A Clinically Applicable Approach to the Classification of B-Cell Non-Hodgkin Lymphomas with Flow Cytometry and Machine Learning

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results
2.1. Database and Models Design
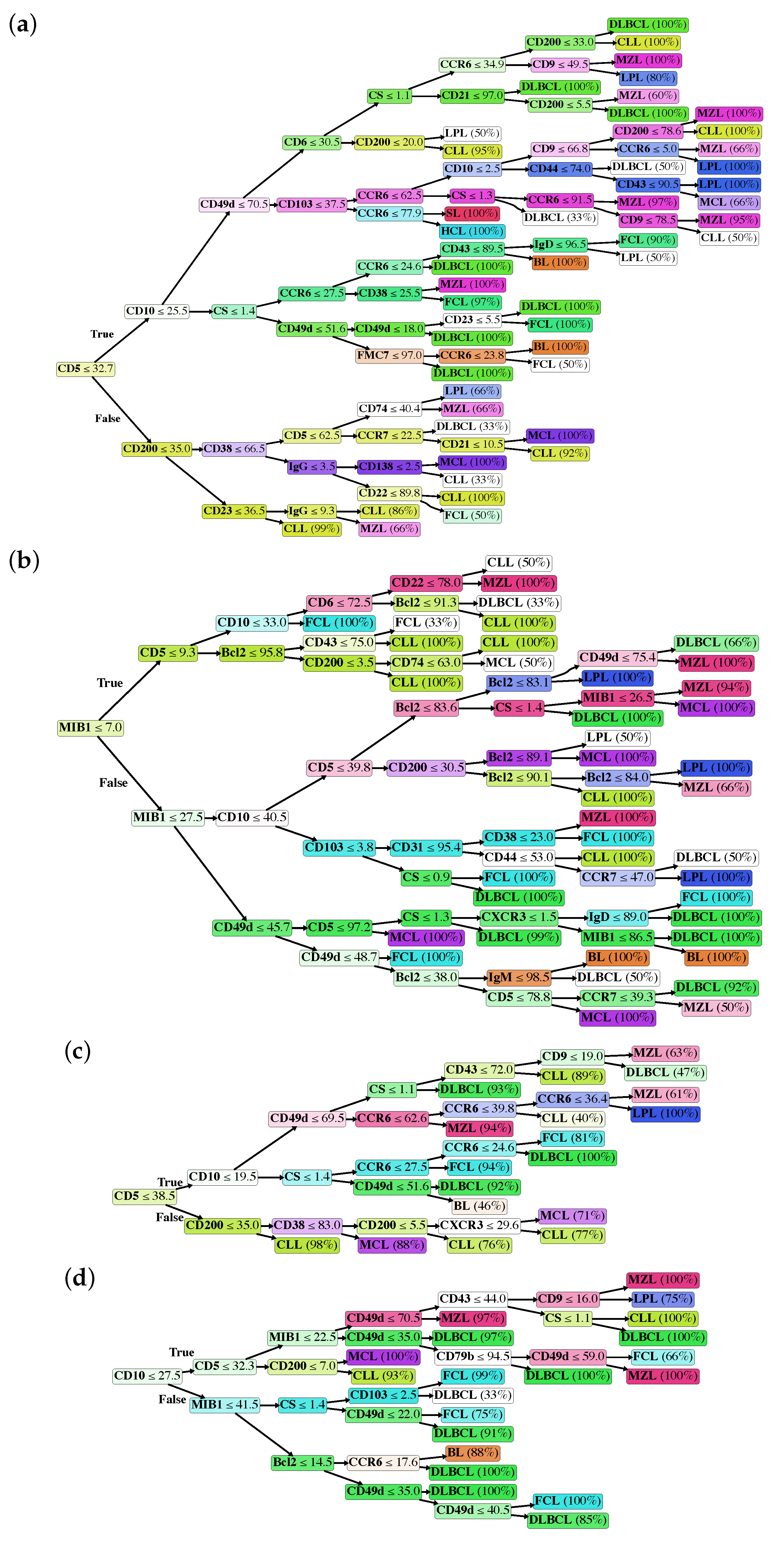
2.2. Classification Trees
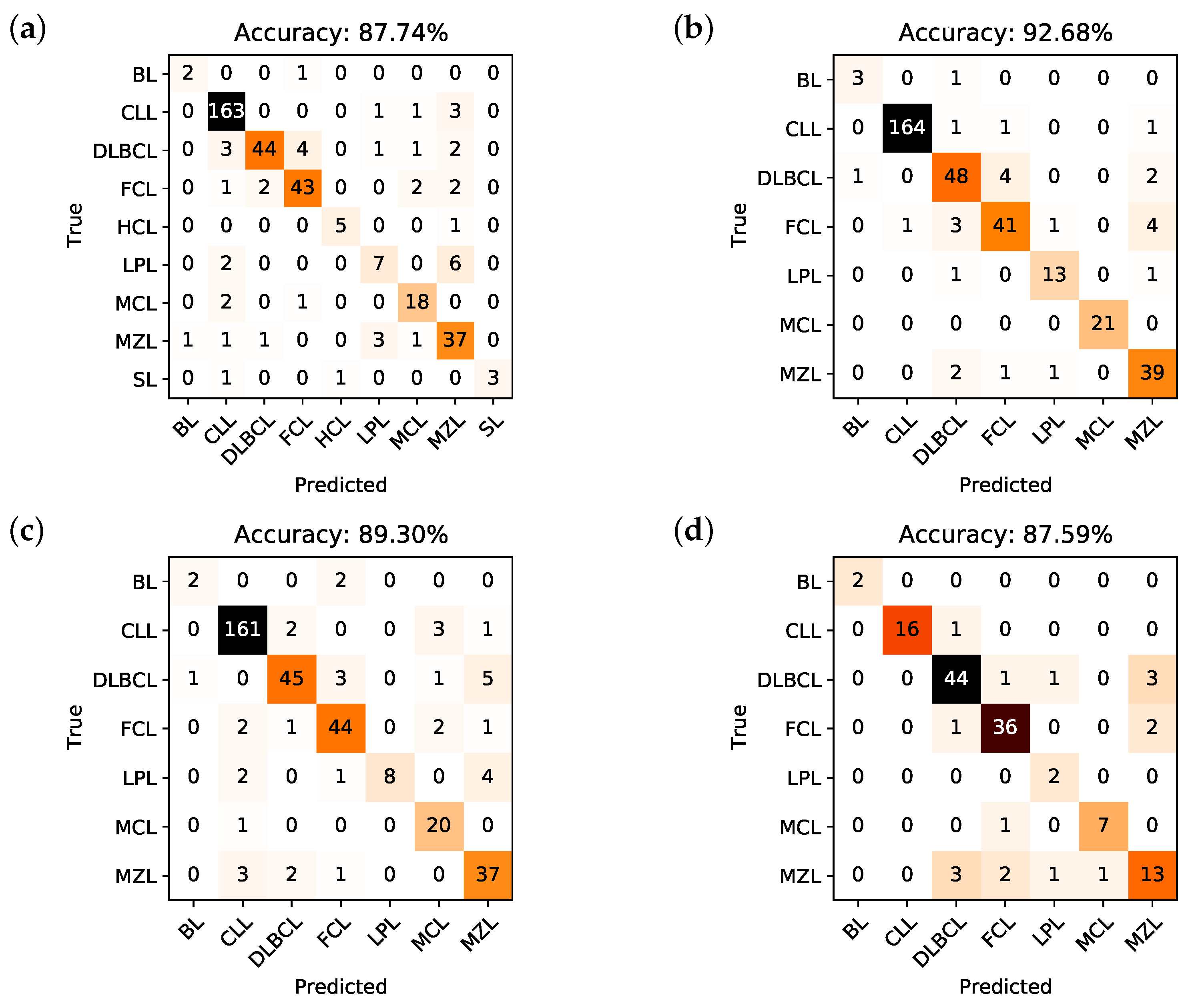
2.3. Performances of the Predictive Models
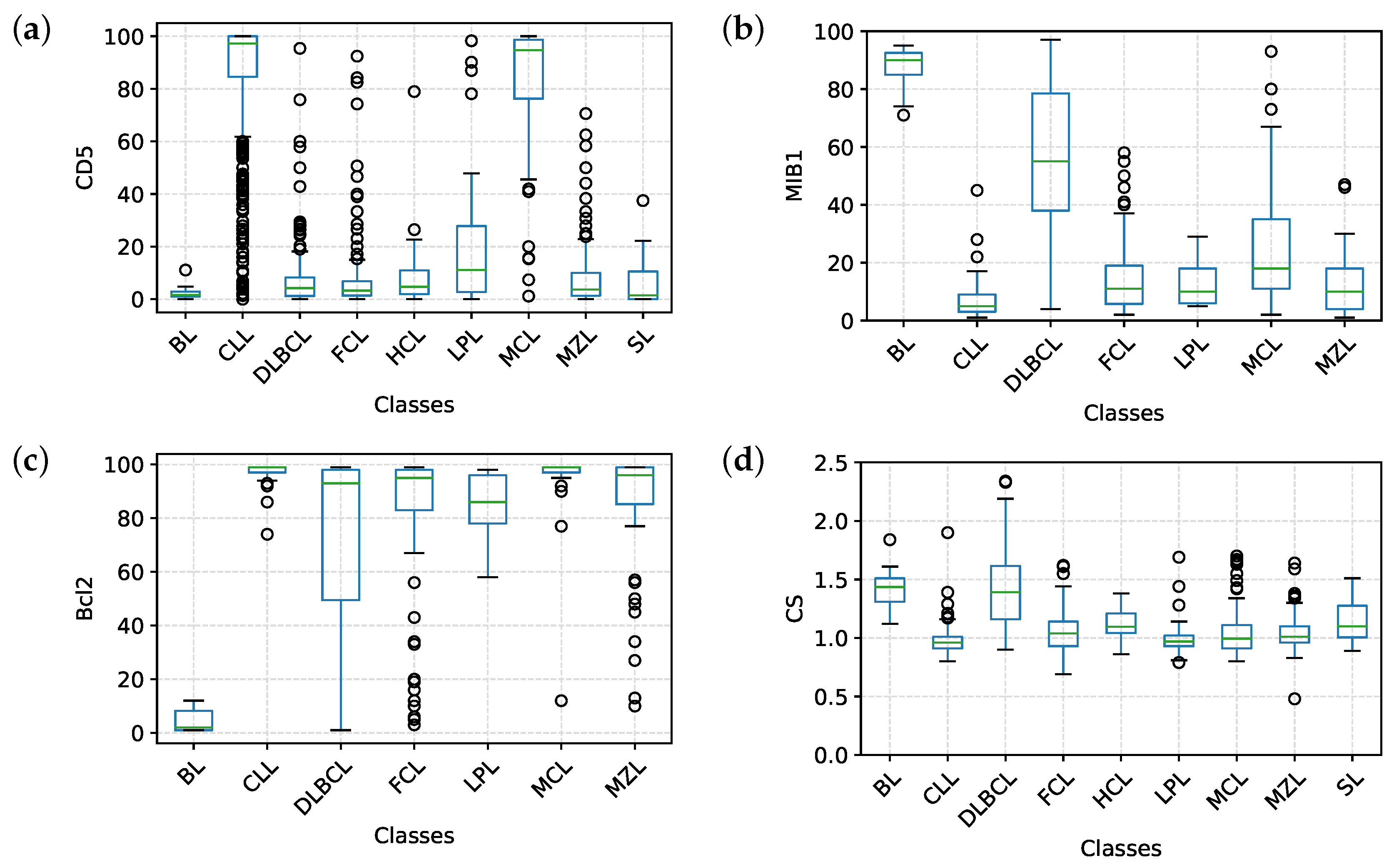
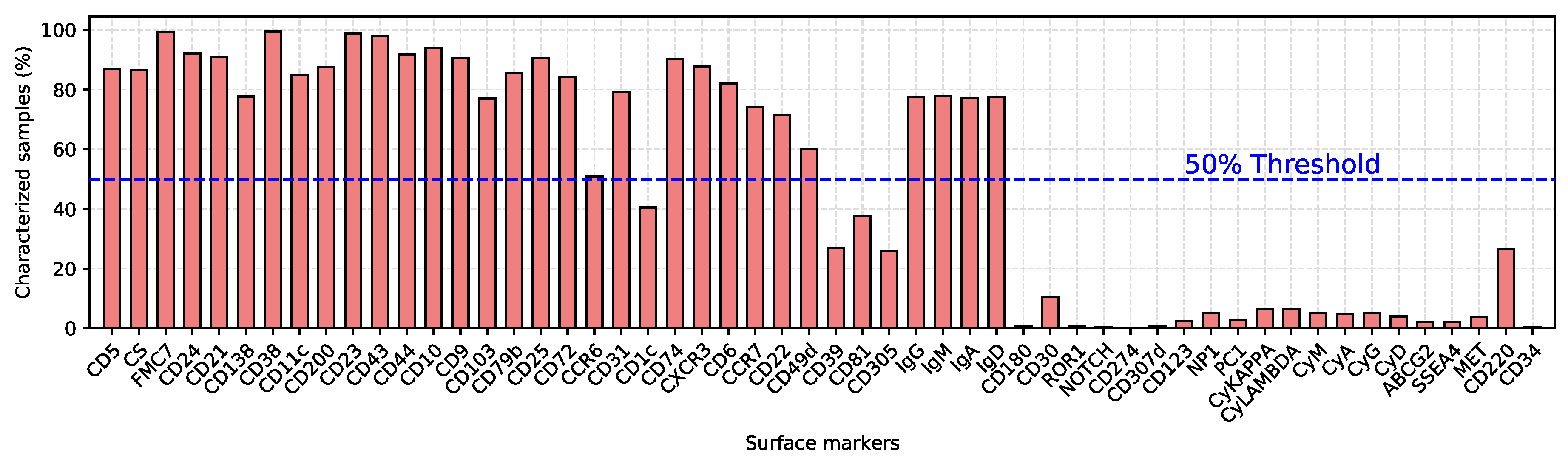
2.4. Analysis of the Predictive Models
3. Discussion
- As mentioned above, markers occur into the tree structure multiple times with different thresholds, applying more fine-grained selections as they approach leaf nodes.
