K.; Polberg, K. Histone deacetylase inhibitors as a new generation of anti-cancer agents. Postepy Hig. Med. Dosw. 2005, 59, 68–74. [Google Scholar]
- Osuka, S.; Takano, S.; Watanabe, S.; Ishikawa, E.; Yamamoto, T.; Matsumura, A. Valproic acid inhibits angiogenesis in vitro and glioma angiogenesis in vivo in the brain. Neurol. Med. Chir. 2012, 52, 186–193. [Google Scholar] [CrossRef]
- VanderMolen, K.M.; McCulloch, W.; Pearce, C.J.; Oberlies, N.H. Romidepsin (Istodax, NSC 630176, FR901228, FK228, depsipeptide): A natural product recently approved for cutaneous T-cell lymphoma. J. Antibiot. 2011, 64, 525–531. [Google Scholar] [CrossRef]
- Kim, E.; Bisson, W.H.; Löhr, C.V.; Williams, D.E.; Ho, E.; Dashwood, R.H.; Rajendran, P. Histone and Non-Histone Targets of Dietary Deacetylase Inhibitors. Curr. Top. Med. Chem. 2016, 16, 714–731. [Google Scholar] [CrossRef] [PubMed]
- Gurzu, S.; Turdean, S.; Kovecsi, A.; Contac, A.O.; Jung, I. Epithelial-mesenchymal, mesenchymal-epithelial, and endothelial-mesenchymal transitions in malignant tumors: An update. World J. Clin. Cases 2015, 3, 393. [Google Scholar] [CrossRef] [PubMed]
- Barriere, G.; Fici, P.; Gallerani, G.; Fabbri, F.; Rigaud, M. Epithelial Mesenchymal Transition: A double-edged sword. Clin. Transl. Med. 2015, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.Y.-J.; Guilford, P.; Thiery, J.P. Early events in cell adhesion and polarity during epithelial-mesenchymal transition. J. Cell Sci. 2012, 125, 4417–4422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Francart, M.-E.; Lambert, J.; Vanwynsberghe, A.M.; Thompson, E.W.; Bourcy, M.; Polette, M.; Gilles, C. Epithelial-mesenchymal plasticity and circulating tumor cells: Travel companions to metastases. Dev. Dyn. 2018, 247, 432–450. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-Y.; Lin, H.-H.; Tang, M.-J.; Wang, Y.-K. Vimentin contributes to epithelial-mesenchymal transition cancer cell mechanics by mediating cytoskeletal organization and focal adhesion maturation. Oncotarget 2015, 6, 15966–15983. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.H.M.; Fang, C.M.; Chuah, L.-H.; Leong, C.O.; Ngai, S.C. E-cadherin: Its dysregulation in carcinogenesis and clinical implications. Crit. Rev. Oncol. Hematol. 2018, 121, 11–22. [Google Scholar] [CrossRef]
- Skrypek, N.; Goossens, S.; De Smedt, E.; Vandamme, N.; Berx, G. Epithelial-to-Mesenchymal Transition: Epigenetic Reprogramming Driving Cellular Plasticity. Trends Genet. 2017, 33, 943–959. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, J.; Chai, K.; Ying, X.; Zhou, B.P. The Role of Snail in EMT and Tumorigenesis. Curr. Cancer Drug Targets 2013, 13, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Gheldof, A.; Hulpiau, P.; van Roy, F.; De Craene, B.; Berx, G. Evolutionary functional analysis and molecular regulation of the ZEB transcription factors. Cell. Mol. Life Sci. 2012, 69, 2527–2541. [Google Scholar] [CrossRef] [PubMed]
- Garg, M. Epithelial, mesenchymal and hybrid epithelial/mesenchymal phenotypes and their clinical relevance in cancer metastasis. Expert Rev. Mol. Med. 2017, 19, e3. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.C.; Zhang, X.H.-F. EMT in Metastasis: Finding the Right Balance. Dev. Cell 2018, 45, 663–665. [Google Scholar] [CrossRef] [PubMed]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Boareto, M.; Debeb, B.G.; Aceto, N.; Farach-Carson, M.C.; Woodward, W.A.; Levine, H. Inflammatory breast cancer: A model for investigating cluster-based dissemination. NPJ Breast Cancer 2017, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.M.; Kuay, K.T.; Koh, P.F.; Asad, M.; Tan, T.Z.; Chung, V.Y.; Lee, S.C.; Thiery, J.P.; Huang, R.-J. An epithelial marker promoter induction screen identifies histone deacetylase inhibitors to restore epithelial differentiation and abolishes anchorage independence growth in cancers. Cell Death Discov. 2016, 2, 16041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagaraja, S.S.; Krishnamoorthy, V.; Raviraj, R.; Paramasivam, A.; Nagarajan, D. Effect of Trichostatin A on radiation induced epithelial-mesenchymal transition in A549 cells. Biochem. Biophys. Res. Commun. 2017, 493, 1534–1541. [Google Scholar] [CrossRef] [PubMed]
