Clinical-Grade Peptide-Based Inhibition of CK2 Blocks Viability and Proliferation of T-ALL Cells and Counteracts IL-7 Stimulation and Stromal Support

 ,
,
Abstract
:1. Introduction
2. Results
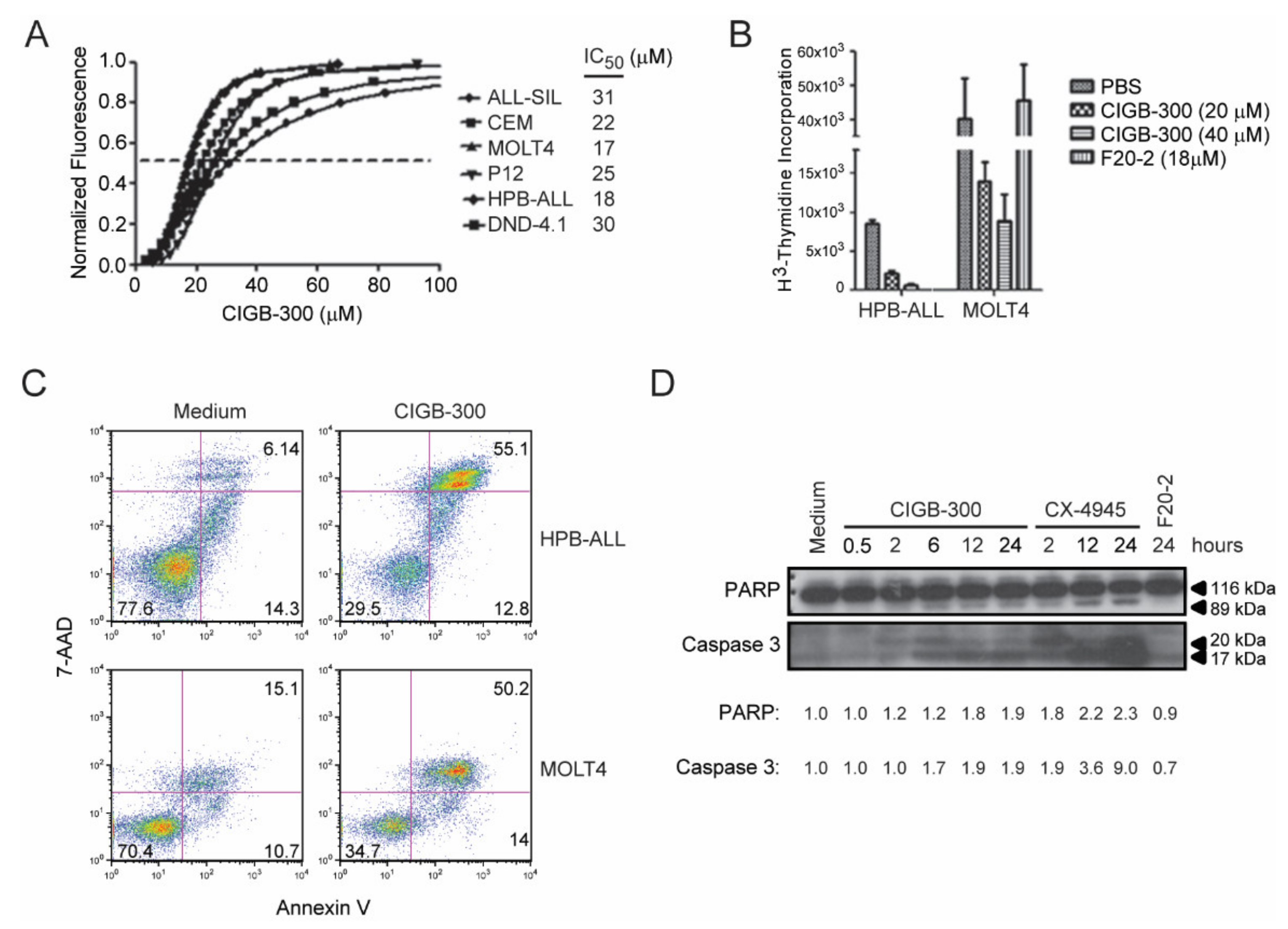
2.1. CIGB-300 Negatively Impacts Viability and Proliferation of T-ALL Cells
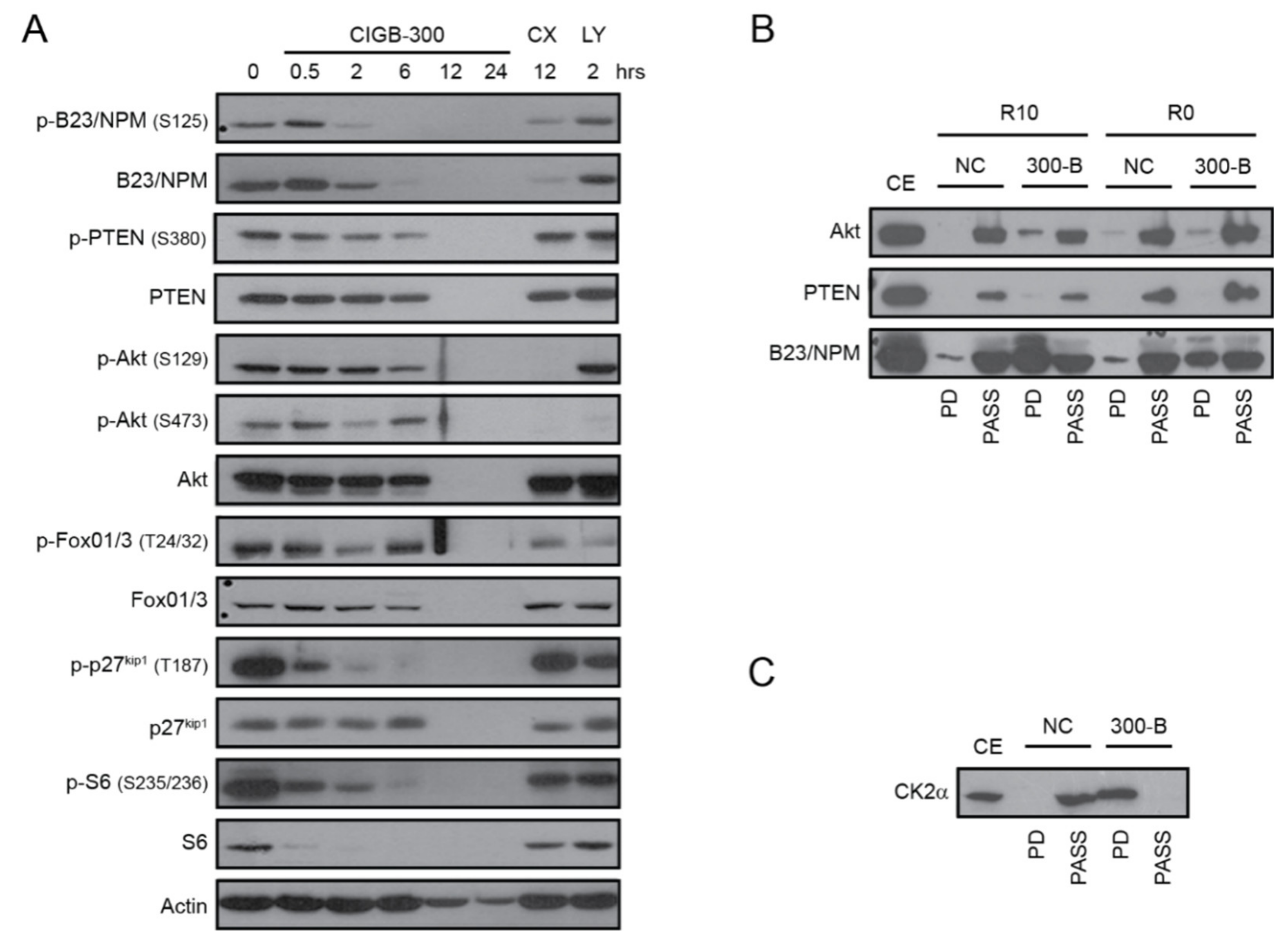
2.2. CIGB-300 Impairs CK2-Mediated Phosphorylation of PTEN, Akt and B23/NPM1
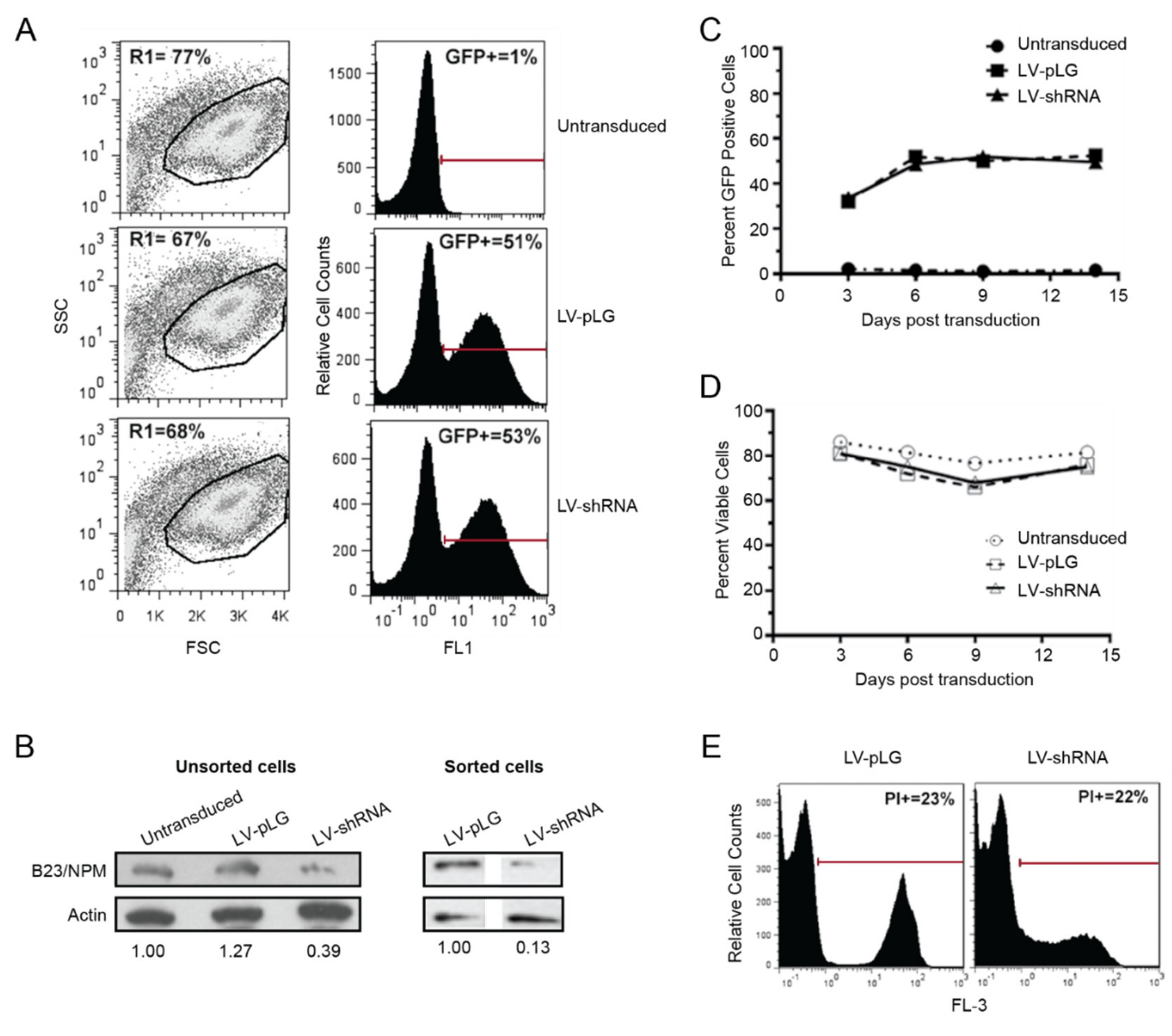
2.3. B23/NPM1 Is not a Major Target for CIGB-300 in T-ALL Cells
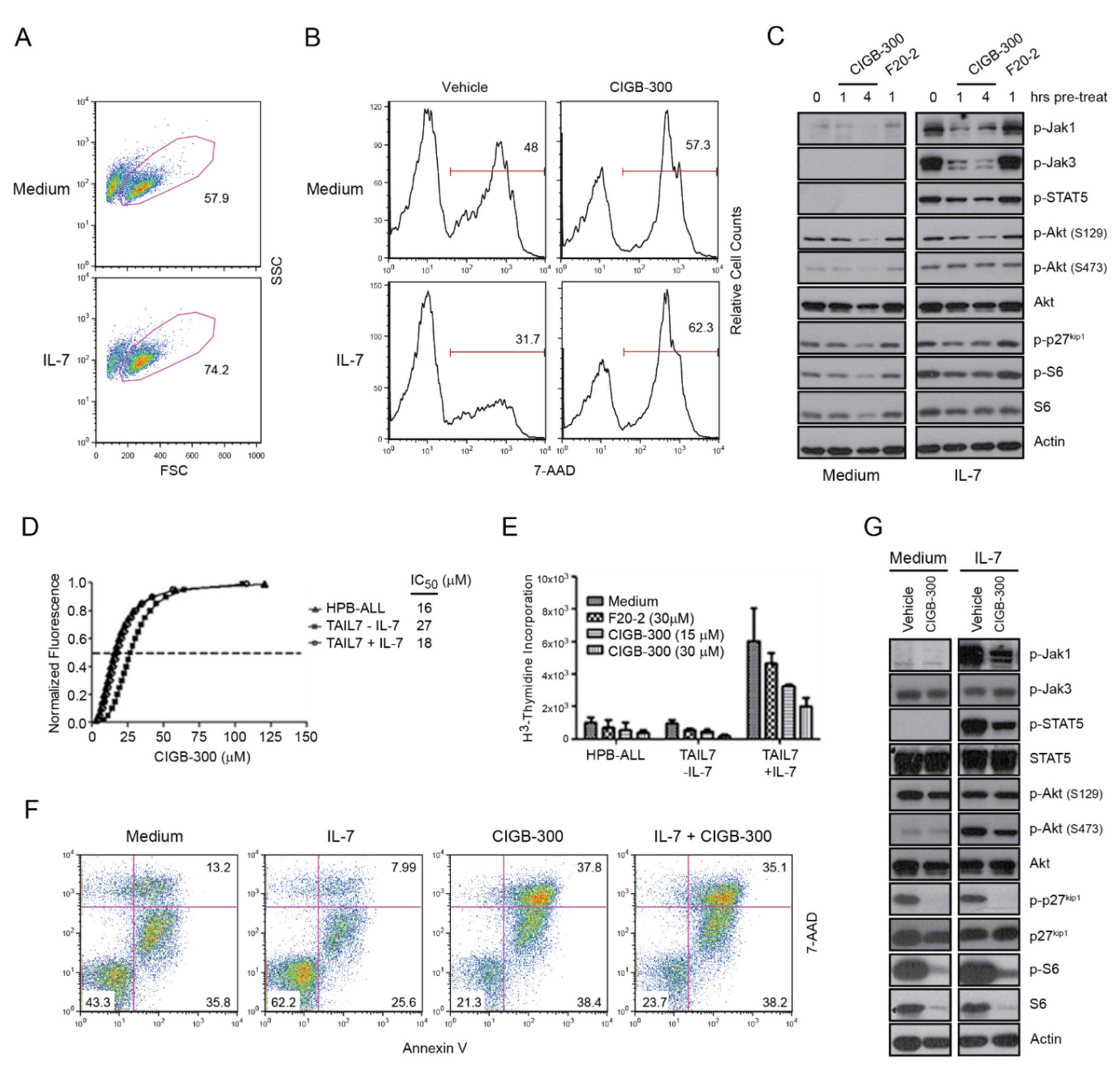
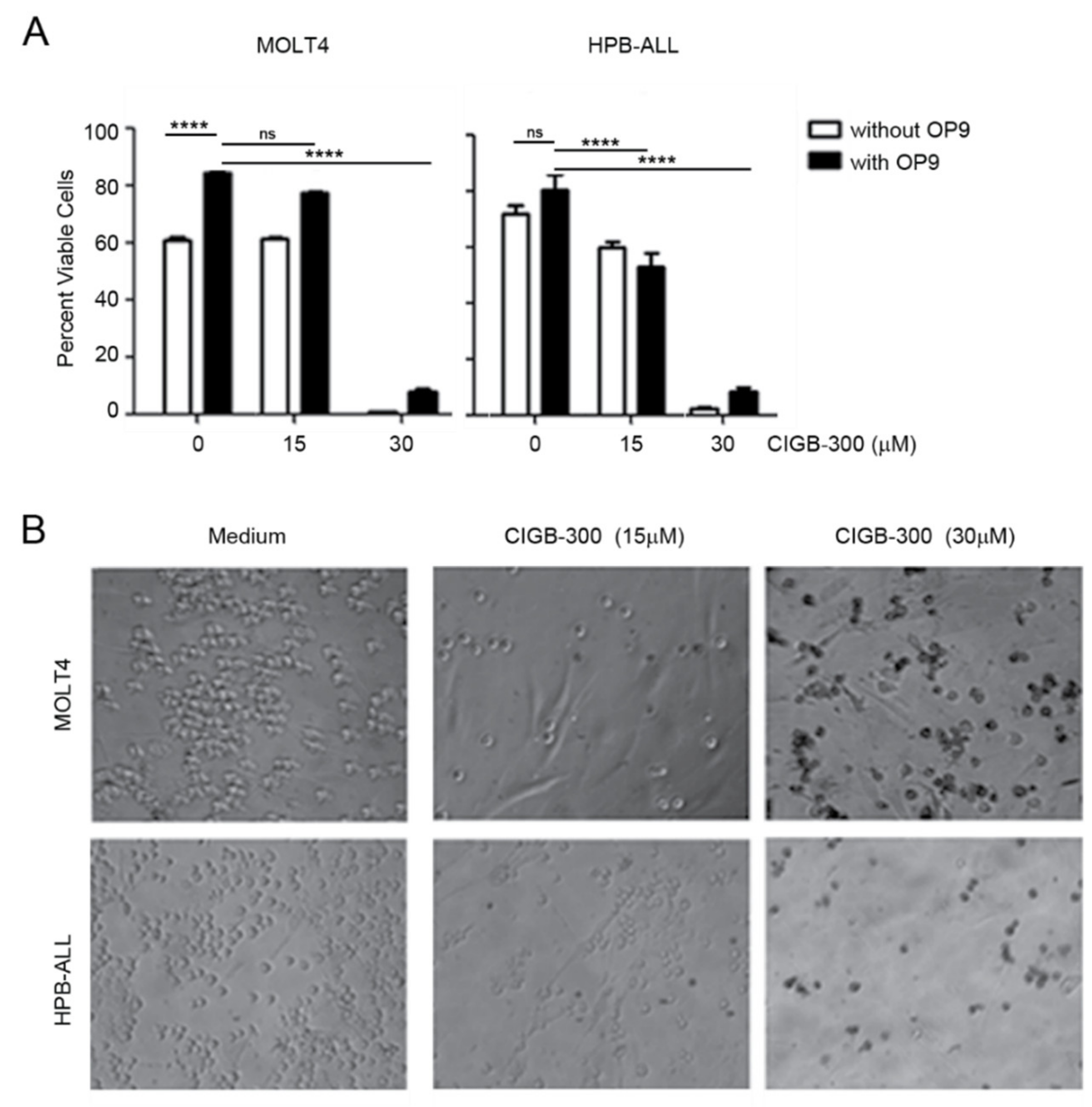
2.4. The Effects of CIGB-300 on T-ALL Cells Are not Reversed by IL-7 Stimulation or Stromal Support
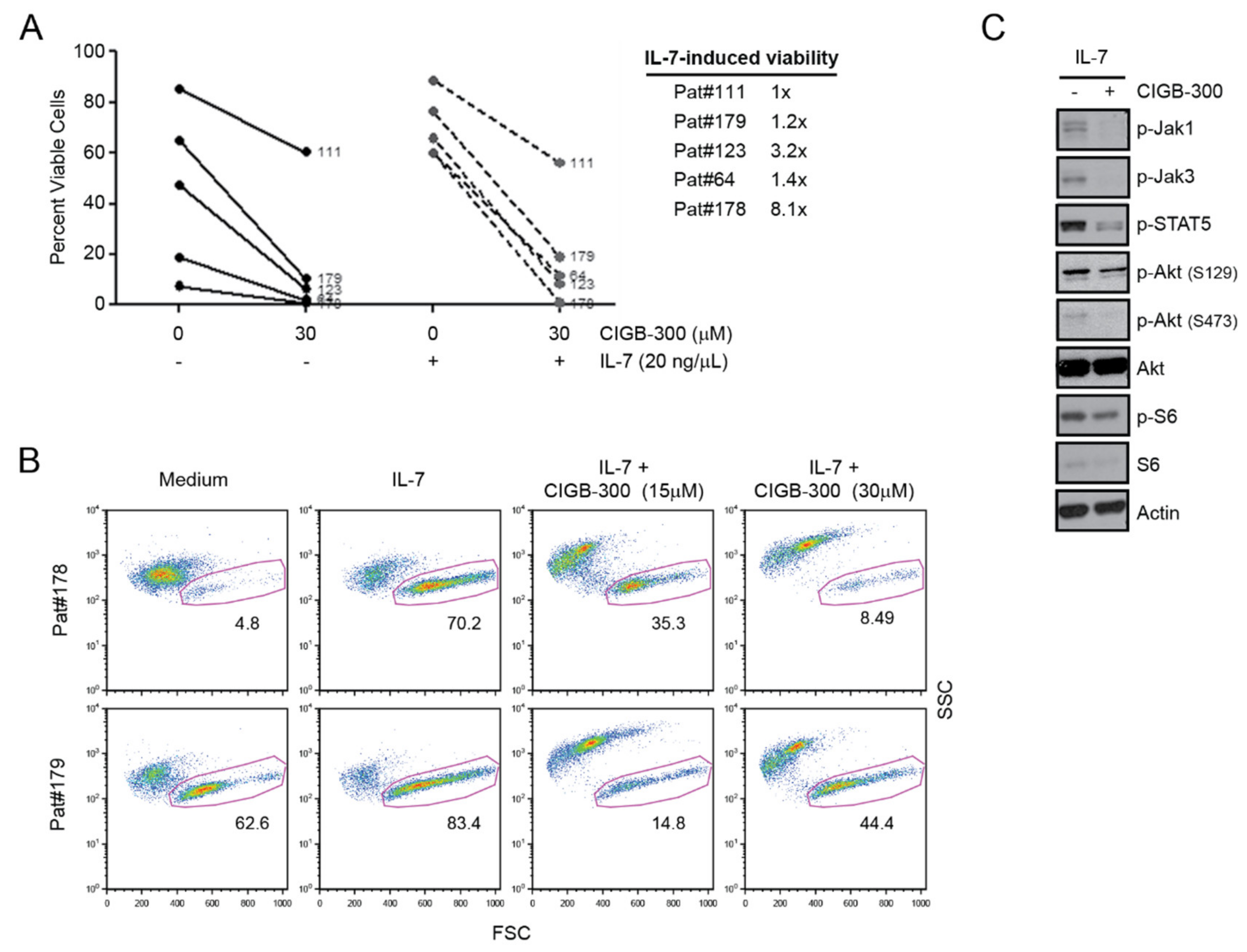
2.5. Primary T-ALL Patient Cells Are Sensitive to CIGB-300 even in the Presence of IL-7
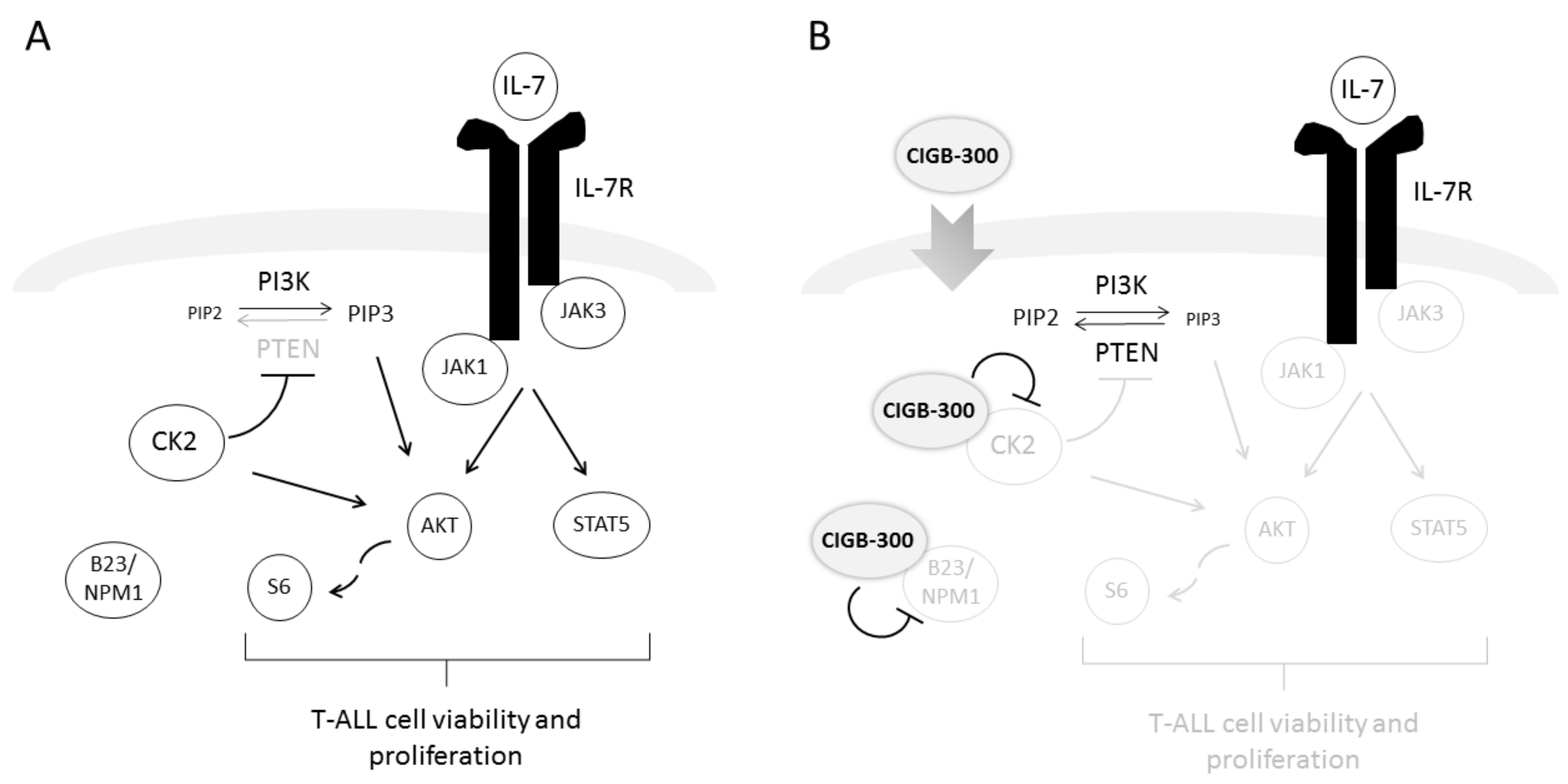
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Patient Samples
4.2. Alamar Blue Assay
4.3. Assessment of Cell Viability
4.4. 3H-Thymidine Incorporation Proliferation Assay
4.5. Signaling Experiments
4.6. Western Blot Analysis
4.7. Pull-Down
4.8. Lentiviral Infection
4.9. Co-Culture Experiments
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brown, V.I.; Seif, A.E.; Reid, G.S.; Teachey, D.T.; Grupp, S.A. Novel molecular and cellular therapeutic targets in acute lymphoblastic leukemia and lymphoproliferative disease. Immunol. Res. 2008, 42, 84–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, A.; Yunes, J.A.; Cardoso, B.A.; Martins, L.R.; Jotta, P.Y.; Abecasis, M.; Nowill, A.E.; Leslie, N.R.; Cardoso, A.A.; Barata, J.T. PTEN posttranslational inactivation and hyperactivation of the PI3K/Akt pathway sustain primary T cell leukemia viability. J. Clin. Investig. 2008, 118, 3762–3774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, M.L.; Akkapeddi, P.; Alcobia, I.; Almeida, A.R.; Cardoso, B.A.; Fragoso, R.; Serafim, T.L.; Barata, J.T. From the outside, from within: Biological and therapeutic relevance of signal transduction in T-cell acute lymphoblastic leukemia. Cell. Signal. 2017, 38, 10–25. [Google Scholar] [CrossRef] [PubMed]
- Bongiovanni, D.; Saccomani, V.; Piovan, E. Aberrant signaling pathways in T-Cell acute lymphoblastic leukemia. Int. J. Mol. Sci. 2017, 18, 1904. [Google Scholar] [CrossRef] [Green Version]
- Girardi, T.; Vicente, C.; Cools, J.; De Keersmaecker, K. The genetics and molecular biology of T-ALL. Blood 2017, 129, 1113–1123. [Google Scholar] [CrossRef]
- Tafuri, A.; Milella, M.; Iacovelli, S.; De Cave, F.; Gregorj, C.; Bergamo, P.; Miele, A.; Licchetta, R.; Konopleva, M.; McCubrey, J.A.; et al. Aberrant proliferative and apoptotic pathways in acute lymphoblastic leukemia (ALL): Molecular therapies to overcome chemo-resistance. In Novel Aspects in Acute Lymphoblastic Leukemia; Faderl, S., Ed.; IntechOpen: London, UK, 2001. [Google Scholar]