- Figure 3a clearly shows that, apart from CD5, CD10 and CD200, the most important parameters to classify B-NHL are CS and, intriguingly, CCR6, CD49d and CD6. CS is generally underused as a classifier in flow cytometry despite its common use as a gate [24,25]. To overcome the problem of the intrinsic variability of the forward scatter as a measure of the cell volume, due to different setups and different instruments, we calculated for each sample the ratio between the forward scatter of neoplastic B-cells and residual T-lymphocytes.CCR6 is the receptor of chemokine CCL20 and of beta-defensins. It is known to be expressed in B-lymphocytes of the mantle and marginal zone, and in their lymphomatous counterparts, but it is not routinely used to classify B-NHL, even though other groups have already reported that it is differentially expressed in various NHL [26]. CD49d is one of the strongest FC-based predictors of overall survival in CLL [27], but limited data are available on its expression in other NHL [28,29]. The importance of these two non-canonical markers, however, could have been overestimated due to missing values (Section 4.1) and to the filling methodology utilized; further studies are needed to confirm these results.Finally, the predictive model also selected CD6, a CD5-related antigen: they are both scavenger-receptor glycoproteins, expressed on normal T-cells and a small subset of normal B-cells [30] and generally highly expressed in CLL and MCL. According to this model, CD6 seems to identify those rare forms of CLL which express low levels of CD5, probably functionally substituting it.
- Some markers which are generally used in a particular setting can have a role in classifying other B-NHL: CD200 and CD23, for example, which are basically used to distinguish CLL from MCL, seem to be differentially expressed in other B-NHL, such as DLBCL or MZL.
4. Materials and Methods
4.1. Flow Cytometry
4.2. Data Collection
4.3. Data Preprocessing
- HCL: Hairy cell leukemia
- SL: Splenic lymphoma/leukemia unclassifiable
- LPL: Lymphoplasmacytic lymphoma
- FCL: Follicular cell lymphoma
- MCL: Mantle cell lymphoma
- BL: Burkitt lymphoma
- CLL: Chronic lymphocytic leukemia, including small lymphocytic lymphoma and high-count CD5+ MBL (CLL-type MBL).
- MZL: Marginal zone lymphoma, including splenic marginal, nodal marginal, and MALT lymphoma.
- DLBCL: Diffuse large B-cell lymphoma, including T-cell/histiocyte-rich large B-cell lymphoma, primary DLBCL of the CNS, primary cutaneous DLBCL, and double/triple hit lymphomas.
4.4. Predictive System
- Maximum depth, with values ranging from 6 to 12;
- Minimum number of samples per split, with values ranging from 2 to 10;
- Minimum number of samples per leaf node, having values in the range 1, 10;
- Maximum number of features criterion (sqrt, log2, or unconstrained);
- Optimal split selection criterion (best or random);
- Split criterion (Gini impurity or information gain).
