Influence of Fibroblasts on Mammary Gland Development, Breast Cancer Microenvironment Remodeling, and Cancer Cell Dissemination

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
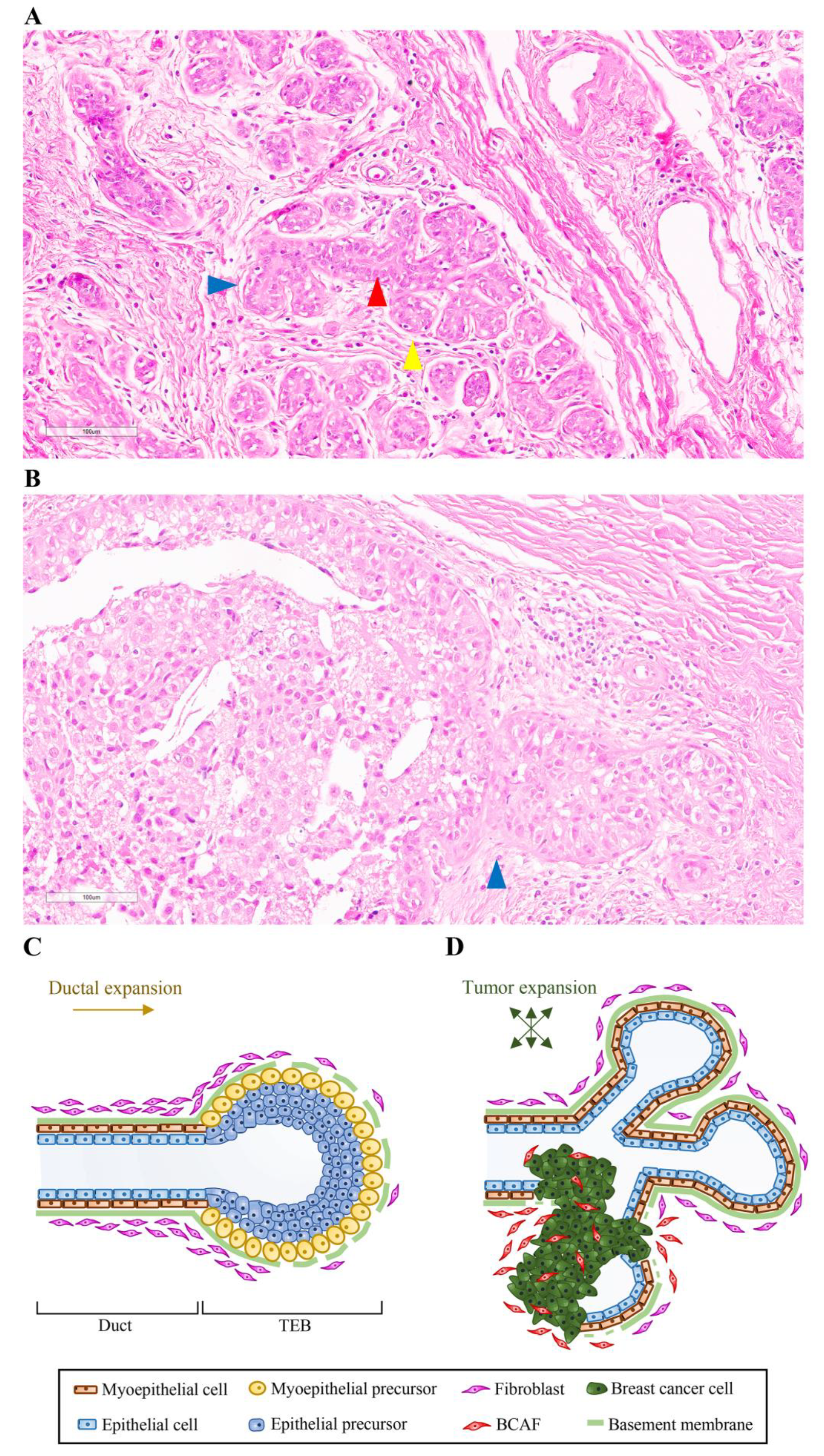
2. Mammary Gland Structure and Development
Influence of Fibroblasts on Mammary Gland Development
3. Hallmarks of Metastatic BCM
4. Influence of Epithelial-Mesenchymal Transition and Autophagy on BC Metastasis and Dissemination
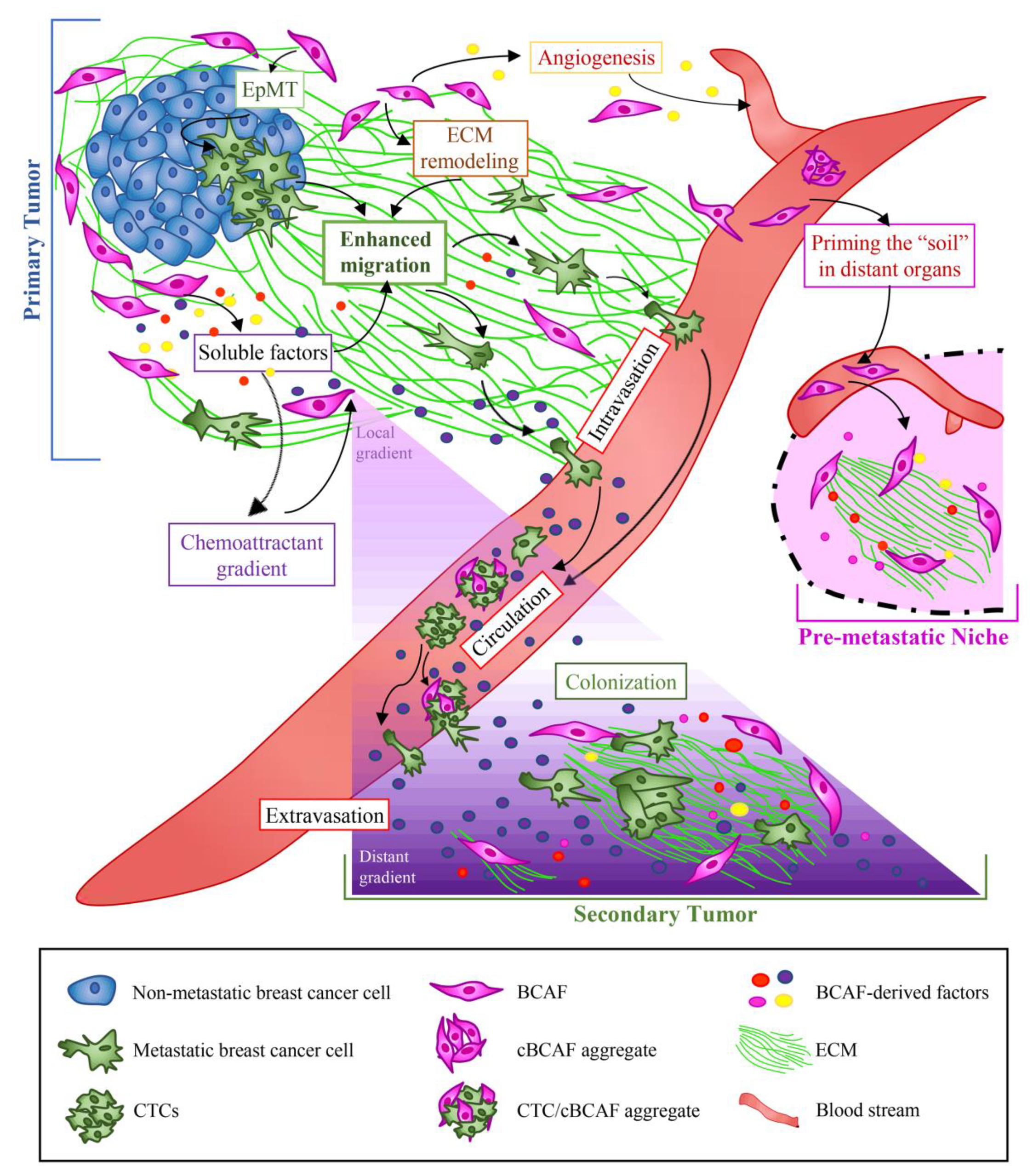
5. Role of BCAFs in BC Cell Dissemination and Metastasis
5.1. Primary, Circulating, and Metastatic BCAFs: The Onset of the Metastatic Voyage
5.2. Role of BCAFs in BC Metastasis through ECM Remodeling
5.3. Role of BCAFs in BC Metastasis via Induction of Angiogenesis
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wolfson, B.; Zhang, Y.; Gernapudi, R.; Duru, N.; Yao, Y.; Lo, P.-K.; Zhou, Q. A High-Fat Diet Promotes Mammary Gland Myofibroblast Differentiation through MicroRNA 140 Downregulation. Mol. Cell. Biol. 2017, 37. [Google Scholar] [CrossRef] [Green Version]
- Koledova, Z. 3D coculture of mammary organoids with fibrospheres: A model for studying epithelial–stromal interactions during mammary branching morphogenesis. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2017; Volume 1612, pp. 107–124. [Google Scholar]
- Polyak, K.; Kalluri, R. The role of the microenvironment in mammary gland development and cancer. Cold Spring Harb. Perspect. Biol. 2010, 2, a003244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jena, M.K.; Jaswal, S.; Kumar, S.; Mohanty, A.K. Molecular mechanism of mammary gland involution: An update. Dev. Biol. 2019, 445, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Jena, M.K.; Mohanty, A.K. New insights of mammary gland during different stages of development. Asian J. Pharm. Clin. Res. 2017, 10, 35–40. [Google Scholar] [CrossRef] [Green Version]
- Monkkonen, T.; Lewis, M.T. New paradigms for the Hedgehog signaling network in mammary gland development and breast Cancer. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 315–332. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Oh, W.; Ulu, A.; Frost, J.A. Minireview: Mouse models of Rho GTPase function in mammary gland development, tumorigenesis, and metastasis. Mol. Endocrinol. 2016, 30, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Macias, H.; Hinck, L. Mammary gland development. Wiley Interdiscip. Rev. Dev. Biol. 2012, 1, 533–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koledova, Z.; Zhang, X.; Streuli, C.; Clarke, R.B.; Klein, O.D.; Werb, Z.; Lu, P. SPRY1 regulates mammary epithelial morphogenesis by modulating EGFR-dependent stromal paracrine signaling and ECM remodeling. Proc. Natl. Acad. Sci. USA 2016, 113, E5731–E5740. [Google Scholar] [CrossRef] [Green Version]
- Inman, J.L.; Robertson, C.; Mott, J.D.; Bissell, M.J. Mammary gland development: Cell fate specification, stem cells and the microenvironment. Development 2015, 142, 1028–1042. [Google Scholar] [CrossRef] [Green Version]
- Lühr, I.; Friedl, A.; Overath, T.; Tholey, A.; Kunze, T.; Hilpert, F.; Sebens, S.; Arnold, N.; Rösel, F.; Oberg, H.H.; et al. Mammary fibroblasts regulate morphogenesis of normal and tumorigenic breast epithelial cells by mechanical and paracrine signals. Cancer Lett. 2012, 325, 175–188. [Google Scholar] [CrossRef] [Green Version]
- Ruocco, M.R.; Avagliano, A.; Granato, G.; Imparato, V.; Masone, S.; Masullo, M.; Nasso, R.; Montagnani, S.; Arcucci, A. Involvement of Breast Cancer-Associated Fibroblasts in Tumor Development, Therapy Resistance and Evaluation of Potential Therapeutic Strategies. Curr. Med. Chem. 2018, 25, 3414–3434. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, S.K.; Srivastava, S.K.; Poosarla, T.; Dyess, D.L.; Holliday, N.P.; Singh, A.P.; Singh, S. Inflammation, immunosuppressive microenvironment and breast cancer: Opportunities for cancer prevention and therapy. Ann. Transl. Med. 2019, 7, 593. [Google Scholar] [CrossRef] [PubMed]
- Avagliano, A.; Ruocco, M.R.; Aliotta, F.; Belviso, I.; Accurso, A.; Masone, S.; Montagnani, S.; Arcucci, A. Mitochondrial Flexibility of Breast Cancers: A Growth Advantage and a Therapeutic Opportunity. Cells 2019, 8, 401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, B.S.; Sarnella, A.; D’Avino, G.; Zannetti, A. Recruitment of stromal cells into tumour microenvironment promote the metastatic spread of breast cancer. Semin. Cancer Biol. 2020, 60, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Li, J.; Zhu, S.; Wu, J.; Chen, C.; Liu, Q.; Wei, W.; Zhang, Y.; Sun, S. Breast cancer subtypes predict the preferential site of distant metastases: A SEER based study. Oncotarget 2017, 8, 27990–27996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makki, J. Diversity of breast carcinoma: Histological subtypes and clinical relevance. Clin. Med. Insights Pathol. 2015, 8, 23–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avagliano, A.; Granato, G.; Ruocco, M.R.; Romano, V.; Belviso, I.; Carfora, A.; Montagnani, S.; Arcucci, A. Metabolic Reprogramming of Cancer Associated Fibroblasts: The Slavery of Stromal Fibroblasts. Biomed. Res. Int. 2018, 2018, 6075403. [Google Scholar] [CrossRef] [Green Version]