- Meggio, F.; Pinna, L.A. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 2003, 17, 349–368. [Google Scholar] [CrossRef]
- Salvi, M.; Sarno, S.; Cesaro, L.; Nakamura, H.; Pinna, L.A. Extraordinary pleiotropy of protein kinase CK2 revealed by weblogo phosphoproteome analysis. Biochim. Biophys. Acta 2009, 1793, 847–859. [Google Scholar] [CrossRef]
- Trembley, J.H.; Wang, G.; Unger, G.; Slaton, J.; Ahmed, K. Protein kinase CK2 in health and disease: CK2: A key player in cancer biology. Cell. Mol. Life Sci. 2009, 66, 1858–1867. [Google Scholar] [CrossRef] [Green Version]
- Barata, J.T. The impact of PTEN regulation by CK2 on PI3K-dependent signaling and leukemia cell survival. Adv. Enzym. Regul. 2011, 51, 37–49. [Google Scholar] [CrossRef]
- Buontempo, F.; McCubrey, J.A.; Orsini, E.; Ruzzene, M.; Cappellini, A.; Lonetti, A.; Evangelisti, C.; Chiarini, F.; Evangelisti, C.; Barata, J.T.; et al. Therapeutic targeting of CK2 in acute and chronic leukemias. Leukemia 2018, 32, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Gomes, A.M.; Soares, M.V.; Ribeiro, P.; Caldas, J.; Povoa, V.; Martins, L.R.; Melao, A.; Serra-Caetano, A.; de Sousa, A.B.; Lacerda, J.F.; et al. Adult B-cell acute lymphoblastic leukemia cells display decreased PTEN activity and constitutive hyperactivation of PI3K/Akt pathway despite high PTEN protein levels. Haematologica 2014, 99, 1062–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, A.; Roolf, C.; Hamed, M.; Gladbach, Y.S.; Sender, S.; Konkolefski, C.; Knubel, G.; Sekora, A.; Fuellen, G.; Vollmar, B.; et al. Combined Casein Kinase II inhibition and epigenetic modulation in acute B-lymphoblastic leukemia. BMC Cancer 2019, 19, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro, S.T.; Tesio, M.; Ribot, J.C.; Macintyre, E.; Barata, J.T.; Silva-Santos, B. Casein kinase 2 controls the survival of normal thymic and leukemic gammadelta T cells via promotion of AKT signaling. Leukemia 2017, 31, 1603–1610. [Google Scholar] [CrossRef] [PubMed]
- Lian, H.; Li, D.; Zhou, Y.; Landesman-Bollag, E.; Zhang, G.; Anderson, N.M.; Tang, K.C.; Roderick, J.E.; Kelliher, M.A.; Seldin, D.C.; et al. CK2 inhibitor CX-4945 destabilizes NOTCH1 and synergizes with JQ1 against human T-acute lymphoblastic leukemic cells. Haematologica 2017, 102, e17–e21. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Song, C.; Ding, Y.; Pan, X.; Ge, Z.; Tan, B.H.; Gowda, C.; Sachdev, M.; Muthusami, S.; Ouyang, H.; et al. Transcriptional Regulation of JARID1B/KDM5B Histone Demethylase by Ikaros, Histone Deacetylase 1 (HDAC1), and Casein Kinase 2 (CK2) in B-cell Acute Lymphoblastic Leukemia. J. Biol. Chem. 2016, 291, 4004–4018. [Google Scholar] [CrossRef] [Green Version]
- Song, C.; Gowda, C.; Pan, X.; Ding, Y.; Tong, Y.; Tan, B.H.; Wang, H.; Muthusami, S.; Ge, Z.; Sachdev, M.; et al. Targeting casein kinase II restores Ikaros tumor suppressor activity and demonstrates therapeutic efficacy in high-risk leukemia. Blood 2015, 126, 1813–1822. [Google Scholar] [CrossRef] [Green Version]