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Campo, E.; Swerdlow, S.H.; Harris, N.L.; Pileri, S.; Stein, H.; Jaffe, E.S. The 2008 WHO classification of lymphoid neoplasms and beyond: Evolving concepts and practical applications. Blood J. Am. Soc. Hematol. 2011, 117, 5019–5032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrena, S.; Almeida, J.; Del Carmen García-Macias, M.; López, A.; Rasillo, A.; Sayagués, J.M.; Rivas, R.A.; Gutiérrez, M.L.; Ciudad, J.; Flores, T.; et al. Flow cytometry immunophenotyping of fine-needle aspiration specimens: Utility in the diagnosis and classification of non-Hodgkin lymphomas. Histopathology 2011, 58, 906–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demurtas, A.; Accinelli, G.; Pacchioni, D.; Godio, L.; Novero, D.; Bussolati, G.; Palestro, G.; Papotti, M.; Stacchini, A. Utility of flow cytometry immunophenotyping in fine-needle aspirate cytologic diagnosis of non-Hodgkin lymphoma: A series of 252 cases and review of the literature. Appl. Immunohistochem. Mol. Morphol. 2010, 18, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Stetler-Stevenson, M. Flow cytometry in lymphoma diagnosis and prognosis: Useful? Best Pract. Res. Clin. Haematol. 2003, 16, 583–597. [Google Scholar] [CrossRef]
- Morse, E.E.; Yamase, H.T.; Greenberg, B.R.; Sporn, J.; Harshaw, S.A.; Kiraly, T.R.; Ziemba, R.A.; Fallon, M.A. The role of flow cytometry in the diagnosis of lymphoma: A critical analysis. Ann. Clin. Lab. Sci. 1994, 24, 6–11. [Google Scholar]
- Kohli, M.; Prevedello, L.M.; Filice, R.W.; Geis, J.R. Implementing machine learning in radiology practice and research. Am. J. Roentgenol. 2017, 208, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Summers, R.M. Machine learning and radiology. Med. Image Anal. 2012, 16, 933–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Xue, D.; Hu, Z.; Chen, H.; Yao, Y.; Zhang, Y.; Li, M.; Wang, Q.; Xu, N. A Survey for Breast Histopathology Image Analysis Using Classical and Deep Neural Networks. In Proceedings of the International Conference on Information Technologies in Biomedicine, Da Nang, Vietnam, 20–22 February 2019; pp. 222–233. [Google Scholar]
- Bayramoglu, N.; Kannala, J.; Heikkilä, J. Deep learning for magnification independent breast cancer histopathology image classification. In Proceedings of the 2016 23rd International Conference on Pattern Recognition (ICPR), Cancun, Mexico, 4–8 December 2016; pp. 2440–2445. [Google Scholar]
- Xiao, D.; Yu, S.; Vignarajan, J.; An, D.; Tay-Kearney, M.L.; Kanagasingam, Y. Retinal hemorrhage detection by rule-based and machine learning approach. In Proceedings of the 2017 39th Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC), Jeju Island, Korea, 11–15 July 2017; pp. 660–663. [Google Scholar]
- Maji, D.; Santara, A.; Ghosh, S.; Sheet, D.; Mitra, P. Deep neural network and random forest hybrid architecture for learning to detect retinal vessels in fundus images. In Proceedings of the 2015 37th Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC), Milano, Italy, 25–29 August 2015; pp. 3029–3032. [Google Scholar]
- Miotto, R.; Li, L.; Kidd, B.A.; Dudley, J.T. Deep patient: An unsupervised representation to predict the future of patients from the electronic health records. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef]
- O’Neill, K.; Aghaeepour, N.; Špidlen, J.; Brinkman, R. Flow cytometry bioinformatics. PLoS Comput. Biol. 2013, 9. [Google Scholar] [CrossRef] [Green Version]