- Da Cunha, B.R.; Domingos, C.; Buzzo Stefanini, A.C.; Henrique, T.; Polachini, G.M.; Castelo-Branco, P.; Tajara, E.H. Cellular interactions in the tumor microenvironment: The role of secretome. J. Cancer 2019, 10, 4574–4587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cacho-Díaz, B.; García-Botello, D.R.; Wegman-Ostrosky, T.; Reyes-Soto, G.; Ortiz-Sánchez, E.; Herrera-Montalvo, L.A. Tumor microenvironment differences between primary tumor and brain metastases. J. Transl. Med. 2020, 18, 1. [Google Scholar] [CrossRef] [Green Version]
- Arcucci, A.; Ruocco, M.R.; Granato, G.; Sacco, A.M.; Montagnani, S. Cancer: An Oxidative Crosstalk Between Solid Tumor Cells and Cancer Associated Fibroblasts. Biomed. Res. Int. 2016; 201. [Google Scholar]
- Tashireva, L.A.; Denisov, E.V.; Gerashchenko, T.S.; Pautova, D.N.; Buldakov, M.A.; Zavyalova, M.V.; Kzhyshkowska, J.; Cherdyntseva, N.V.; Perelmuter, V.M. Intratumoral heterogeneity of macrophages and fibroblasts in breast cancer is associated with the morphological diversity of tumor cells and contributes to lymph node metastasis. Immunobiology 2017, 222, 631–640. [Google Scholar] [CrossRef]
- Tower, H.; Ruppert, M.; Britt, K. The immune microenvironment of breast cancer progression. Cancers 2019, 11, 1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Hao, Q.; Cao, W.; Vadgama, J.V.; Wu, Y. Celecoxib in breast cancer prevention and therapy. Cancer Manag. Res. 2018, 10, 4653–4667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ao, Z.; Shah, S.H.; Machlin, L.M.; Parajuli, R.; Miller, P.C.; Rawal, S.; Williams, A.J.; Cote, R.J.; Lippman, M.E.; Datar, R.H.; et al. Identification of cancer-associated fibroblasts in circulating blood from patients with metastatic breast cancer. Cancer Res. 2015, 75, 4681–4687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigelt, B.; Peterse, J.L.; Van’t Veer, L.J. Breast cancer metastasis: Markers and models. Nat. Rev. Cancer 2005, 5, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Pellacani, D.; Tan, S.; Lefort, S.; Eaves, C.J. Transcriptional regulation of normal human mammary cell heterogeneity and its perturbation in breast cancer. EMBO J. 2019, 38, e100330. [Google Scholar] [CrossRef] [PubMed]
- Conklin, M.W.; Keely, P.J. Why the stroma matters in breast cancer: Insights into breast cancer patient outcomes through the examination of stromal biomarkers. Cell Adhes. Migr. 2012, 6, 249–260. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, S.; Pickup, M.W.; Weaver, V.M. From transformation to metastasis: Deconstructing the extracellular matrix in breast cancer. Cancer Metastasis Rev. 2016, 35, 655–667. [Google Scholar] [CrossRef]
- Brownfield, D.G.; Venugopalan, G.; Lo, A.; Mori, H.; Tanner, K.; Fletcher, D.A.; Bissell, M.J. Patterned collagen fibers orient branching mammary epithelium through distinct signaling modules. Curr. Biol. 2013, 23, 703–709. [Google Scholar] [CrossRef] [Green Version]
- Ingman, W.V.; Wyckoff, J.; Gouon-Evans, V.; Condeelis, J.; Pollard, J.W. Macrophages promote collagen fibrillogenesis around terminal end buds of the developing mammary gland. Dev. Dyn. 2006, 235, 3222–3229. [Google Scholar] [CrossRef]
- Peuhu, E.; Kaukonen, R.; Lerche, M.; Saari, M.; Guzmán, C.; Rantakari, P.; De Franceschi, N.; Wärri, A.; Georgiadou, M.; Jacquemet, G.; et al. SHARPIN regulates collagen architecture and ductal outgrowth in the developing mouse mammary gland. EMBO J. 2017, 36, 165–182. [Google Scholar] [CrossRef]
- Cichon, M.A.; Degnim, A.C.; Visscher, D.W.; Radisky, D.C. Microenvironmental influences that drive progression from benign breast disease to invasive breast cancer. J. Mammary Gland Biol. Neoplasia 2010, 15, 389–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewald, A.J.; Brenot, A.; Duong, M.; Chan, B.S.; Werb, Z. Collective Epithelial Migration and Cell Rearrangements Drive Mammary Branching Morphogenesis. Dev. Cell 2008, 14, 570–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paine, I.S.; Lewis, M.T. The Terminal End Bud: The Little Engine that Could. J. Mammary Gland Biol. Neoplasia 2017, 22, 93–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCave, E.J.; Cass, C.A.P.; Burg, K.J.L.; Booth, B.W. The normal microenvironment directs mammary gland development. J. Mammary Gland Biol. Neoplasia 2010, 15, 291–299. [Google Scholar] [CrossRef]
- Mallepell, S.; Krust, A.; Chambon, P.; Brisken, C. Paracrine signaling through the epithelial estrogen receptor α is required for proliferation and morphogenesis in the mammary gland. Proc. Natl. Acad. Sci. USA 2006, 103, 2196–2201. [Google Scholar] [CrossRef] [Green Version]
- Brisken, C.; Park, S.; Vass, T.; Lydon, J.P.; O’Malley, B.W.; Weinberg, R.A. A paracrine role for the epithelial progesterone receptor in mammary gland development. Proc. Natl. Acad. Sci. USA 1998, 95, 5076–5081. [Google Scholar] [CrossRef] [Green Version]
- Russo, J.; Russo, I.H. Development of the human breast. Maturitas 2004, 49, 2–15. [Google Scholar] [CrossRef]
- Morsing, M.; Klitgaard, M.C.; Jafari, A.; Villadsen, R.; Kassem, M.; Petersen, O.W.; Rønnov-Jessen, L. Evidence of two distinct functionally specialized fibroblast lineages in breast stroma. Breast Cancer Res. 2016, 18, 108. [Google Scholar] [CrossRef] [Green Version]