- Silva, A.; Jotta, P.Y.; Silveira, A.B.; Ribeiro, D.; Brandalise, S.R.; Yunes, J.A.; Barata, J.T. Regulation of PTEN by CK2 and Notch1 in primary T-cell acute lymphoblastic leukemia: Rationale for combined use of CK2- and gamma-secretase inhibitors. Haematologica 2010, 95, 674–678. [Google Scholar] [CrossRef] [Green Version]
- Kelliher, M.A.; Seldin, D.C.; Leder, P. Tal-1 induces T cell acute lymphoblastic leukemia accelerated by casein kinase IIalpha. EMBO J. 1996, 15, 5160–5166. [Google Scholar] [CrossRef]
- Torres, J.; Pulido, R. The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. Implications for PTEN stability to proteasome-mediated degradation. J. Biol. Chem. 2001, 276, 993–998. [Google Scholar] [CrossRef] [Green Version]
- Di Maira, G.; Salvi, M.; Arrigoni, G.; Marin, O.; Sarno, S.; Brustolon, F.; Pinna, L.A.; Ruzzene, M. Protein kinase CK2 phosphorylates and upregulates Akt/PKB. Cell Death Differ. 2005, 12, 668–677. [Google Scholar] [CrossRef]
- Li, Z.; Song, C.; Ouyang, H.; Lai, L.; Payne, K.J.; Dovat, S. Cell cycle-specific function of Ikaros in human leukemia. Pediatr. Blood Cancer 2012, 59, 69–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Qin, H.; Frank, S.J.; Deng, L.; Litchfield, D.W.; Tefferi, A.; Pardanani, A.; Lin, F.T.; Li, J.; Sha, B.; et al. A CK2-dependent mechanism for activation of the JAK-STAT signaling pathway. Blood 2011, 118, 156–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aparicio-Siegmund, S.; Sommer, J.; Monhasery, N.; Schwanbeck, R.; Keil, E.; Finkenstadt, D.; Pfeffer, K.; Rose-John, S.; Scheller, J.; Garbers, C. Inhibition of protein kinase II (CK2) prevents induced signal transducer and activator of transcription (STAT) 1/3 and constitutive STAT3 activation. Oncotarget 2014, 5, 2131–2148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melao, A.; Spit, M.; Cardoso, B.A.; Barata, J.T. Optimal interleukin-7 receptor-mediated signaling, cell cycle progression and viability of T-cell acute lymphoblastic leukemia cells rely on casein kinase 2 activity. Haematologica 2016, 101, 1368–1379. [Google Scholar] [CrossRef] [Green Version]
- Barata, J.T.; Durum, S.K.; Seddon, B. Flip the coin: IL-7 and IL-7R in health and disease. Nat. Immunol. 2019, 20, 1584–1593. [Google Scholar] [CrossRef]
- Buontempo, F.; Orsini, E.; Martins, L.R.; Antunes, I.; Lonetti, A.; Chiarini, F.; Tabellini, G.; Evangelisti, C.; Evangelisti, C.; Melchionda, F.; et al. Cytotoxic activity of the casein kinase 2 inhibitor CX-4945 against T-cell acute lymphoblastic leukemia: Targeting the unfolded protein response signaling. Leukemia 2014, 28, 543–553. [Google Scholar] [CrossRef]
- Buontempo, F.; Orsini, E.; Lonetti, A.; Cappellini, A.; Chiarini, F.; Evangelisti, C.; Evangelisti, C.; Melchionda, F.; Pession, A.; Bertaina, A.; et al. Synergistic cytotoxic effects of bortezomib and CK2 inhibitor CX-4945 in acute lymphoblastic leukemia: Turning off the prosurvival ER chaperone BIP/Grp78 and turning on the pro-apoptotic NF-kappaB. Oncotarget 2016, 7, 1323–1340. [Google Scholar] [CrossRef] [Green Version]