- Rahim, A.; Meskas, J.; Drissler, S.; Yue, A.; Lorenc, A.; Laing, A.; Saran, N.; White, J.; Abeler-Dörner, L.; Hayday, A.; et al. High throughput automated analysis of big flow cytometry data. Methods 2018, 134, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Angeletti, C. A method for the interpretation of flow cytometry data using genetic algorithms. J. Pathol. Inform. 2018, 9, 16. [Google Scholar] [CrossRef] [PubMed]
- Ko, B.S.; Wang, Y.F.; Li, J.L.; Li, C.C.; Weng, P.F.; Hsu, S.C.; Hou, H.A.; Huang, H.H.; Yao, M.; Lin, C.T.; et al. Clinically validated machine learning algorithm for detecting residual diseases with multicolor flow cytometry analysis in acute myeloid leukemia and myelodysplastic syndrome. EBioMedicine 2018, 37, 91–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bashashati, A.; Lo, K.; Gottardo, R.; Gascoyne, R.D.; Weng, A.; Brinkman, R. A pipeline for automated analysis of flow cytometry data: Preliminary results on lymphoma sub-type diagnosis. In Proceedings of the 2009 Annual International Conference of the IEEE Engineering in Medicine and Biology Society, Minneapolis, MN, USA, 3–6 September 2009; pp. 4945–4948. [Google Scholar]
- Zare, H.; Bashashati, A.; Kridel, R.; Aghaeepour, N.; Haffari, G.; Connors, J.M.; Gascoyne, R.D.; Gupta, A.; Brinkman, R.R.; Weng, A.P. Automated analysis of multidimensional flow cytometry data improves diagnostic accuracy between mantle cell lymphoma and small lymphocytic lymphoma. Am. J. Clin. Pathol. 2012, 137, 75–85. [Google Scholar] [CrossRef]
- Höllein, A.; Zhao, M.; Schabath, R.; Haferlach, T.; Haferlach, C.; Krawitz, P.; Kern, W. An Artificial Intelligence (AI) Approach for Automated Flow Cytometric Diagnosis of B-Cell Lymphoma. Blood 2018, 132, 2856–2856. [Google Scholar] [CrossRef]
- Kern, W.; Elsner, F.; Zhao, M.; Mallesh, N.; Schabath, R.; Haferlach, C.; Krawitz, P.; Lueling, H.; Haferlach, T. An Artificial Neural Network Providing Highly Reliable Decision Support in a Routine Setting for Classification of B-Cell Neoplasms Based on Flow Cytometric Raw Data. Blood 2019. [Google Scholar] [CrossRef]
- Mallesh, N.; Zhao, M.; Elsner, F.; Lueling, H.; Schabath, R.; Haferlach, C.; Haferlach, T.; Krawitz, P.; Kern, W. Knowledge Transfer between Artificial Neural Networks for Different Multicolor Flow Cytometry Protocols Improves Classification Performance for Rare B-Cell Neoplasm Subtypes. Blood 2019. [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Ichinohasama, R.; DeCoteau, J.F.; Myers, J.; Kadin, M.E.; Sawai, T.; Ooya, K. Three-color flow cytometry in the diagnosis of malignant lymphoma based on the comparative cell morphology of lymphoma cells and reactive lymphocytes. Leukemia 1997, 11, 1891–1903. [Google Scholar] [CrossRef] [Green Version]
- Manocha, S.; Matrai, Z.; Osthoff, M.; Carter, A.; Pettitt, A.R. Correlation between cell size and CD38 expression in chronic lymphocytic leukaemia. Leuk. Lymphoma 2003, 44, 797–800. [Google Scholar] [CrossRef]
- Dürig, J.; Schmücker, U.; Dührsen, U. Differential expression of chemokine receptors in B cell malignancies. Leukemia 2001, 15, 752–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulian, P.; Shanafelt, T.D.; Fegan, C.; Zucchetto, A.; Cro, L.; Nückel, H.; Baldini, L.; Kurtova, A.V.; Ferrajoli, A.; Burger, J.A.; et al. CD49d is the strongest flow cytometry–based predictor of overall survival in chronic lymphocytic leukemia. J. Clin. Oncol. 2014, 32, 897. [Google Scholar] [CrossRef] [PubMed]
- Matos, D.; Rizzatti, E.G.; Garcia, A.; Gallo, D.; Falcão, R.P. Adhesion molecule profiles of B-cell non-Hodgkin’s lymphomas in the leukemic phase. Braz. J. Med Biol. Res. 2006, 39, 1349–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, W.G.; Singleton, T.P.; Schnitzer, B.; Ross, C.W.; Stoolman, L.M. Adhesion molecule expression in CD5-negative/CD10-negative chronic B-cell leukemias: Comparison with non-Hodgkin’s lymphomas and CD5-positive B-cell chronic lymphocytic leukemia. Hum. Pathol. 2001, 32, 66–73. [Google Scholar] [CrossRef]