- Hutson, S.W.; Cowen, P.N.; Bird, C.C. Morphometric studies of age related changes in normal human breast and their significance for evolution of mammary cancer. J. Clin. Pathol. 1985, 38, 281–287. [Google Scholar] [CrossRef] [Green Version]
- Henson, D.E.; Tarone, R.E. On the Possible Role of Involution in the Natural History of Breast Cancer. Cancer 1993, 7, 2154–2156. [Google Scholar] [CrossRef]
- Figueroa, J.D.; Pfeiffer, R.M.; Patel, D.A.; Linville, L.; Brinton, L.A.; Gierach, G.L.; Yang, X.R.; Papathomas, D.; Visscher, D.; Mies, C.; et al. Terminal Duct Lobular Unit Involution of the Normal Breast: Implications for Breast Cancer Etiology. J. Natl. Cancer Inst. 2014, 106, dju286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milanese, T.R.; Hartmann, L.C.; Sellers, T.A.; Frost, M.H.; Vierkant, R.A.; Maloney, S.D.; Pankratz, V.S.; Degnim, A.C.; Vachon, C.M.; Reynolds, C.A.; et al. Age-related lobular involution and risk of breast cancer. J. Natl. Cancer Inst. 2006, 98, 1600–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, C.J.; Khaled, W.T. Mammary development in the embryo and adult: A journey of morphogenesis and commitment. Development 2008, 135, 995–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rybinski, B.; Franco-Barraza, J.; Cukierman, E. The wound healing, chronic fibrosis, and cancer progression triad. Physiol. Genom. 2014, 46, 223–244. [Google Scholar] [CrossRef] [PubMed]
- DeClerck, Y.A. Desmoplasia: A response or a niche? Cancer Discov. 2012, 2, 772–774. [Google Scholar] [CrossRef] [Green Version]
- Seo, B.R.; Bhardwaj, P.; Choi, S.; Gonzalez, J.; Eguiluz, R.C.A.; Wang, K.; Mohanan, S.; Morris, P.G.; Du, B.; Zhou, X.K.; et al. Obesity-dependent changes in interstitial ECM mechanics promote breast tumorigenesis. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Scherer, P.E. Adipocyte-derived endotrophin promotes malignant tumor progression. J. Clin. Invest. 2012, 122, 4243–4256. [Google Scholar] [CrossRef] [Green Version]
- Kojima, Y.; Acar, A.; Eaton, E.N.; Mellody, K.T.; Scheel, C.; Ben-Porath, I.; Onder, T.T.; Wang, Z.C.; Richardson, A.L.; Weinberg, R.A.; et al. Autocrine TGF-β and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc. Natl. Acad. Sci. USA 2010, 107, 20009–20014. [Google Scholar] [CrossRef] [Green Version]
- Unsworth, A.; Anderson, R.; Britt, K. Stromal fibroblasts and the immune microenvironment: Partners in mammary gland biology and pathology? J. Mammary Gland Biol. Neoplasia 2014, 19, 169–182. [Google Scholar] [CrossRef]
- Jackson, D.; Bresnick, J.; Rosewell, I.; Crafton, T.; Poulsom, R.; Stamp, G.; Dickson, C. Fibroblast growth factor receptor signalling has a role in lobuloalveolar development of the mammary gland. J. Cell Sci. 1997, 110, 1261–1268. [Google Scholar]
- Schedin, P.; Mitrenga, T.; McDaniel, S.; Kaeck, M. Mammary ECM Composition and function are altered by reproductive state. Mol. Carcinog. 2004, 41, 207–220. [Google Scholar] [CrossRef]
- Talhouk, R.S.; Bissell, M.J.; Werb, Z. Coordinated expression of extracellular matrix-degrading proteinases and their inhibitors regulates mammary epithelial function during involution. J. Cell Biol. 1992, 118, 1271–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.J.; Jung, H.S.; Lu, P. Pleiotropic functions of fibroblast growth factor signaling in embryonic mammary gland development. J. Mammary Gland Biol. Neoplasia 2013, 18, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Soriano, J.V.; Pepper, M.S.; Orci, L.; Montesano, R. Roles of Hepatocyte Growth Factor/Scatter Factor and Transforming Growth Factor-β1 in Mammary Gland Ductal Morphogenesis. J. Mammary Gland Biol. Neoplasia 1998, 3, 133–150. [Google Scholar] [CrossRef]
- Pond, A.C.; Bin, X.; Batts, T.; Roarty, K.; Hilsenbeck, S.; Rosen, J.M. Fibroblast growth factor receptor signaling is essential for normal mammary gland development and stem cell function. Stem Cells 2013, 31, 178–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, P.; Ewald, A.J.; Martin, G.R.; Werb, Z. Genetic mosaic analysis reveals FGF receptor 2 function in terminal end buds during mammary gland branching morphogenesis. Dev. Biol. 2008, 321, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Xian, W.; Schwertfeger, K.L.; Vargo-Gogola, T.; Rosen, J.M. Pleiotropic effects of FGFR1 on cell proliferation, survival, and migration in a 3D mammary epithelial cell model. J. Cell Biol. 2005, 171, 663–673. [Google Scholar] [CrossRef]
- Sumbal, J.; Koledova, Z. FGF signaling in mammary gland fibroblasts regulates multiple fibroblast functions and mammary epithelial morphogenesis. Development 2019, 146. [Google Scholar] [CrossRef]
- Sternlicht, M.D.; Kouros-Mehr, H.; Lu, P.; Werb, Z. Hormonal and local control of mammary branching morphogenesis. Differentiation 2006, 74, 365–381. [Google Scholar] [CrossRef] [Green Version]
- Kawazoe, T.; Taniguchi, K. The Sprouty/Spred family as tumor suppressors: Coming of age. Cancer Sci. 2019, 110, 1525–1535. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Qiao, G.; Lu, P. Modulation of fibroblast growth factor signaling is essential for mammary epithelial morphogenesis. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, T.; Yao, L.; Han, Y.; Hao, P.; Lu, P. Quantitative phosphoproteomics reveals system-wide phosphorylation network altered by spry in mouse mammary stromal fibroblasts. Int. J. Mol. Sci. 2019, 20, 5400. [Google Scholar] [CrossRef] [PubMed]