- Park, I.-H.; Kang, J.-H.; Shin, J.-M.; Lee, H.-M. Trichostatin A Inhibits Epithelial Mesenchymal Transition Induced by TGF-β1 in Airway Epithelium. PLoS ONE 2016, 11, e0162058. [Google Scholar] [CrossRef]
- Mateen, S.; Raina, K.; Agarwal, C.; Chan, D.; Agarwal, R. Silibinin synergizes with histone deacetylase and DNA methyltransferase inhibitors in upregulating E-cadherin expression together with inhibition of migration and invasion of human non-small cell lung cancer cells. J. Pharmacol. Exp. Ther. 2013, 345, 206–214. [Google Scholar] [CrossRef]
- Noguchi, S.; Eitoku, M.; Moriya, S.; Kondo, S.; Kiyosawa, H.; Watanabe, T.; Suganuma, N. Regulation of Gene Expression by Sodium Valproate in Epithelial-to-Mesenchymal Transition. Lung 2015, 193, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, M.; Jiang, X.; Mei, X.; Liu, X. Histone deacetylase inhibitor SAHA-induced epithelial–mesenchymal transition by upregulating Slug in lung cancer cells. Anticancer Drugs 2018, 29, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Lyu, H.; Liu, H.; Shi, X.; Song, Y.; Liu, B. Downregulation of the long noncoding RNA GAS5-AS1 contributes to tumor metastasis in non-small cell lung cancer. Sci. Rep. 2016, 6, 31093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Chang, Y.; Cao, P. CCR7 preservation via histone deacetylase inhibition promotes epithelial-mesenchymal transition of hepatocellular carcinoma cells. Exp. Cell Res. 2018, 371, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Liu, H.; Liu, Z.-G.; Wang, H.-S.; Zhang, F.; Wang, H.; Zhang, J.; Chen, J.-J.; Huang, H.-J.; Tan, Y.; et al. Histone deacetylase inhibitors upregulate Snail via Smad2/3 phosphorylation and stabilization of Snail to promote metastasis of hepatoma cells. Cancer Lett. 2018, 420, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Wang, J.; Zheng, T.; Song, R.; Liang, Y.; Bhatta, N.; Yin, D.; Pan, S.; Liu, J.; Jiang, H.; et al. LBH589 Inhibits proliferation and metastasis of hepatocellular carcinoma via inhibition of gankyrin/STAT3/Akt pathway. Mol. Cancer 2013, 12, 114. [Google Scholar] [CrossRef] [PubMed]
- Soukupova, J.; Bertran, E.; Peñuelas-Haro, I.; Urdiroz-Urricelqui, U.; Borgman, M.; Kohlhof, H.; Fabregat, I. Resminostat induces changes in epithelial plasticity of hepatocellular carcinoma cells and sensitizes them to sorafenib-induced apoptosis. Oncotarget 2017, 8, 110367–110379. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lee, E.; Ji, M.; Park, S. HDAC inhibitors, trichostatin A and valproic acid, increase E-cadherin and vimentin expression but inhibit migration and invasion of cholangiocarcinoma cells. Oncol. Rep. 2018, 40, 346–354. [Google Scholar] [CrossRef]
- Mishra, V.K.; Wegwitz, F.; Kosinsky, R.L.; Sen, M.; Baumgartner, R.; Wulff, T.; Siveke, J.T.; Schildhaus, H.-U.; Najafova, Z.; Kari, V.; et al. Histone deacetylase class-I inhibition promotes epithelial gene expression in pancreatic cancer cells in a BRD4- and MYC-dependent manner. Nucleic Acids Res. 2017, 45, 6334–6349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polireddy, K.; Dong, R.; McDonald, P.R.; Wang, T.; Luke, B.; Chen, P.; Broward, M.; Roy, A.; Chen, Q. Targeting Epithelial-Mesenchymal Transition for Identification of Inhibitors for Pancreatic Cancer Cell Invasion and Tumor Spheres Formation. PLoS ONE 2016, 11, e0164811. [Google Scholar] [CrossRef] [PubMed]