- Padgaonkar, A.; Rechkoblit, O.; Vasquez-Del Carpio, R.; Pallela, V.; Venkata Subbaiah, D.; Cosenza, S.C.; Baker, S.J.; Ramana Reddy, M.V.; Aggarwal, A.; Reddy, E.P. Targeting protein kinase CK2 and CDK4/6 pathways with a multi-kinase inhibitor ON108110 suppresses pro-survival signaling and growth in mantle cell lymphoma and T-acute lymphoblastic leukemia. Oncotarget 2018, 9, 37753–37765. [Google Scholar] [CrossRef]
- Siddiqui-Jain, A.; Drygin, D.; Streiner, N.; Chua, P.; Pierre, F.; O‘Brien, S.E.; Bliesath, J.; Omori, M.; Huser, N.; Ho, C.; et al. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010, 70, 10288–10298. [Google Scholar] [CrossRef] [Green Version]
- Pierre, F.; Chua, P.C.; O‘Brien, S.E.; Siddiqui-Jain, A.; Bourbon, P.; Haddach, M.; Michaux, J.; Nagasawa, J.; Schwaebe, M.K.; Stefan, E.; et al. Discovery and SAR of 5-(3-chlorophenylamino)benzo[c][2,6]naphthyridine-8-carboxylic acid (CX-4945), the first clinical stage inhibitor of protein kinase CK2 for the treatment of cancer. J. Med. Chem. 2011, 54, 635–654. [Google Scholar] [CrossRef]
- Perea, S.E.; Reyes, O.; Puchades, Y.; Mendoza, O.; Vispo, N.S.; Torrens, I.; Santos, A.; Silva, R.; Acevedo, B.; Lopez, E.; et al. Antitumor effect of a novel proapoptotic peptide that impairs the phosphorylation by the protein kinase 2 (casein kinase 2). Cancer Res. 2004, 64, 7127–7129. [Google Scholar] [CrossRef] [Green Version]
- Perea, S.E.; Reyes, O.; Baladron, I.; Perera, Y.; Farina, H.; Gil, J.; Rodriguez, A.; Bacardi, D.; Marcelo, J.L.; Cosme, K.; et al. CIGB-300, a novel proapoptotic peptide that impairs the CK2 phosphorylation and exhibits anticancer properties both in vitro and in vivo. Mol. Cell. Biochem. 2008, 316, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Perera, Y.; Costales, H.C.; Diaz, Y.; Reyes, O.; Farina, H.G.; Mendez, L.; Gomez, R.E.; Acevedo, B.E.; Gomez, D.E.; Alonso, D.F.; et al. Sensitivity of tumor cells towards CIGB-300 anticancer peptide relies on its nucleolar localization. J. Pept. Sci. 2012, 18, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Perea, S.E.; Baladron, I.; Garcia, Y.; Perera, Y.; Lopez, A.; Soriano, J.L.; Batista, N.; Palau, A.; Hernandez, I.; Farina, H.; et al. CIGB-300, a synthetic peptide-based drug that targets the CK2 phosphoaceptor domain. Translational and clinical research. Mol. Cell. Biochem. 2011, 356, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Solares, A.M.; Santana, A.; Baladron, I.; Valenzuela, C.; Gonzalez, C.A.; Diaz, A.; Castillo, D.; Ramos, T.; Gomez, R.; Alonso, D.F.; et al. Safety and preliminary efficacy data of a novel casein kinase 2 (CK2) peptide inhibitor administered intralesionally at four dose levels in patients with cervical malignancies. BMC Cancer 2009, 9, 146. [Google Scholar] [CrossRef] [Green Version]