- Maennling, A.E.; Tur, M.K.; Niebert, M.; Klockenbring, T.; Zeppernick, F.; Gattenlöhner, S.; Meinhold-Heerlein, I.; Hussain, A.F. Molecular targeting therapy against egfr family in breast cancer: Progress and future potentials. Cancers 2019, 11, 5400. [Google Scholar] [CrossRef] [Green Version]
- Andrae, J.; Gallini, R.; Betsholtz, C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008, 22, 1276–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammer, A.M.; Sizemore, G.M.; Shukla, V.C.; Avendano, A.; Sizemore, S.T.; Chang, J.J.; Kladney, R.D.; Cuitiño, M.C.; Thies, K.A.; Verfurth, Q.; et al. Stromal PDGFR-α Activation Enhances Matrix Stiffness, Impedes Mammary Ductal Development, and Accelerates Tumor Growth. Neoplasia 2017, 19, 496–508. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Minnier, J.; Burchard, J.; Chiotti, K.; Spellman, P.; Schedin, P. Physiologically activated mammary fibroblasts promote postpartum mammary cancer. JCI Insight 2017, 2, e89206. [Google Scholar] [CrossRef]
- Tian, Z.; Tang, J.; Yang, Q.; Li, X.; Zhu, J.; Wu, G. Atypical ubiquitin-binding protein SHARPIN promotes breast cancer progression. Biomed. Pharmacother. 2019, 119, 109414. [Google Scholar] [CrossRef]
- Kasirer-Friede, A.; Tjahjono, W.; Eto, K.; Shattil, S.J. SHARPIN at the nexus of integrin, immune, and inflammatory signaling in human platelets. Proc. Natl. Acad. Sci. USA 2019, 116, 4983–4988. [Google Scholar] [CrossRef] [Green Version]
- Pouwels, J.; DeFranceschi, N.; Rantakari, P.; Auvinen, K.; Karikoski, M.; Mattila, E.; Potter, C.; Sundberg, J.P.; Hogg, N.; Gahmberg, C.G.; et al. SHARPIN regulates uropod detachment in migrating lymphocytes. Cell Rep. 2013, 5, 619–628. [Google Scholar] [CrossRef]
- Rantala, J.K.; Pouwels, J.; Pellinen, T.; Veltel, S.; Laasola, P.; Mattila, E.; Potter, C.S.; Duffy, T.; Sundberg, J.P.; Kallioniemi, O.; et al. SHARPIN is an endogenous inhibitor of β1-integrin activation. Nat. Cell Biol. 2011, 13, 1315–1324. [Google Scholar] [CrossRef] [Green Version]
- Peuhu, E.; Salomaa, S.I.; De Franceschi, N.; Potter, C.S.; Sundberg, J.P.; Pouwels, J. Integrin beta 1 inhibition alleviates the chronic hyperproliferative dermatitis phenotype of SHARPIN-deficient mice. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerche, M.; Elosegui-Artola, A.; Kechagia, J.Z.; Guzmán, C.; Georgiadou, M.; Andreu, I.; Gullberg, D.; Roca-Cusachs, P.; Peuhu, E.; Ivaska, J. Integrin Binding Dynamics Modulate Ligand-Specific Mechanosensing in Mammary Gland Fibroblasts. iScience 2020, 23, 100907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deome, K.B.; Faulkin, L.J., Jr.; Bern, H.A.; Blair, P.B. Development of Mammary Tumors from Hyperplastic Alveolar Nodules Transplanted Into Gland-Free Mammary Fat Pads of Female C3H Mice. Cancer Res. 1959, 19, 515–520. [Google Scholar]
- Kuperwasser, C.; Chavarria, T.; Wu, M.; Magrane, G.; Gray, J.W.; Carey, L.; Richardson, A.; Weinberg, R.A. Reconstruction of functionally normal and malignant human breast tissues in mice. Proc. Natl. Acad. Sci. USA 2004, 101, 4966–4971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Egeblad, M.; Rasch, M.G.; Weaver, V.M. Dynamic interplay between the collagen scaffold and tumor evolution. Curr. Opin. Cell Biol. 2010, 22, 697–706. [Google Scholar] [CrossRef] [Green Version]
- Coleman, W.B. Breast Ductal Carcinoma in Situ: Precursor to Invasive Breast Cancer. Am. J. Pathol. 2019, 189, 942–945. [Google Scholar] [CrossRef] [Green Version]
- Eiro, N.; Gonzalez, L.O.; Fraile, M.; Cid, S.; Schneider, J.; Vizoso, F.J. Breast cancer tumor stroma: Cellular components, phenotypic heterogeneity, intercellular communication, prognostic implications and therapeutic opportunities. Cancers 2019, 11, 664. [Google Scholar] [CrossRef] [Green Version]
- Sappino, A.P.; Skalli, O.; Jackson, B.; Schürch, W.; Gabbiani, G. Smooth-muscle differentiation in stromal cells of malignant and non-malignant breast tissues. Int. J. Cancer 1988, 41, 707–712. [Google Scholar] [CrossRef]
- Rønnov-Jessen, L.; Petersen, O.W.; Koteliansky, V.E.; Bissell, M.J. The origin of the myofibroblasts in breast cancer: Recapitulation of tumor environment in culture unravels diversity and implicates converted fibroblasts and recruited smooth muscle cells. J. Clin. Invest. 1995, 95, 859–873. [Google Scholar] [CrossRef] [Green Version]
- Vaughan, M.B.; Howard, E.W.; Tomasek, J.J. Transforming growth factor-β1 promotes the morphological and functional differentiation of the myofibroblast. Exp. Cell Res. 2000, 257, 180–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuelten, C.H.; Busch, J.I.; Tang, B.; Flanders, K.C.; Oshima, A.; Sutton, E.; Karpova, T.S.; Roberts, A.B.; Wakefield, L.M.; Niederhuber, J.E. Transient tumor-fibroblast interactions increase tumor cell malignancy by a TGF-b Mediated Mechanism in a Mouse Xenograft Model of Breast Cancer. PLoS ONE 2010, 5, e9832. [Google Scholar] [CrossRef] [PubMed]