- Meidhof, S.; Brabletz, S.; Lehmann, W.; Preca, B.-T.; Mock, K.; Ruh, M.; Schüler, J.; Berthold, M.; Weber, A.; Burk, U.; et al. ZEB1-associated drug resistance in cancer cells is reversed by the class I HDAC inhibitor mocetinostat. EMBO Mol. Med. 2015, 7, 831–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarova, D.; Bordonaro, M. ZEB1 Mediates Drug Resistance and EMT in p300-Deficient CRC. J. Cancer 2017, 8, 1453–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-H.; Lee, C.-H.; Liou, J.-P.; Teng, C.-M.; Pan, S.-L. Molecular mechanisms underlying the antitumor activity of (E)-N-hydroxy-3-(1-(4-methoxyphenylsulfonyl)-1,2,3,4-tetrahydroquinolin-6-yl)acrylamide in human colorectal cancer cells in vitro and in vivo. Oncotarget 2015, 6, 35991–36002. [Google Scholar] [CrossRef] [PubMed]
- Kiweler, N.; Brill, B.; Wirth, M.; Breuksch, I.; Laguna, T.; Dietrich, C.; Strand, S.; Schneider, G.; Groner, B.; Butter, F.; et al. The histone deacetylases HDAC1 and HDAC2 are required for the growth and survival of renal carcinoma cells. Arch. Toxicol. 2018, 92, 2227–2243. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.Y.; Kee, H.J.; Kurz, T.; Hansen, F.K.; Ryu, Y.; Kim, G.R.; Lin, M.Q.; Jin, L.; Piao, Z.H.; Jeong, M.H. Class I HDACs specifically regulate E-cadherin expression in human renal epithelial cells. J. Cell. Mol. Med. 2016, 20, 2289–2298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skowron, M.A.; Sathe, A.; Romano, A.; Hoffmann, M.J.; Schulz, W.A.; van Koeveringe, G.A.; Albers, P.; Nawroth, R.; Niegisch, G. Applying the chicken embryo chorioallantoic membrane assay to study treatment approaches in urothelial carcinoma. Urol. Oncol. Semin. Orig. Investig. 2017, 35, 544.e11–544.e23. [Google Scholar] [CrossRef]
- Elshafae, S.M.; Kohart, N.A.; Altstadt, L.A.; Dirksen, W.P.; Rosol, T.J. The Effect of a Histone Deacetylase Inhibitor (AR-42) on Canine Prostate Cancer Growth and Metastasis. Prostate 2017, 77, 776–793. [Google Scholar] [CrossRef] [PubMed]
- McLeod, A.B.; Stice, J.P.; Wardell, S.E.; Alley, H.M.; Chang, C.; McDonnell, D.P. Validation of histone deacetylase 3 as a therapeutic target in castration-resistant prostate cancer. Prostate 2018, 78, 266–277. [Google Scholar] [CrossRef]
- Sakamoto, T.; Kobayashi, S.; Yamada, D.; Nagano, H.; Tomokuni, A.; Tomimaru, Y.; Noda, T.; Gotoh, K.; Asaoka, T.; Wada, H.; et al. A Histone Deacetylase Inhibitor Suppresses Epithelial-Mesenchymal Transition and Attenuates Chemoresistance in Biliary Tract Cancer. PLoS ONE 2016, 11, e0145985. [Google Scholar] [CrossRef]
- Wu, S.; Luo, Z.; Yu, P.-J.; Xie, H.; He, Y.-W. Suberoylanilide hydroxamic acid (SAHA) promotes the epithelial mesenchymal transition of triple negative breast cancer cells via HDAC8/FOXA1 signals. Biol. Chem. 2016, 397. [Google Scholar] [CrossRef]
- Debeb, B.G.; Lacerda, L.; Xu, W.; Larson, R.; Solley, T.; Atkinson, R.; Sulman, E.P.; Ueno, N.T.; Krishnamurthy, S.; Reuben, J.M.; et al. Histone deacetylase inhibitors stimulate dedifferentiation of human breast cancer cells through WNT/β-catenin signaling. Stem Cells 2012, 30, 2366–2377. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, L.V.; Tate, C.R.; Segar, H.C.; Burks, H.E.; Phamduy, T.B.; Hoang, V.; Elliott, S.; Gilliam, D.; Pounder, F.N.; Anbalagan, M.; et al. Suppression of triple-negative breast cancer metastasis by pan-DAC inhibitor panobinostat via inhibition of ZEB family of EMT master regulators. Breast Cancer Res. Treat. 