- Osorio, L.M.; De Santiago, A.; Aguilar-SantelisesHå, M.; Mellstedt, K.; Jondal, M. CD6 ligation modulates the Bcl-2/Bax ratio and protects chronic lymphocytic leukemia B cells from apoptosis induced by anti-IgM. Blood J. Am. Soc. Hematol. 1997, 89, 2833–2841. [Google Scholar] [CrossRef] [Green Version]
- Kalogeraki, A.; Tzardi, M.; Panagiotides, I.; Koutsoubi, K.; Bolioti, S.; Rontogianni, D.; Stefanaki, K.; Zois, E.; Karidi, E.; Darivianaki, K.; et al. MIB1 (Ki-67) expression in non-Hodgkin’s lymphomas. Anticancer Res. 1997, 17, 487–491. [Google Scholar]
- Ali, A.E.; Morgen, E.K.; Geddie, W.R.; Boerner, S.L.; Massey, C.; Bailey, D.J.; da Cunha Santos, G. Classifying B-cell non-Hodgkin lymphoma by using MIB-1 proliferative index in fine-needle aspirates. Cancer Cytopathol. 2010, 118, 166–172. [Google Scholar] [CrossRef]
- He, X.; Chen, Z.; Fu, T.; Jin, X.; Yu, T.; Liang, Y.; Zhao, X.; Huang, L. Ki-67 is a valuable prognostic predictor of lymphoma but its utility varies in lymphoma subtypes: Evidence from a systematic meta-analysis. BMC Cancer 2014, 14, 153. [Google Scholar] [CrossRef]
- Pezzella, F.; Tse, A.; Cordell, J.; Pulford, K.; Gatter, K.; Mason, D. Expression of the bcl-2 oncogene protein is not specific for the 14; 18 chromosomal translocation. Am. J. Pathol. 1990, 137, 225. [Google Scholar]
- Nguyen, P.L.; Harris, N.L.; Ritz, J.; Robertson, M.J. Expression of CD95 antigen and Bcl-2 protein in non-Hodgkin’s lymphomas and Hodgkin’s disease. Am. J. Pathol. 1996, 148, 847. [Google Scholar]
- Laane, E.; Tani, E.; Björklund, E.; Elmberger, G.; Everaus, H.; Skoog, L.; Porwit-MacDonald, A. Flow cytometric immunophenotyping including Bcl-2 detection on fine needle aspirates in the diagnosis of reactive lymphadenopathy and non-Hodgkin’s lymphoma. Cytom. Part Clin. Cytom. J. Int. Soc. Anal. Cytol. 2005, 64, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.; Arber, D.; Chang, K.; Wilson, C.; Weiss, L. Frequency of bcl-2 expression in non-Hodgkin’s lymphoma: A study of 778 cases with comparison of marginal zone lymphoma and monocytoid B-cell hyperplasia. Mod. Pathol. 1998, 11, 864–869. [Google Scholar] [PubMed]
- Hermine, O.; Haioun, C.; Lepage, E.; d’Agay, M.F.; Briere, J.; Lavignac, C.; Fillet, G.; Salles, G.; Marolleau, J.P.; Diebold, J.; et al. Prognostic significance of bcl-2 protein expression in aggressive non-Hodgkin’s lymphoma. Groupe d’Etude des Lymphomes de l’Adulte (GELA). Blood 1996, 87, 265–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, N.A.; Slack, G.W.; Savage, K.J.; Connors, J.M.; Ben-Neriah, S.; Rogic, S.; Scott, D.W.; Tan, K.L.; Steidl, C.; Sehn, L.H.; et al. Concurrent expression of MYC and BCL2 in diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J. Clin. Oncol. 2012, 30, 3452. [Google Scholar] [CrossRef] [PubMed]
- Gelman, A.; Hill, J. Data Analysis Using Regression and Multilevel/Hierarchical Models; Cambridge University Press: Cambridge, UK, 2006. [Google Scholar]
- Breiman, L.; Friedman, J.; Stone, C.J.; Olshen, R.A. Classification and Regression Trees; CRC Press: Boca Raton, FL, USA, 1984. [Google Scholar]
- McKinney, W. Pandas: A foundational Python library for data analysis and statistics. Python High Perform. Sci. Comput. 2011, 14, 1–9. [Google Scholar]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Neyman, J. On the two different aspects of the representative method: The method of stratified sampling and the method of purposive selection. In Breakthroughs in Statistics; Springer: New York, NY, USA, 1992; pp. 123–150. [Google Scholar]