- Soriano-García, J.L.; López-Díaz, A.; Solares-Asteasuainzarra, M.; Baladrón-Castrillo, I.; Batista-Albuerne, N.; García-García, I.; González-Méndez, L.; Perera-Negrín, Y.; Valenzuela-Silva, C.M.; Pedro, A.P.; et al. Pharmacological and safety evaluation of CIGB-300, a casein kinase 2 inhibitor peptide, administered intralesionally to patients with cervical cancer stage IB2/II. J. Cancer Res. Ther. 2013, 1, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Batista-Albuerne, N.; González-Méndez, L.; García-García, I.; Fernández Sánchez, E.; García-Diegues, R.; Torre-Santos, A.; Martín-Bauta, Y.; Raíces-Cruz, I.; Valenzuela-Silva, C.; Reyes-Nicolás, V.; et al. Phase I Study of CIGB-300 Administered Intravenously in Patients with Relapsed/Refractory Solid Tumors. J. Med. Oncol. 2018, 1, 55175989. [Google Scholar]
- Masuda, M.; Motoji, T.; Oshimi, K.; Mizoguchi, H. Effects of interleukin-7 on proliferation of hematopoietic malignant cells. Exp. Hematol. 1990, 18, 965–967. [Google Scholar]
- Digel, W.; Schmid, M.; Heil, G.; Conrad, P.; Gillis, S.; Porzsolt, F. Human interleukin-7 induces proliferation of neoplastic cells from chronic lymphocytic leukemia and acute leukemias. Blood 1991, 78, 753–759. [Google Scholar] [CrossRef] [Green Version]
- Barata, J.T.; Boussiotis, V.A.; Yunes, J.A.; Ferrando, A.A.; Moreau, L.A.; Veiga, J.P.; Sallan, S.E.; Look, A.T.; Nadler, L.M.; Cardoso, A.A. IL-7-dependent human leukemia T-cell line as a valuable tool for drug discovery in T-ALL. Blood 2004, 103, 1891–1900. [Google Scholar] [CrossRef] [Green Version]
- Barata, J.T.; Cardoso, A.A.; Nadler, L.M.; Boussiotis, V.A. Interleukin-7 promotes survival and cell cycle progression of T-cell acute lymphoblastic leukemia cells by down-regulating the cyclin-dependent kinase inhibitor p27(kip1). Blood 2001, 98, 1524–1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scupoli, M.T.; Perbellini, O.; Krampera, M.; Vinante, F.; Cioffi, F.; Pizzolo, G. Interleukin 7 requirement for survival of T-cell acute lymphoblastic leukemia and human thymocytes on bone marrow stroma. Haematologica 2007, 92, 264–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, A.; Laranjeira, A.B.; Martins, L.R.; Cardoso, B.A.; Demengeot, J.; Yunes, J.A.; Seddon, B.; Barata, J.T. IL-7 contributes to the progression of human T-cell acute lymphoblastic leukemias. Cancer Res. 2011, 71, 4780–4789. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Garcia, S.; Mosquera, M.; Fuentes, P.; Palumbo, T.; Escudero, A.; Perez-Martinez, A.; Ramirez, M.; Corcoran, A.E.; Toribio, M.L. IL-7R is essential for leukemia-initiating cell activity of T-cell acute lymphoblastic leukemia. Blood 2019, 134, 2171–2182. [Google Scholar] [CrossRef] [PubMed]