2014, 145, 593–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, R.K.; Kurzrock, R.; Shankar, S. MS-275 Sensitizes TRAIL-Resistant Breast Cancer Cells, Inhibits Angiogenesis and Metastasis, and Reverses Epithelial-Mesenchymal Transition In vivo. Mol. Cancer Ther. 2010, 9, 3254–3266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, F.; Sun, G.; Zhong, M.; Yu, Y.; Brewer, M.A. Anticancer efficacy of cisplatin and trichostatin A or 5-aza-2’-deoxycytidine on ovarian cancer. Br. J. Cancer 2013, 108, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Bruzzese, F.; Leone, A.; Rocco, M.; Carbone, C.; Piro, G.; Caraglia, M.; Di Gennaro, E.; Budillon, A. HDAC inhibitor vorinostat enhances the antitumor effect of gefitinib in squamous cell carcinoma of head and neck by modulating ErbB receptor expression and reverting EMT. J. Cell. Physiol. 2011, 226, 2378–2390. [Google Scholar] [CrossRef] [PubMed]
- Kanamoto, A.; Ninomiya, I.; Harada, S.; Tsukada, T.; Okamoto, K.; Nakanuma, S.; Sakai, S.; Makino, I.; Kinoshita, J.; Hayashi, H.; et al. Valproic acid inhibits irradiation-induced epithelial-mesenchymal transition and stem cell-like characteristics in esophageal squamous cell carcinoma. Int. J. Oncol. 2016, 49, 1859–1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, E.J.; Cho, B.J.; Lee, D.J.; Hwang, Y.H.; Chun, S.H.; Kim, H.H.; Kim, I.A. Enhanced cytotoxic effect of radiation and temozolomide in malignant glioma cells: Targeting PI3K-AKT-mTOR signaling, HSP90 and histone deacetylases. BMC Cancer 2014, 14, 17. [Google Scholar] [CrossRef] [PubMed]
- Shankar, S.; Nall, D.; Tang, S.-N.; Meeker, D.; Passarini, J.; Sharma, J.; Srivastava, R.K. Resveratrol inhibits pancreatic cancer stem cell characteristics in human and KrasG12D transgenic mice by inhibiting pluripotency maintaining factors and epithelial-mesenchymal transition. PLoS ONE 2011, 6, e16530. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Hishikawa, K.; Marumo, T.; Fujita, T. Inhibition of Histone Deacetylase Activity Suppresses Epithelial-to-Mesenchymal Transition Induced by TGF-beta1 in Human Renal Epithelial Cells. J. Am. Soc. Nephrol. 2007, 18, 58–65. [Google Scholar] [CrossRef]
- Ruscetti, M.; Dadashian, E.L.; Guo, W.; Quach, B.; Mulholland, D.J.; Park, J.W.; Tran, L.M.; Kobayashi, N.; Bianchi-Frias, D.; Xing, Y.; et al. HDAC inhibition impedes epithelial–mesenchymal plasticity and suppresses metastatic, castration-resistant prostate cancer. Oncogene 2016, 35, 3781–3795. [Google Scholar] [CrossRef]
- Sarkar, S.; Goldgar, S.; Byler, S.; Rosenthal, S.; Heerboth, S. Demethylation and re-expression of epigenetically silenced tumor suppressor genes: Sensitization of cancer cells by combination therapy. Epigenomics 2013, 5, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Kazanets, A.; Shorstova, T.; Hilmi, K.; Marques, M.; Witcher, M. Epigenetic silencing of tumor suppressor genes: Paradigms, puzzles, and potential. Biochim. Biophys. Acta Rev. Cancer 2016, 1865, 275–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefter, L.P.; Dima, S.; Sunamura, M.; Furukawa, T.; Sato, Y.; Abe, M.; Chivu, M.; Popescu, I.; Horii, A. Transcriptional silencing of ETS-1 efficiently suppresses angiogenesis of pancreatic cancer. Cancer Gene Ther. 2009, 16, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Avgustinova, A.; Symeonidi, A.; Castellanos, A.; Urdiroz-Urricelqui, U.; Solé-Boldo, L.; Martín, M.; Pérez-Rodríguez, I.; Prats, N.; Lehner, B.; Supek, F.; et al. Loss of G9a preserves mutation patterns but increases chromatin accessibility, genomic instability and aggressiveness in skin tumours. Nat. Cell Biol. 2018, 20, 1400–1409. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
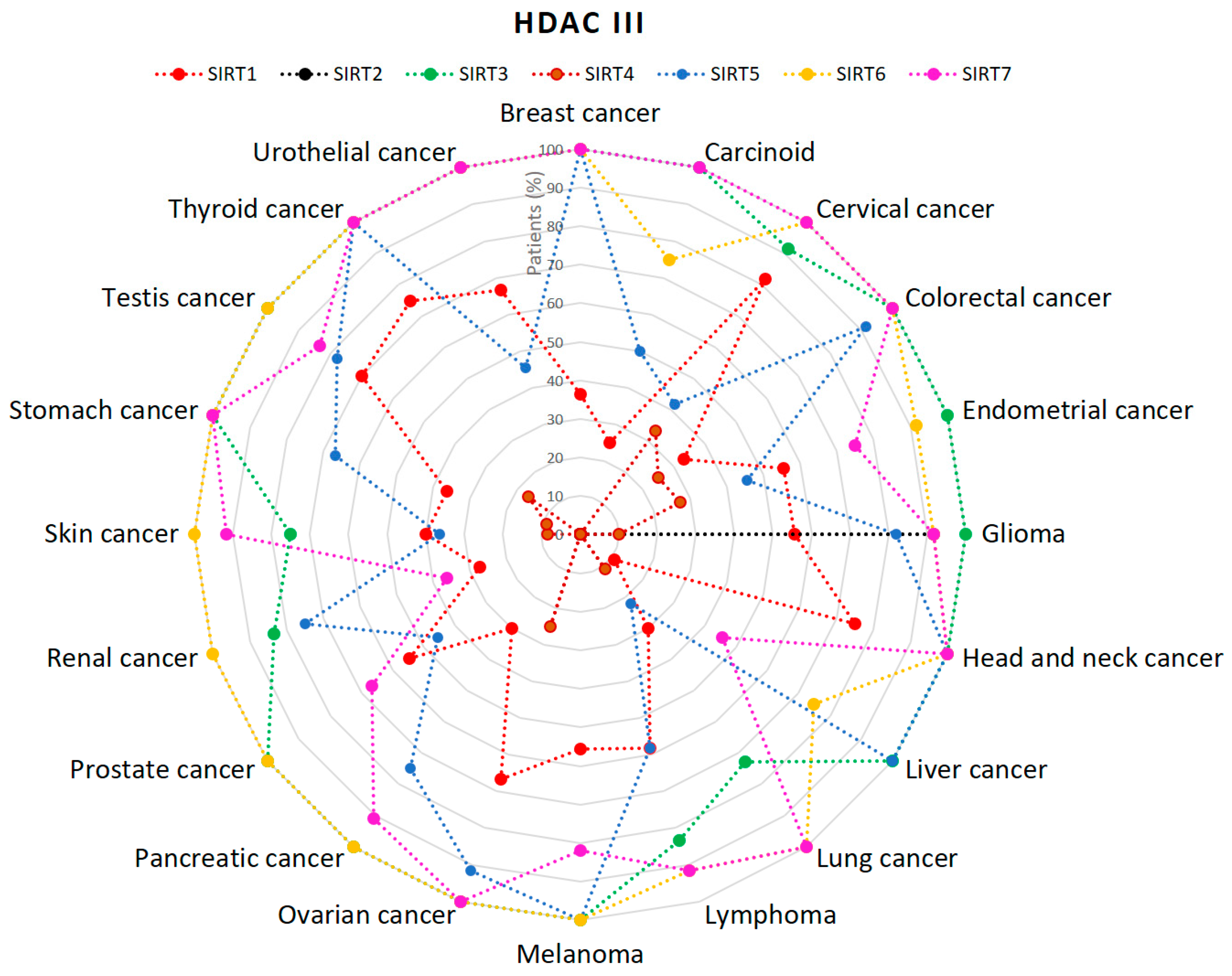{kind=link}
{kind=link}
{kind=link}
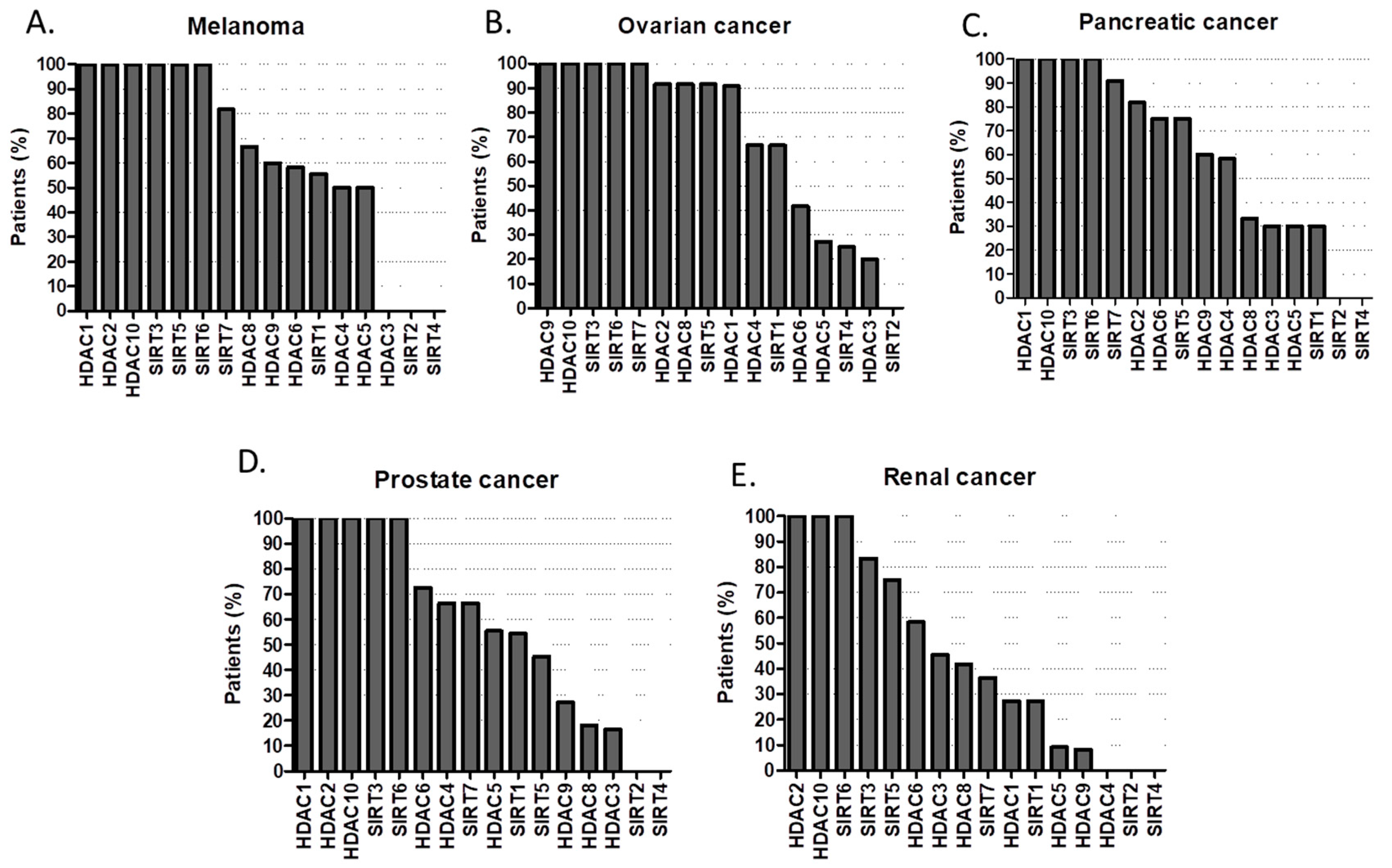{kind=link}