- Arcucci, A.; Ruocco, M.R.; Albano, F.; Granato, G.; Romano, V.; Corso, G.; Bancone, C.; De Vendittis, E.; Della Corte, A.; Montagnani, S. Analysis of extracellular superoxide dismutase and Akt in ascending aortic aneurysm with tricuspid or bicuspid aortic valve. Eur. J. Histochem. 2014, 58, 200–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arcucci, A.; Ruocco, M.R.; Amatruda, N.; Riccio, A.; Tarantino, G.; Albano, F.; Mele, V.; Montagnani, S. Analysis of Extracellular Superoxide Dismutase in Fibroblasts from Patients With Systemic Sclerosis. J. Biol. Regul. Homeost. Agents 2011, 25, 647–654. [Google Scholar]
- Denicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–110. [Google Scholar] [CrossRef]
- Wilmanski, T.; Zhou, X.; Zheng, W.; Shinde, A.; Donkin, S.S.; Wendt, M.; Burgess, J.R.; Teegarden, D. Inhibition of pyruvate carboxylase by 1α,25-dihydroxyvitamin D promotes oxidative stress in early breast cancer progression. Cancer Lett. 2017, 411, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-β signaling in fibrosis. Growth Factors 2011, 29, 196–202. [Google Scholar] [CrossRef] [Green Version]
- Casey, T.M.; Eneman, J.; Crocker, A.; White, J.; Tessitore, J.; Stanley, M.; Harlow, S.; Bunn, J.Y.; Weaver, D.; Muss, H.; et al. Cancer associated fibroblasts stimulated by transforming growth factor beta1 (TGF-β1) increase invasion rate of tumor cells: A population study. Breast Cancer Res. Treat. 2008, 110, 39–49. [Google Scholar] [CrossRef]
- Shangguan, L.; Ti, X.; Krause, U.; Hai, B.; Zhao, Y.; Yang, Z.; Liu, F. Inhibition of TGF-β/Smad signaling by BAMBI blocks differentiation of human mesenchymal stem cells to carcinoma-associated fibroblasts and abolishes their protumor effects. Stem Cells 2012, 30, 2810–2819. [Google Scholar] [CrossRef]
- Goulet, C.R.; Bernard, G.; Tremblay, S.; Chabaud, S.; Bolduc, S.; Pouliot, F. Exosomes induce fibroblast differentiation into cancer-associated fibroblasts through TGFb signaling. Mol. Cancer Res. 2018, 16, 1196–1204. [Google Scholar] [CrossRef] [Green Version]
- Hinz, B.; Celetta, G.; Tomasek, J.J.; Gabbiani, G.; Chaponnier, C. Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol. Biol. Cell 2001, 12, 2730–2741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, A.; Gu, F.; Guo, X.; Zhang, X.; Fu, L. Breast cancer-associated fibroblasts: Their roles in tumor initiation, progression and clinical applications. Front. Med. 2016, 10, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Buchsbaum, R.J.; Oh, S.Y. Breast cancer-associated fibroblasts: Where we are and where we need to go. Cancers 2016, 8, 19. [Google Scholar] [CrossRef] [Green Version]
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef]
- Pantel, K.; Brakenhoff, R.H. Dissecting the metastatic cascade. Nat. Rev. Cancer 2004, 4, 448–456. [Google Scholar] [CrossRef]
- Kalluri, R. EMT: When epithelial cells decide to become mesenchymal-like cells. J. Clin. Invest. 2009, 119, 1417–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yilmaz, M.; Christofori, G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009, 28, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Melzer, C.; von der Ohe, J.; Hass, R. Breast Carcinoma: From Initial Tumor Cell Detachment to Settlement at Secondary Sites. Biomed. Res. Int. 2017, 2017, 8534371. [Google Scholar] [CrossRef]
- Jain, R.K.; Carmeliet, P. SnapShot: Tumor angiogenesis. Cell 2012, 149. [Google Scholar] [CrossRef] [Green Version]
- Raica, M.; Ribatti, D. Targeting Tumor Lymphangiogenesis: An Update. Curr. Med. Chem. 2010, 17, 698–708. [Google Scholar] [CrossRef]
- Alitalo, A.; Detmar, M. Interaction of tumor cells and lymphatic vessels in cancer progression. Oncogene 2012, 31, 4499–4508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginter, P.S.; Karagiannis, G.S.; Entenberg, D.; Lin, Y.; Condeelis, J.; Jones, J.G.; Oktay, M.H. Tumor microenvironment of metastasis (TMEM) doorways are restricted to the blood vessel endothelium in both primary breast cancers and their lymph node metastases. Cancers 2019, 11, 1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karagiannis, G.S.; Condeelis, J.S.; Oktay, M.H. Chemotherapy-induced metastasis: Mechanisms and translational opportunities. Clin. Exp. Metastasis 2018, 35, 269–284. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, G.S.; Goswami, S.; Jones, J.G.; Oktay, M.H.; Condeelis, J.S. Signatures of breast cancer metastasis at a glance. J. Cell Sci. 2016, 129, 1751–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oktay, M.H.; Jones, J.G. TMEM: A novel breast cancer dissemination marker for the assessment of metastatic risk. Biomark. Med. 2015, 9, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Gertler, F.; Condeelis, J. Metastasis: Tumor cells becoming MENAcing. Trends Cell Biol. 2011, 21, 81–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harney, A.S.; Arwert, E.N.; Entenberg, D.; Wang, Y.; Guo, P.; Qian, B.Z.; Oktay, M.H.; Pollard, J.W.; Jones, J.G.; Condeelis, J.S. Real-time imaging reveals local, transient vascular permeability, and tumor cell intravasation stimulated by TIE2hi macrophage–derived VEGFA. Cancer Discov. 2015, 5, 932–943. [Google Scholar] [CrossRef] [Green Version]