- Perera, Y.; Farina, H.G.; Gil, J.; Rodriguez, A.; Benavent, F.; Castellanos, L.; Gomez, R.E.; Acevedo, B.E.; Alonso, D.F.; Perea, S.E. Anticancer peptide CIGB-300 binds to nucleophosmin/B23, impairs its CK2-mediated phosphorylation, and leads to apoptosis through its nucleolar disassembly activity. Mol. Cancer Ther. 2009, 8, 1189–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, L.R.; Perera, Y.; Lucio, P.; Silva, M.G.; Perea, S.E.; Barata, J.T. Targeting chronic lymphocytic leukemia using CIGB-300, a clinical-stage CK2-specific cell-permeable peptide inhibitor. Oncotarget 2014, 5, 258–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro, D.; Melao, A.; van Boxtel, R.; Santos, C.I.; Silva, A.; Silva, M.C.; Cardoso, B.A.; Coffer, P.J.; Barata, J.T. STAT5 is essential for IL-7-mediated viability, growth, and proliferation of T-cell acute lymphoblastic leukemia cells. Blood Adv. 2018, 2, 2199–2213. [Google Scholar] [CrossRef]
- Martins, L.R.; Lucio, P.; Melao, A.; Antunes, I.; Cardoso, B.A.; Stansfield, R.; Bertilaccio, M.T.; Ghia, P.; Drygin, D.; Silva, M.G.; et al. Activity of the clinical-stage CK2-specific inhibitor CX-4945 against chronic lymphocytic leukemia. Leukemia 2014, 28, 179–182. [Google Scholar] [CrossRef]
- Kim, J.S.; Eom, J.I.; Cheong, J.W.; Choi, A.J.; Lee, J.K.; Yang, W.I.; Min, Y.H. Protein kinase CK2alpha as an unfavorable prognostic marker and novel therapeutic target in acute myeloid leukemia. Clin. Cancer Res. 2007, 13, 1019–1028. [Google Scholar] [CrossRef] [Green Version]
- Borgo, C.; Cesaro, L.; Salizzato, V.; Ruzzene, M.; Massimino, M.L.; Pinna, L.A.; Donella-Deana, A. Aberrant signalling by protein kinase CK2 in imatinib-resistant chronic myeloid leukaemia cells: Biochemical evidence and therapeutic perspectives. Mol. Oncol. 2013, 7, 1103–1115. [Google Scholar] [CrossRef]
- Morotti, A.; Carra, G.; Panuzzo, C.; Crivellaro, S.; Taulli, R.; Guerrasio, A.; Saglio, G. Protein Kinase CK2: A targetable BCR-ABL partner in philadelphia positive leukemias. Adv. Hematol. 2015, 2015, 612567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arriazu, E.; Vicente, C.; Pippa, R.; Peris, I.; Martinez-Balsalobre, E.; Garcia-Ramirez, P.; Marcotegui, N.; Igea, A.; Alignani, D.; Rifon, J.; et al. A new regulatory mechanism of protein phosphatase 2A activity via SET in acute myeloid leukemia. Blood Cancer J. 2020, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Perera, Y.; Ramos, Y.; Padrón, G.; Caballero, E.; Guirola, O.; Caligiuri, L.G.; Lorenzo, N.; Gottardo, F.; Farina, H.G.; Filhol, O.; et al. CIGB-300 anti-cancer peptide regulates the protein kinase CK2-dependent phosphoproteome. Mol. Cell. Biochem. 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Barata, J.T.; Keenan, T.D.; Silva, A.; Nadler, L.M.; Boussiotis, V.A.; Cardoso, A.A. Common gamma chain-signaling cytokines promote proliferation of T-cell acute lymphoblastic leukemia. Haematologica 2004, 89, 1459–1467. [Google Scholar] [PubMed]