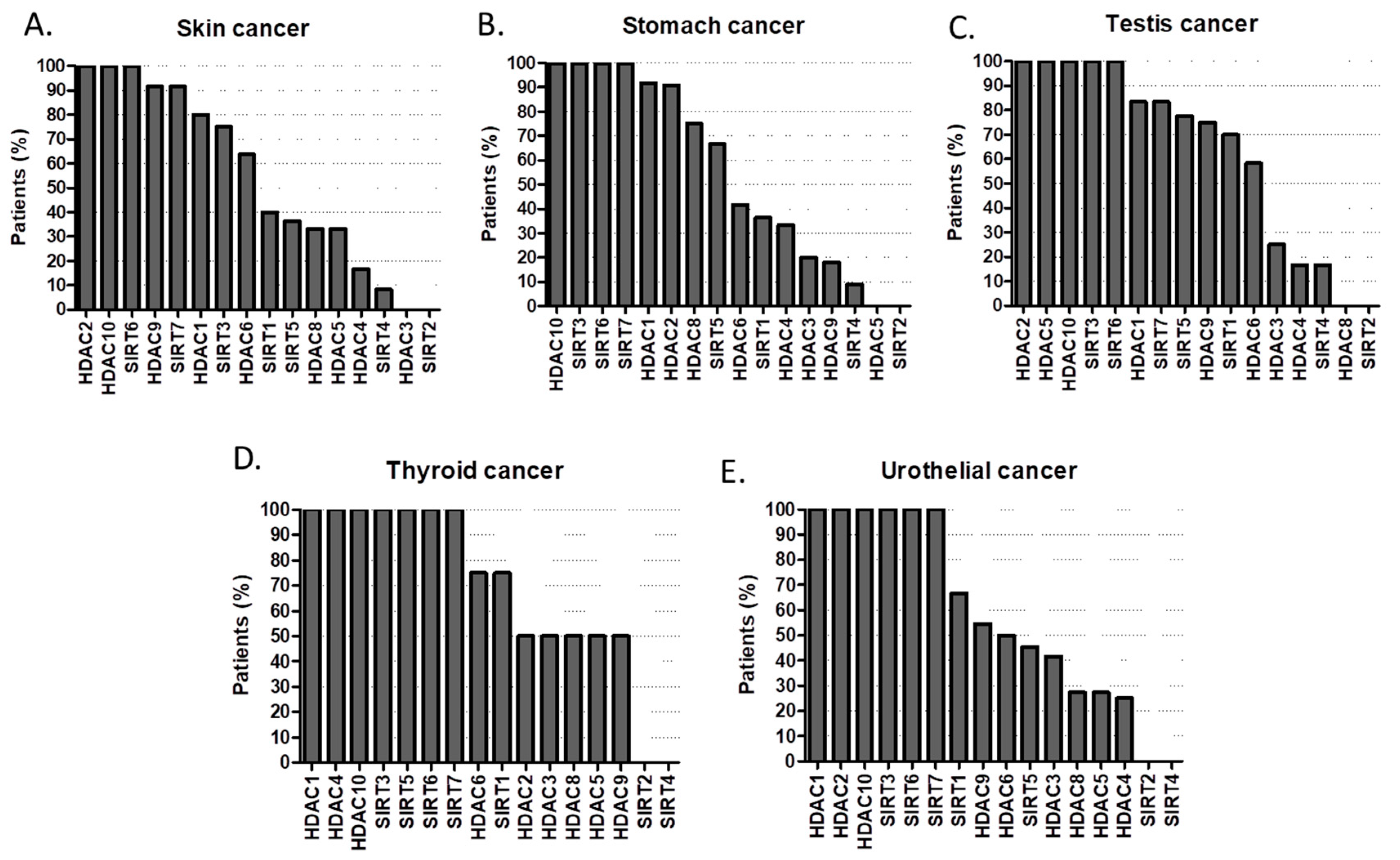{kind=link}
{kind=link}
| Class of HDI | HDI | HDAC Targets | Ref. |
|---|---|---|---|
| Short chain fatty acid | Phenylbutyrate (PBA) | Pan-inhibitor | [84] |
| Sodium butyrate (NaB) | I, IIa | [85] | |
| Butyrate | I, IIa | [83] | |
| Valproic acid | I, IIa | [86] | |
| Hydroxamic acid–derived compounds | Vorinostat (SAHA) | Pan-inhibitor | [87] |
| Belinostat (PXD-101) | Pan-inhibitor | [88] | |
| Resminostat (4SC-201) | Pan-inhibitor | [83] | |
| Panobinostat (LBH589) | I, II | [83] | |
| Trochostatin A (TSA) | I, II | [24] | |
| Benzamides | Entinostat (MS-275) | I | [89] |
| Mocetinostat (MGCD103) | I | [90] | |
| Domatinostat (4SC-202) | I | [80] | |
| Cyclic peptides | Romidepsin (FK228) | I | [91] |
| Apicidin (CAS183506-66-3) | I | [83] |
| Type of Cancer | HDI (Individually or in Combination) | Experimental Model | Type of Treatment | E-cadherin | B-catenin | N-cadherin | Vimentin | Transcription Factors | Changes in Morphology | Migration and Invasion | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Lung cancer | SAHA | A549 cells in vitro | cells treated with SAHA vs. untreated cells | ↓ | → | N/A | ↑ | ↑SLUG | from cobblestone to mesenchymal spindle-like | ↑migration | [118] |
| Lung cancer | TSA | A549 cells in vitro | irradiated cells treated with TSA vs. irradiated cells | ↑ | ↑ | ↓ | ↓ | ↓SNAIL, ZEB | reduction of mesenchymal-like phenotype | ↓migration | [114] [115] |
| Lung cancer | TSA + silibinin | H1299 cells in vitro | cells treated with TSA and silibinin vs. cells treated with silibinin | ↑ | N/A | N/A | N/A | ↓ZEB1 | N/A | ↓migration and invasion | [116] |
| Lung cancer | VPA | A549 cells in vitro | cells treated with VPA vs. untreated cells | ↑ | N/A | N/A | N/A | N/A | reduction of spindle-like morphology | N/A | [117] |
| Hepatocellular carcinoma | TSA | HepG2 cells, Huh7 cells in vitro | cells treated with TSA vs. untreated cells | ↓ | N/A | ↑ | ↑ | ↑SNAIL, TWIST | N/A | ↑migration and invasion | [120] |
| Hepatocellular carcinoma | VPA | HepG2 cells, Huh7 cells in vitro | cells treated with VPA vs. untreated cells | ↓ | N/A | ↑ | ↑ | ↑SNAIL, TWIST | N/A | ↑migration and invasion | [120] |
| Hepatocellular carcinoma | SAHA | HepG2 cells in vitro | cells treated with SAHA vs. untreated cells | ↓ | N/A | ↑ | ↑ | ↑SNAIL, TWIST | N/A | ↑migration and invasion | [120] |
| Hepatocellular carcinoma | MS-275 | HepG2 cells in vitro | cells treated with MS-275 vs. untreated cells | ↓ | N/A | ↑ | ↑ | ↑SNAIL, TWIST | N/A | ↑migration and invasion | [120] |
| Hepatocellular carcinoma | SAHA | HepG2 cells, QGY-7703 cells in vitro; mouse in vivo | cells treated with SAHA vs. untreated cells | N/A | N/A | ↑ | ↑ | ↑SNAIL through SMAD2/3 phosphorylation | changes of phenotype were detected | ↑invasion | [121] |
| Hepatocellular carcinoma | NaB | HepG2 cells/QGY-7703 cells in vitro; mouse in vivo | cells treated with NaB vs. untreated cells | N/A | N/A | N/A | ↑ | ↑SNAIL through SMAD2/3 phosphorylation | N/A | ↑invasion | [121] |
| Hepatocellular carcinoma | LBH589 | HepG2 cells in vitro | cells treated with LBH589 vs. untreated cells | ↑ | N/A | ↓ | ↓ | ↓TWIST1 | N/A | ↓invasion | [122] |
| Hepatocellular carcinoma | RAS2410 | Hep3B, HLE, HLF cells in vitro | cells treated with RAS2410 vs. untreated cells | ↑ | N/A | ↓ | ↓ | →TWIST, SNAI1 | N/A | ↓migration and invasion | [123] |
| Cholangiocarcinoma | VPA | HuCC-T1 cells in vitro | cells treated with VPA vs. untreated cells | → | N/A | N/A | → | N/A | no changes | ↓migration and invasion | [124] |
| Cholangiocarcinoma | TSA | HuCC-T1 cells in vitro | cells treated with TSA vs. untreated cells | ↑ | N/A | N/A | ↑ | N/A | no changes | ↓migration and invasion | [124] |