- Bengio, Y.; Grandvalet, Y. No unbiased estimator of the variance of k-fold cross-validation. J. Mach. Learn. Res. 2004, 5, 1089–1105. [Google Scholar]
- Tenace, V.; Calimera, A. Quasi-exact logic functions through classification trees. Integration 2018, 63, 248–255. [Google Scholar] [CrossRef]
- Breiman, L. Some properties of splitting criteria. Mach. Learn. 1996, 24, 41–47. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, J.R. Induction of decision trees. Mach. Learn. 1986, 1, 81–106. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| B-NHL | Accuracy | Sensitivity | Specificity | PPV | NPV |
|---|---|---|---|---|---|
| Model I | |||||
| BL | 99.45 | 66.67 | 99.73 | 66.67 | 99.73 |
| CLL | 95.90 | 97.01 | 94.96 | 94.21 | 97.42 |
| DLBCL | 96.18 | 80.00 | 99.04 | 93.62 | 96.56 |
| FCL | 96.45 | 86.00 | 98.10 | 87.76 | 97.79 |
| HCL | 99.45 | 83.32 | 99.71 | 83.32 | 99.71 |
| LPL | 96.45 | 46.67 | 98.57 | 58.32 | 97.75 |
| MCL | 97.81 | 85.70 | 98.54 | 78.26 | 99.12 |
| MZL | 94.28 | 84.09 | 95.67 | 72.54 | 97.78 |
| SL | 99.45 | 60.00 | 100.00 | 100.00 | 99.45 |
| Model II | |||||
| BL | 99.43 | 75.00 | 99.71 | 75.00 | 99.71 |
| CLL | 98.87 | 98.20 | 99.46 | 99.39 | 98.42 |
| DLBCL | 95.76 | 87.26 | 97.32 | 85.70 | 97.65 |
| FCL | 95.76 | 82.00 | 98.03 | 87.23 | 97.07 |
| LPL | 98.87 | 86.67 | 99.40 | 86.67 | 99.40 |
| MCL | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 |
| MZL | 96.62 | 90.70 | 97.43 | 82.98 | 98.70 |
| Model III | |||||
| BL | 99.15 | 50.00 | 99.71 | 66.67 | 99.43 |
| CLL | 96.06 | 96.40 | 95.73 | 95.26 | 96.76 |
| DLBCL | 95.76 | 81.81 | 98.32 | 90.00 | 96.71 |
| FCL | 96.34 | 88.00 | 97.70 | 86.26 | 98.03 |
| LPL | 98.03 | 53.32 | 100.00 | 100.00 | 97.98 |
| MCL | 98.03 | 95.23 | 98.20 | 76.92 | 99.70 |
| MZL | 95.20 | 86.04 | 96.46 | 77.07 | 98.04 |
| Model IV | |||||
| BL | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 |
| CLL | 99.26 | 94.12 | 100.00 | 100.00 | 99.17 |
| DLBCL | 92.70 | 89.79 | 94.31 | 89.79 | 94.31 |
| FCL | 94.89 | 92.31 | 95.92 | 90.00 | 96.90 |
| LPL | 98.54 | 100.00 | 98.51 | 50.00 | 100.00 |
| MCL | 98.54 | 87.50 | 99.21 | 87.50 | 99.21 |
| MZL | 91.23 | 65.00 | 95.73 | 72.21 | 94.12 |
| B-NHL Category | Total Samples | Blood Samples | Non-Blood Samples |
|---|---|---|---|
| CLL | 670 | 602 | 68 |
| FCL | 199 | 43 | 156 |
| SL | 19 | 17 | 2 |
| MZL | 174 | 94 | 80 |
| DLBCL | 220 | 25 | 195 |
| LPL | 60 | 53 | 7 |
| MCL | 83 | 51 | 32 |
| HCL | 26 | 26 | 0 |
| BL | 14 | 4 | 10 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaidano, V.; Tenace, V.; Santoro, N.; Varvello, S.; Cignetti, A.; Prato, G.; Saglio, G.; De Rosa, G.; Geuna, M. A Clinically Applicable Approach to the Classification of B-Cell Non-Hodgkin Lymphomas with Flow Cytometry and Machine Learning. Cancers 2020, 12, 1684. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061684
Gaidano V, Tenace V, Santoro N, Varvello S, Cignetti A, Prato G, Saglio G, De Rosa G, Geuna M. A Clinically Applicable Approach to the Classification of B-Cell Non-Hodgkin Lymphomas with Flow Cytometry and Machine Learning. Cancers. 2020; 12(6):1684. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061684
Chicago/Turabian StyleGaidano, Valentina, Valerio Tenace, Nathalie Santoro, Silvia Varvello, Alessandro Cignetti, Giuseppina Prato, Giuseppe Saglio, Giovanni De Rosa, and Massimo Geuna. 2020. "A Clinically Applicable Approach to the Classification of B-Cell Non-Hodgkin Lymphomas with Flow Cytometry and Machine Learning" Cancers 12, no. 6: 1684. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061684