- Sparano, J.A.; Gray, R.; Oktay, M.H.; Entenberg, D.; Rohan, T.; Xue, X.; Donovan, M.; Peterson, M.; Shuber, A.; Hamilton, D.A.; et al. A metastasis biomarker (MetaSite BreastTM Score) is associated with distant recurrence in hormone receptor-positive, HER2-negative early-stage breast cancer. NPJ Breast Cancer 2017, 3. [Google Scholar] [CrossRef] [Green Version]
- Walker, C.; Mojares, E.; Del Río Hernández, A. Role of extracellular matrix in development and cancer progression. Int. J. Mol. Sci. 2018, 19, 3028. [Google Scholar] [CrossRef] [Green Version]
- Tamimi, S.O.; Ahmed, A. Stromal changes in invasive breast carcinoma: An ultrastructural study. J. Pathol. 1987, 153, 163–170. [Google Scholar] [CrossRef]
- Insua-Rodríguez, J.; Oskarsson, T. The extracellular matrix in breast cancer. Adv. Drug Deliv. Rev. 2016, 97, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.K.; Huang, S.; Lu, W.; Lev, D.C.; Price, J.E. Vascular endothelial growth factor expression promotes the growth of breast cancer brain metastases in nude mice. Clin. Exp. Metastasis 2004, 21, 107–118. [Google Scholar] [CrossRef]
- Nduom, E.K.; Yang, C.; Merrill, M.J.; Zhuang, Z.; Lonser, R.R. Characterization of the blood-brain barrier of metastatic and primary malignant neoplasms. J. Neurosurg. 2013, 119, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Uzunalli, G.; Dieterly, A.M.; Kemet, C.M.; Weng, H.Y.; Soepriatna, A.H.; Goergen, C.J.; Shinde, A.B.; Wendt, M.K.; Lyle, L.T. Dynamic transition of the blood-brain barrier in the development of non-small cell lung cancer brain metastases. Oncotarget 2019, 10, 6334–6348. [Google Scholar] [CrossRef] [Green Version]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.T.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix Crosslinking Forces Tumor Progression by Enhancing Integrin Signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [Green Version]
- Ramaswamy, S.; Ross, K.N.; Lander, E.S.; Golub, T.R. A molecular signature of metastasis in primary solid tumors. Nat. Genet. 2003, 33, 49–54. [Google Scholar] [CrossRef]
- Acerbi, I.; Cassereau, L.; Dean, I.; Shi, Q.; Au, A.; Park, C.; Chen, Y.Y.; Liphardt, J.; Hwang, E.S.; Weaver, V.M. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integr. Biol. 2015, 7, 1120–1134. [Google Scholar] [CrossRef] [Green Version]
- Vallet, S.D.; Ricard-Blum, S. Lysyl oxidases: From enzyme activity to extracellular matrix cross-links. Essays Biochem. 2019, 63, 349–364. [Google Scholar]
- Erler, J.T.; Giaccia, A.J. Lysyl oxidase mediates hypoxic control of metastasis. Cancer Res. 2006, 66, 10238–10241. [Google Scholar] [CrossRef] [Green Version]
- Mäki, J.M.; Sormunen, R.; Lippo, S.; Kaarteenaho-Wiik, R.; Soininen, R.; Myllyharju, J. Lysyl oxidase is essential for normal development and function of the respiratory system and for the integrity of elastic and collagen fibers in various tissues. Am. J. Pathol. 2005, 167, 927–936. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Hou, Y.; Yang, G.; Wang, X.; Tang, S.; Du, Y.E.; Yang, L.; Yu, T.; Zhang, H.; Zhou, M.; et al. Stromal miR-200s contribute to breast cancer cell invasion through CAF activation and ECM remodeling. Cell Death Differ. 2016, 23, 132–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, T.R.; Gartland, A.; Erler, J.T. Lysyl oxidase, a targetable secreted molecule involved in cancer metastasis. Cancer Res. 2016, 76, 188–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schito, L.; Rey, S. Hypoxic pathobiology of breast cancer metastasis. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Rundqvist, H.; Johnson, R.S. Tumour oxygenation: Implications for breast cancer prognosis. J. Intern. Med. 2013, 274, 105–112. [Google Scholar] [CrossRef]
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef] [Green Version]
- Dales, J.P.; Garcia, S.; Meunier-Carpentier, S.; Andrac-Meyer, L.; Haddad, O.; Lavaut, M.N.; Allasia, C.; Bonnier, P.; Charpin, C. Overexpression of hypoxia-inducible factor HIF-1α predicts early relapse in breast cancer: Retrospective study in a series of 745 patients. Int. J. Cancer 2005, 116, 734–739. [Google Scholar] [CrossRef]
- Winter, S.C.; Buffa, F.M.; Silva, P.; Miller, C.; Valentine, H.R.; Turley, H.; Shah, K.A.; Cox, G.J.; Corbridge, R.J.; Homer, J.J.; et al. Relation of a hypoxia metagene derived from head and neck cancer to prognosis of multiple cancers. Cancer Res. 2007, 67, 3441–3449. [Google Scholar] [CrossRef] [Green Version]
- Cox, T.R.; Rumney, R.M.H.; Schoof, E.M.; Perryman, L.; Høye, A.M.; Agrawal, A.; Bird, D.; Latif, N.A.; Forrest, H.; Evans, H.R.; et al. The hypoxic cancer secretome induces pre-metastatic bone lesions through lysyl oxidase. Nature 2015, 522, 106–110. [Google Scholar] [CrossRef] [Green Version]
- Psaila, B.; Lyden, D. The metastatic niche: Adapting the foreign soil. Nat. Rev. Cancer 2009, 9, 285–293. [Google Scholar] [CrossRef]
- Smithen, D.A.; Leung, L.M.H.; Challinor, M.; Lawrence, R.; Tang, H.; Niculescu-Duvaz, D.; Pearce, S.P.; McLeary, R.; Lopes, F.; Aljarah, M.; et al. 2-Aminomethylene-5-sulfonylthiazole Inhibitors of Lysyl Oxidase (LOX) and LOXL2 Show Significant Efficacy in Delaying Tumor Growth. J. Med. Chem. 2020, 63, 2308–2324. [Google Scholar] [CrossRef]