| Cholangiocarcinoma | VPA + gemcitabine | HuCC-T1 cells in vitro | cells treated with VPA and gemcitabine vs. cells treated gemcitabine | ↑ | N/A | N/A | ↑ | N/A | from spindle to rectangular caused by gemcitabine | ↓migration and invasion | [124] |
| Cholangiocarcinoma | TSA + gemcitabine | HuCC-T1 cells in vitro | cells treated with TSA and gemcitabine vs. cells treated gemcitabine | ↑ | N/A | N/A | ↑ | N/A | from spindle to rectangular caused by gemcitabine | ↓migration and invasion | [124] |
| Pancreatic cancer | 4SC-202 | Panc1 cells L3.6 cells in vitro | TGF-β1 pretreated Panc1 cells treated with 4SC-202 vs. untreated cells in vitro; mice with implanted L3.6 cells in vivo | ↓ | N/A | ↑ | ↓ | ↓ZEB1, SNAIL1 | N/A | N/A | [125] |
| Pancreatic cancer | BSI | Panc1 cells in vitro | Panc1 cells treated with BSI vs. untreated cells in vitro | ↑ | N/A | ↓ | N/A | ↓SNAIL | tumor spheres formation is unchanged but their size is significantly decreased | ↓migration and invasion | [126] |
| Pancreatic cancer | MGCD103 + gemcitabine | Panc1 cells, hPaca-1 derived tumor cells in vitro | Panc1 cells, hPaca-1 derived tumor cells treated with MGCD103 and gemcitabine vs. gemcitabine treated cells in vitro | ↑ | N/A | N/A | N/A | ↓ZEB1 | N/A | N/A | [127] |
| Pancreatic cancer | SAHA | Pancreatic CSCs | pancreatic CSCs treated with SAHA vs. untreated cells in vitro | ↑ | N/A | ↓ | N/A | ↓ZEB, SNAIL, SLUG | N/A | ↓invasion | [12] |
| Colorectal cancer | TSA | SW480 cells in vitro | cells treated with TSA vs. untreated cells | ↑ | N/A | N/A | ↓ | ↓SLUG | N/A | ↓migration and invasion | [121] |
| Colorectal cancer | VPA | SW480 cells in vitro | cells treated with VPA vs. untreated cells | ↓ | N/A | ↑ | ↑ | ↑SNAIL | N/A | ↑migration and invasion | [128] |
| Colorectal cancer | VPA | HCT116 cells in vitro | cells treated with VPA vs. untreated cells | ↓ | N/A | ↑ | ↑ | ↑SNAIL | N/A | ↑migration and invasion | [128] |
| Colorectal cancer | Compound 11 | HCT116 cells in vitro | cells treated with compound 11 vs. untreated cells | ↑ | ↓ | ↓ | ↓ | N/A | N/A | ↓migration | [129] |
| Colorectal cancer | Compound 11 | HT29 cells in vitro | cells treated with compound 11 vs. untreated cells | N/A | N/A | N/A | N/A | N/A | N/A | ↓migration | [129] |
| Colorectal cancer | Compound 11 | HCT116 xenograft model in vivo | mice treated compound 11 vs. untreated mice | ↑ | N/A | ↓ | ↓ | N/A | N/A | ↓migration | [129] |
| Colorectal cancer | TSA | HT29, SW480, DLD1, HTC116 cells in vitro | cells treated with TSA vs. untreated cells | ↓ | N/A | N/A | ↑ | N/A | altered to spindle like morphology | →migration, ↑invasion only in DLD1 cells | [15] |
| Colorectal cancer | VPA | HT29, SW480, DLD1, HTC116 cells in vitro | cells treated with VPA vs. untreated cells | ↓ | N/A | N/A | ↑ | N/A | altered to spindle like morphology | →migration, ↑invasion in DLD1 and SW480 cells | [15] |
| Colorectal cancer | TGF-β1 | HT29, SW480, DLD1, HTC116 cells in vitro | cells treated with TGF-β1 vs. untreated cells | ↓ | N/A | N/A | ↑ | N/A | altered to spindle like morphology | ↑invasion only in DLD1 cells | [15] |
| Colorectal cancer | TSA+ TGF-β1 | HT29, SW480, DLD1, HTC116 cells in vitro | cells treated with TSA and TGF-β1 vs. untreated cells | ↓ | N/A | N/A | ↑ | N/A | altered to spindle like morphology | HT29 N/A, SW480 ↑migration, LDL1 →invasion, HTC116 N/A | [15] |
| Colorectal cancer | VPA + TGF-β1 | HT29, SW480, DLD1, HTC116 cells in vitro | cells treated with VPA and TGF-β1 vs. untreated cells | ↓ | N/A | N/A | ↑ | N/A | altered to spindle like morphology | HT29 N/A, SW480 ↑migration, LDL1 ↑migration, →invasion, HTC116 N/A | [15] |