- Akkapeddi, P.; Fragoso, R.; Hixon, J.A.; Ramalho, A.S.; Oliveira, M.L.; Carvalho, T.; Gloger, A.; Matasci, M.; Corzana, F.; Durum, S.K.; et al. A fully human anti-IL-7Ralpha antibody promotes antitumor activity against T-cell acute lymphoblastic leukemia. Leukemia 2019, 33, 2155–2168. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, S.; Abundis, C.; Boccalatte, F.; Mehrotra, P.; Chiang, M.Y.; Yui, M.A.; Wang, L.; Zhang, H.; Zollman, A.; Bonfim-Silva, R.; et al. Therapeutic targeting of the E3 ubiquitin ligase SKP2 in T-ALL. Leukemia 2019, 33, 2155–2168. [Google Scholar] [CrossRef] [Green Version]
- Zenatti, P.P.; Ribeiro, D.; Li, W.; Zuurbier, L.; Silva, M.C.; Paganin, M.; Tritapoe, J.; Hixon, J.A.; Silveira, A.B.; Cardoso, B.A.; et al. Oncogenic IL7R gain-of-function mutations in childhood T-cell acute lymphoblastic leukemia. Nat. Genet. 2011, 43, 932–939. [Google Scholar] [CrossRef]
- Delgado-Martin, C.; Meyer, L.K.; Huang, B.J.; Shimano, K.A.; Zinter, M.S.; Nguyen, J.V.; Smith, G.A.; Taunton, J.; Winter, S.S.; Roderick, J.R.; et al. JAK/STAT pathway inhibition overcomes IL7-induced glucocorticoid resistance in a subset of human T-cell acute lymphoblastic leukemias. Leukemia 2017, 31, 2568–2576. [Google Scholar] [CrossRef]
- Meyer, L.K.; Huang, B.J.; Delgado-Martin, C.; Roy, R.P.; Hechmer, A.; Wandler, A.M.; Vincent, T.L.; Fortina, P.; Olshen, A.B.; Wood, B.L.; et al. Glucocorticoids paradoxically facilitate steroid resistance in T cell acute lymphoblastic leukemias and thymocytes. J. Clin. Investig. 2020, 130, 863–876. [Google Scholar] [CrossRef] [Green Version]
- Richard, J.P.; Melikov, K.; Brooks, H.; Prevot, P.; Lebleu, B.; Chernomordik, L.V. Cellular uptake of unconjugated TAT peptide involves clathrin-dependent endocytosis and heparan sulfate receptors. J. Biol. Chem. 2005, 280, 15300–15306. [Google Scholar] [CrossRef] [Green Version]
- Galovic, M.; Xu, D.; Areces, L.B.; van der Kammen, R.; Innocenti, M. Interplay between N-WASP and CK2 optimizes clathrin-mediated endocytosis of EGFR. J. Cell Sci. 2011, 124, 2001–2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henriques, C.M.; Rino, J.; Nibbs, R.J.; Graham, G.J.; Barata, J.T. IL-7 induces rapid clathrin-mediated internalization and JAK3-dependent degradation of IL-7Ralpha in T cells. Blood 2010, 115, 3269–3277. [Google Scholar] [CrossRef]
- Calvo, J.; Fahy, L.; Uzan, B.; Pflumio, F. Desperately seeking a home marrow niche for T-cell acute lymphoblastic leukaemia. Adv. Biol. Regul. 2019, 74, 100640. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, T.M.; Zuniga-Pflucker, J.C. Induction of T cell development from hematopoietic progenitor cells by delta-like-1 in vitro. Immunity 2002, 17, 749–756. [Google Scholar] [CrossRef] [Green Version]
- Sanda, T.; Tyner, J.W.; Gutierrez, A.; Ngo, V.N.; Glover, J.; Chang, B.H.; Yost, A.; Ma, W.; Fleischman, A.G.; Zhou, W.; et al. TYK2-STAT1-BCL2 pathway dependence in T-cell acute lymphoblastic leukemia. Cancer Discov. 2013, 3, 564–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Pierce, L.J.; Spangrude, G.J. Distinct roles of IL-7 and stem cell factor in the OP9-DL1 T-cell differentiation culture system. Exp. Hematol. 2006, 34, 1730–1740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, T.-C.; Hayball, M. CalcuSyn for Windows, Multiple-drug dose-effect analyzer and manual. In Biosoft; Cambridge Place: Cambridge, UK, 1996. [Google Scholar]