- Chang, J.; Lucas, M.C.; Leonte, L.E.; Garcia-Montolio, M.; Singh, L.B.; Findlay, A.D.; Deodhar, M.; Foot, J.S.; Jarolimek, W.; Timpson, P.; et al. Pre-clinical evaluation of small molecule LOXL2 inhibitors in breast cancer. Oncotarget 2017, 8, 26066–26078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Liao, S.; Diop-Frimpong, B.; Chen, W.; Goel, S.; Naxerova, K.; Ancukiewicz, M.; Boucher, Y.; Jain, R.K.; Xu, L. TGF-β blockade improves the distribution and efficacy of therapeutics in breast carcinoma by normalizing the tumor stroma. Proc. Natl. Acad. Sci. USA 2012, 109, 16618–16623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zormpas-Petridis, K.; Boult, J.K.R.; Reeves, E.L.; Heindl, A.; Vinci, M.; Lopes, F.; Cummings, C.; Springer, C.J.; Chesler, L.; et al. Investigating the contribution of collagen to the tumor biomechanical phenotype with noninvasive magnetic resonance elastography. Cancer Res. 2019, 79, 5874–5883. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, T.; Fässler, R.; Hohenester, E. Laminin: The crux of basement membrane assembly. J. Cell Biol. 2004, 164, 959–963. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.G.; Gao, M.Q.; Choi, Y.P.; Kang, S.; Park, H.R.; Kang, K.S.; Cho, N.H. Invasive breast cancer induces laminin-332 upregulation and integrin β4 neoexpression in myofibroblasts to confer an anoikis-resistant phenotype during tissue remodeling. Breast Cancer Res. 2012, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, T.-C.; Yang, C.-H.; Cheng, L.-H.; Chang, W.-T.; Lin, Y.-R.; Cheng, H.-C. Fibronectin in Cancer: Friend or Foe. Cells 2019, 9, 27. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Garcia, B.; Eiró, N.; Marín, L.; González-Reyes, S.; González, L.O.; Lamelas, M.L.; Vizoso, F.J. Expression and prognostic significance of fibronectin and matrix metalloproteases in breast cancer metastasis. Histopathology 2014, 64, 512–522. [Google Scholar] [CrossRef]
- Balanis, N.; Wendt, M.K.; Schiemann, B.J.; Wang, Z.; Schiemann, W.P.; Carlin, C.R. Epithelial to Mesenchymal Transition Promotes Breast Cancer Progression via a Fibronectin-dependent STAT3 Signaling Pathway. J. Biol. Chem. 2013, 288, 17954–17967. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Wu, F.; Seo, B.R.; Fischbach, C.; Chen, W.; Hsu, L.; Gourdon, D. Breast cancer cells alter the dynamics of stromal fibronectin-collagen interactions. Matrix Biol. 2017, 60–61, 86–95. [Google Scholar] [CrossRef]
- Maeda, T.; Desouky, J.; Friedl, A. Syndecan-1 expression by stromal fibroblasts promotes breast carcinoma growth in vivo and stimulates tumor angiogenesis. Oncogene 2006, 25, 1408–1412. [Google Scholar] [CrossRef] [Green Version]
- Stanley, M.J.; Stanley, M.W.; Sanderson, R.D.; Zera, R. Syndecan-1 Expression Is Induced in the Stroma of Infiltrating Breast Carcinoma. Am. J. Clin. Pathol. 1999, 112. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Mosher, R.; Seo, S.; Beebe, D.; Friedl, A. Syndecan-1 in breast cancer stroma fibroblasts regulates extracellular matrix fiber organization and carcinoma cell motility. Am. J. Pathol. 2011, 178, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Ni, W.; Cui, C.; Fang, L.; Xuan, Y. Tenascin C is a prognostic determinant and potential cancer-associated fibroblasts marker for breast ductal carcinoma. Exp. Mol. Pathol. 2017, 102, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Degen, M.; Brellier, F.; Schenk, S.; Driscoll, R.; Zaman, K.; Stupp, R.; Tornillo, L.; Terracciano, L.; Chiquet-Ehrismann, R.; Rüegg, C.; et al. Tenascin-W, a new marker of cancer stroma, is elevated in sera of colon and breast cancer patients. Int. J. Cancer 2008, 122, 2454–2461. [Google Scholar] [CrossRef]
- Degen, M.; Brellier, F.; Kain, R.; Ruiz, C.; Terracciano, L.; Orend, G.; Chiquet-Ehrismann, R. Tenascin-W is a novel marker for activated tumor stroma in low-grade human breast cancer and influences cell behavior. Cancer Res. 2007, 67, 9169–9179. [Google Scholar] [CrossRef] [Green Version]
- Le Bras, G.F.; Taubenslag, K.J.; Andl, C.D. The regulation of cell-cell adhesion during epithelial-mesenchymal transition, motility and tumor progression. Cell Adhes. Migr. 2012, 6, 365–373. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Bueno, G.; Portillo, F.; Cano, A. Transcriptional regulation of cell polarity in EMT and cancer. Oncogene 2008, 27, 6958–6969. [Google Scholar] [CrossRef] [Green Version]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell 2018, 45, 681–695. [Google Scholar] [CrossRef] [Green Version]
- Tsai, J.H.; Yang, J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013, 27, 2192–2206. [Google Scholar] [CrossRef] [Green Version]