| Renal cancer | VPA | Renca cells in vitro, mice in vivo | cells treated with VPA vs. untreated cells | ↓ | ↓ | N/A | ↓ | ↑TWIST1, ↓TWIST2 →SNAIL1, SNAIL2 | interspace between cells after HDIs treatment | ↓migration | [130] |
| Renal cancer | MS-275 | Renca cells in vitro, mice in vivo | cells treated with MS-275 vs. untreated cells | ↓ | ↓ | N/A | N/A | N/A | interspace between cells after HDIs treatment | ↓migration | [130] |
| Renal cancer | TSA | HK2 cells in vitro | TGF-β1-pretreated HK2 cells treated with TSA vs. TGF-β1-treated HK2 cells | ↑ | N/A | → | N/A | N/A | N/A | N/A | [131] |
| Renal cancer | TSA | RPTEC cells in vitro | TGF-β1-pretreated RPTEC cells treated with TSA vs. untreated RPTEC cells | ↑ | N/A | N/A | N/A | →SMAD2, SMAD3 | from cuboidal to elongated form | N/A | [131] |
| Urothelial cancer | CDDP+SAHA | RT-112 and T-24 cells in cell culture or implanted on the chicken chorioallantoic membrane (CAM) | cells implanted on the CAM treated with CDDP + SAHA vs. cells treated with CDDP | N/A | N/A | N/A | N/A | N/A | CAM tumor reduction | [132] | |
| Urothelial cancer | CDDP+Romidepsin | RT-112 and T-24 cells in cell culture or implanted on the chicken chorioallantoic membrane (CAM) | cells implanted on the CAM treated CDDP+Romidepsin vs. cells treated with CDDP | N/A | N/A | N/A | N/A | N/A | CAM tumor reduction | [132] | |
| Prostate cancer | AR-42 | Ace-1 cells in vitro | cells treated with AR-42 vs. untreated cells | ↓ | → | ↓ | → | ↓TWIST, MYOF, ↑SNAIL, SLUG, PTEN, FAK, ZEB1 | reduction of spindle like morphology | ↓migration and invasion | [133] |
| Prostate cancer | AR-42 | nude mice with implanted Ace-1 cells in vivo | mice with Ace-1 cells treated AR-42 vs. untreated mice | N/A | N/A | N/A | N/A | N/A | irregular shape of cell after AR42 treatment | ↓reduction of bone metastasis | [133] |
| Prostate cancer | SAHA, TSA, RGFP966 | LNCaP cells in vitro | cells treated with HDIs vs. untreated cells | N/A | N/A | N/A | ↑SAHA, TSA; →RGFP966 | ↓NKX1, FOXA1; ↑SLUG, ZEB1 (SAHA, TSA), →SLUG, ZEB1 (RGFP966) | N/A | ↑ migration (SAHA), N/A (TSA), →migration (RGFP99) | [134] |
| Prostate cancer | TSA | PC3 cells in vitro | cells treated with TSA vs. untreated cells | ↑ | N/A | N/A | ↓ | ↓SLUG | N/A | ↓migration and invasion | [13] |
| Prostate cancer | VPA | PC3 cells in vitro | cells treated with VPA vs. untreated cells | ↑ | N/A | N/A | N/A | N/A | N/A | ↓migration | [11] |
| Breast cancer | SAHA | MzChA-1 and TFK-1 cells in vitro | cells treated with SAHA pretreated with TGF-β1 vs. cells treated with TGF-β1 | ↑ | N/A | ↓ | ↓ | inhibition of p-SMAD2, p-SMAD3 and SMAD4 nuclear translocation induced by TGF-β1 | reduction of changes from valvate-like- to spindle-like shapes caused by TGF-β1 | N/A | [135] |
| Breast cancer | SAHA | MDA-MB-231 and BT-549 cells in vitro | cells treated with SAHA vs. untreated cells | ↓ | N/A | ↑ | ↑ | →SNAIL, SLUG, TWIST and ZEB expression and translocation | N/A | ↑migration | [136] |
| Breast cancer | SAHA, VPA | MDA-MB-231 and SUM159 cells in vitro | ed with VPA or SAHA vs. untreated cells | not detected | N/A | ↑ | ↑ | ↓FOXC3, ZEB1 ↑SNAIL2, TWIST1 | ↑sphere formation | ↑migration | [137] |
| Breast cancer | LBH589 | MDA-MB-231 and BT-549 cells in vitro | cell treated with LBH589 vs. untreated cells | ↑ | N/A | ↓ | ↓ | ↓ZEB1, ZEB2 | more epithelial phenotype | ↓migration and invasion | [138] |
| Breast cancer | LBH589 | MCF7 cells in vitro | cell treated with LBH589 vs. untreated cells | → | N/A | N/A | → | →ZEB1, ZEB2 | more epithelial phenotype | ↓migration and invasion | [138] |
| Breast cancer | MS-275 | MDA-MB-231 and Hs578T cells in vitro | cells treated with MS-275 vs. untreated cells | ↑ | N/A | ↓ | ↓ | ↓SNAIL, TWIST | more epithelial phenotype | ↓migration | [14] |