- Boussiotis, V.A.; Freeman, G.J.; Berezovskaya, A.; Barber, D.L.; Nadler, L.M. Maintenance of human T cell anergy: Blocking of IL-2 gene transcription by activated Rap1. Science 1997, 278, 124–128. [Google Scholar] [CrossRef]
- Toledo, J.R.; Prieto, Y.; Oramas, N.; Sanchez, O. Polyethylenimine-based transfection method as a simple and effective way to produce recombinant lentiviral vectors. Appl. Biochem. Biotechnol. 2009, 157, 538–544. [Google Scholar] [CrossRef]
- Tiscornia, G.; Singer, O.; Verma, I.M. Production and purification of lentiviral vectors. Nat. Protoc. 2006, 1, 241–245. [Google Scholar] [CrossRef]
- O’Doherty, U.; Swiggard, W.J.; Malim, M.H. Human immunodeficiency virus type 1 spinoculation enhances infection through virus binding. J. Virol. 2000, 74, 10074–10080. [Google Scholar] [CrossRef] [Green Version]
- Holmes, R.; Zuniga-Pflucker, J.C. The OP9-DL1 system: Generation of T-lymphocytes from embryonic or hematopoietic stem cells in vitro. Cold Spring Harb. Protoc. 2009, 2009, 5156. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subgroup | Immunophenotype | ||
|---|---|---|---|
| T-ALL cell lines | ALL-SIL | TLX1 | CD3− CD4+ CD8+ |
| CEM | TAL1 | CD3− CD4+ CD8− | |
| MOLT4 | TAL1 | CD3− CD4+ CD8+ | |
| P12-ICHIKAWA | LMO2 | CD3− CD4+ CD8− | |
| HPB-ALL | TLX3 | CD3+ CD4+ CD8+ | |
| DND4.1 | TLX3 | CD3+ CD4+ CD8− | |
| Primary T-ALL samples | Pat#64 | LMO2 | CD3− CD4+ CD8+ |
| Pat#111 | LMO2-LYL1 | CD3− CD4− CD8− | |
| Pat#123 | ND 1 | CD3− CD4− CD8− | |
| Pat#178 | TAL1 | CD3− CD4+ CD8+ | |
| Pat#179 | ND 1 | CD3− CD4− CD8− |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perera, Y.; Melão, A.; Ramón, A.C.; Vázquez, D.; Ribeiro, D.; Perea, S.E.; Barata, J.T. Clinical-Grade Peptide-Based Inhibition of CK2 Blocks Viability and Proliferation of T-ALL Cells and Counteracts IL-7 Stimulation and Stromal Support. Cancers 2020, 12, 1377. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061377
Perera Y, Melão A, Ramón AC, Vázquez D, Ribeiro D, Perea SE, Barata JT. Clinical-Grade Peptide-Based Inhibition of CK2 Blocks Viability and Proliferation of T-ALL Cells and Counteracts IL-7 Stimulation and Stromal Support. Cancers. 2020; 12(6):1377. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061377
Chicago/Turabian StylePerera, Yasser, Alice Melão, Ailyn C. Ramón, Dania Vázquez, Daniel Ribeiro, Silvio E. Perea, and João T. Barata. 2020. "Clinical-Grade Peptide-Based Inhibition of CK2 Blocks Viability and Proliferation of T-ALL Cells and Counteracts IL-7 Stimulation and Stromal Support" Cancers 12, no. 6: 1377. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061377