| Breast cancer | MS-275 | Balb c nude mice implanted with TRAIL resistant MDA-MB-468 cells in vivo | mice treated MS-275 vs. untreated mice | ↑ | N/A | N/A | ↓ | ↓ZEB1, SNAIL, SLUG | N/A | N/A | [139] |
| Breast cancer | MS-275+TRAIL | Balb c nude mice implanted with TRAIL resistant MDA-MB-468 cells in vivo | mice treated MS-275+TRAIL vs. mice treated TRAIL only | ↑ | N/A | N/A | ↓ | ↓ZEB1, SNAIL, SLUG | N/A | N/A | [139] |
| Ovarian cancer | TSA | SKOV3 cells in vitro | cells treated with TSA vs. untreated cells | ↓ | N/A | N/A | ↓ | N/A | N/A | ↓migration | [140] |
| Ovarian cancer | TSA+cisplatin | SKOV3 cells in vitro | cells treated with TSA + cisplatin vs. untreated cells | ↓ | N/A | N/A | ↓ | N/A | N/A | ↓migration | [140] |
| Ovarian cancer | TSA+cisplatin | Mice with HEY injected cells in vivo | mice treated with cisplatin followed by TSA vs. untreated mice | ↑ | N/A | N/A | ↓ | ↓SNAIL, SLUG, TWIST | N/A | N/A | [140] |
| Head and neck cancer | SAHA | Hep-2 and KB cells in vitro | cells treated with SAHA vs. untreated cells | ↑ | ↑ | N/A | ↓ | N/A | reduction of the spindle like morphology | ↓migration and invasion | [141] |
| Head and neck cancer | VPA | TE9 cells pretreated with TGF-β1 or irradiation in vitro | cells treated with VPA and TGF-β1 or irradiation before vs. cells treated with TGF-β1 or irradiation | ↑ | N/A | N/A | ↓ | ↓SMAD2 and SMAD3 phosphorylation, ↓TWIST, SNAIL, SLUG | reduction of spindle like morphology caused by TGF-β1 or irradiation | ↓migration and invasion | [142] |
| Malignant glioma | LBH589+irradiation | U251 cells in vitro | cells treated with LBH589+irradiation vs. untreated cells | ↑ | N/A | N/A | N/A | N/A | reduction of vasculogenic mimicry formation | ↓migration and invasion | [143] |
| HDI | ↑EMT | ↓EMT | Unclear Mechanism |
|---|---|---|---|
| VPA | Hepatocellular carcinoma [120], breast [112], colorectal cancer [128] | Lung [117], prostate [11], head and neck cancer [142] | Renal cancer [130] |
| SAHA | Hepatocellular carcinoma [120], lung [118], breast cancer [136] | Pancreatic [12], head and neck cancer [141] | - |
| TSA | Hepatocellular carcinoma [120], colorectal cancer [128] | Lung [114,115], prostate cancer [13] | Cholangiocarcinoma [124], ovarian cancer [140] |
| MS-275 | Hepatocellular carcinoma [120] | Breast cancer [14] | Renal cancer [130] |
| LBH589 | - | Hepatocellular carcinoma [122], breast cancer [14] | - |
| RAS2410 | - | Hepatocellular carcinoma [123] | - |
| 4SC-202 | - | - | Pancreatic cancer [125] |
| AR-42 | - | - | Prostate cancer [133] |
| NaB | Hepatocellular carcinoma [121] | - | - |
| BSI | - | Pancreatic cancer [126] | - |
| Compound 11 | - | Colorectal cancer [129] | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wawruszak, A.; Kalafut, J.; Okon, E.; Czapinski, J.; Halasa, M.; Przybyszewska, A.; Miziak, P.; Okla, K.; Rivero-Muller, A.; Stepulak, A. Histone Deacetylase Inhibitors and Phenotypical Transformation of Cancer Cells. Cancers 2019, 11, 148. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11020148
Wawruszak A, Kalafut J, Okon E, Czapinski J, Halasa M, Przybyszewska A, Miziak P, Okla K, Rivero-Muller A, Stepulak A. Histone Deacetylase Inhibitors and Phenotypical Transformation of Cancer Cells. Cancers. 2019; 11(2):148. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11020148
Chicago/Turabian StyleWawruszak, Anna, Joanna Kalafut, Estera Okon, Jakub Czapinski, Marta Halasa, Alicja Przybyszewska, Paulina Miziak, Karolina Okla, Adolfo Rivero-Muller, and Andrzej Stepulak. 2019. "Histone Deacetylase Inhibitors and Phenotypical Transformation of Cancer Cells" Cancers 11, no. 2: 148. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11020148