- Albano, F.; Chiurazzi, F.; Mimmi, S.; Vecchio, E.; Pastore, A.; Cimmino, C.; Frieri, C.; Iaccino, E.; Pisano, A.; Golino, G.; et al. The expression of inhibitor of bruton’s tyrosine kinase gene is progressively up regulated in the clinical course of chronic lymphocytic leukaemia conferring resistance to apoptosis. Cell Death Dis. 2018, 9, 13. [Google Scholar] [CrossRef] [Green Version]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinde, A.; Libring, S.; Alpsoy, A.; Abdullah, A.; Schaber, J.A.; Solorio, L.; Wendt, M.K. Autocrine fibronectin inhibits breast cancer metastasis. Mol. Cancer Res. 2018, 16, 1579–1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinde, A.; Paez, J.S.; Libring, S.; Hopkins, K.; Solorio, L.; Wendt, M.K. Transglutaminase-2 facilitates extracellular vesicle-mediated establishment of the metastatic niche. Oncogenesis 2020, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, M.; Brooks, M.; Wicha, M. Epithelial-Mesenchymal Plasticity of Breast Cancer Stem Cells: Implications for Metastasis and Therapeutic Resistance. Curr. Pharm. Des. 2015, 21, 1301–1310. [Google Scholar] [CrossRef] [Green Version]
- Libring, S.; Shinde, A.; Chanda, M.K.; Nuru, M.; George, H.; Saleh, A.M.; Abdullah, A.; Kinzer-Ursem, T.L.; Calve, S.; Wendt, M.K.; et al. The dynamic relationship of breast cancer cells and fibroblasts in fibronectin accumulation at primary and metastatic tumor sites. Cancers 2020, 12, 1270. [Google Scholar] [CrossRef] [PubMed]
- Sleeman, J.P. The metastatic niche and stromal progression. Cancer Metastasis Rev. 2012, 31, 429–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef]
- Pisano, A.; Albano, F.; Vecchio, E.; Renna, M.; Scala, G.; Quinto, I.; Fiume, G. Revisiting bacterial ubiquitin ligase effectors: Weapons for host exploitation. Int. J. Mol. Sci. 2018, 19, 3576. [Google Scholar] [CrossRef] [Green Version]
- Clarke, A.J.; Simon, A.K. Autophagy in the renewal, differentiation and homeostasis of immune cells. Nat. Rev. Immunol. 2019, 19, 170–183. [Google Scholar] [CrossRef]
- Münz, C. Autophagy proteins in antigen processing for presentation on MHC molecules. Immunol. Rev. 2016, 272, 17–27. [Google Scholar] [CrossRef]
- Molina, M.L.; García-Bernal, D.; Martinez, S.; Valdor, R. Autophagy in the immunosuppressive perivascular microenvironment of glioblastoma. Cancers 2020, 12, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, E.; Mehnert, J.M.; Chan, C.S. Autophagy, Metabolism, and Cancer. Clin. Cancer Res. 2015, 21, 5037–5046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, H.R.; Jo, Y.H.; Kim, J.; Shin, Y.; Kim, S.S.; Choi, T.G. Roles of autophagy in oxidative stress. Int. J. Mol. Sci. 2020, 21, 3289. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.W.; Lee, S.H. The roles of autophagy in cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisano, A.; Ceglia, S.; Palmieri, C.; Vecchio, E.; Fiume, G.; De Laurentiis, A.; Mimmi, S.; Falcone, C.; Iaccino, E.; Scialdone, A.; et al. CRL3IBTK regulates the tumor suppressor Pdcd4 through ubiquitylation coupled to proteasomal degradation. J. Biol. Chem. 2015, 290, 13958–13971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.F.; Shi, Y.H.; Ding, Z.B.; Ke, A.W.; Gu, C.Y.; Hui, B.; Zhou, J.; Qiu, S.J.; Dai, Z.; Fan, J. Autophagy inhibition suppresses pulmonary metastasis of HCC in mice via impairing anoikis resistance and colonization of HCC cells. Autophagy 2013, 9, 2056–2068. [Google Scholar] [CrossRef] [Green Version]
- Akalay, I.; Janji, B.; Hasmim, M.; Noman, M.Z.; André, F.; De Cremoux, P.; Bertheau, P.; Badoual, C.; Vielh, P.; Larsen, A.K.; et al. Epithelial-to-mesenchymal transition and autophagy induction in breast carcinoma promote escape from t-cell-mediated lysis. Cancer Res. 2013, 73, 2418–2427. [Google Scholar] [CrossRef] [Green Version]
- You, L.; Wang, Z.; Li, H.; Shou, J.; Jing, Z.; Xie, J.; Sui, X.; Pan, H.; Han, W. The role of STAT3 in autophagy. Autophagy 2015, 11, 729–739. [Google Scholar] [CrossRef] [Green Version]
- Shen, S.; Niso-Santano, M.; Adjemian, S.; Takehara, T.; Malik, S.A.; Minoux, H.; Souquere, S.; Mariño, G.; Lachkar, S.; Senovilla, L.; et al. Cytoplasmic STAT3 Represses Autophagy by Inhibiting PKR Activity. Mol. Cell 2012, 48, 667–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, H.M.; Yu, C.R.; Dambuza, I.; Marrero, B.; Egwuagu, C.E. STAT3 protein interacts with class O forkhead transcription factors in the cytoplasm and regulates nuclear/cytoplasmic localization of FoxO1 and FoxO3a proteins in CD4+ T cells. J. Biol. Chem. 2012, 287, 30436–30443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires, B.R.B.; Mencalha, A.L.; Ferreira, G.M.; De Souza, W.F.; Morgado-Díaz, J.A.; Maia, A.M.; Corrêa, S.; Abdelhay, E.S.F.W. NF-kappaB is involved in the regulation of EMT genes in breast cancer cells. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiume, G.; Rossi, A.; de Laurentiis, A.; Falcone, C.; Pisano, A.; Vecchio, E.; Pontoriero, M.; Scala, I.; Scialdone, A.; Masci, F.F.; et al. Eukaryotic Initiation Factor 4H Is under Transcriptional Control of p65/NF-κB. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Xiong, H.; Liu, D.; Hill, C.; Ertay, A.; Li, J.; Zou, Y.; Miller, P.; White, E.; Downward, J.; et al. Autophagy inhibition specifically promotes epithelial-mesenchymal transition and invasion in RAS-mutated cancer cells. Autophagy 2019, 15, 886–899. [Google Scholar] [CrossRef] [PubMed]
- Gugnoni, M.; Sancisi, V.; Manzotti, G.; Gandolfi, G.; Ciarrocchi, A. Autophagy and epithelial–mesenchymal transition: An intricate interplay in cancer. Cell Death Dis. 2016, 7, e2520. [Google Scholar] [CrossRef] [PubMed]
- Hardy, S.D.; Shinde, A.; Wang, W.H.; Wendt, M.K.; Geahlen, R.L. Regulation of epithelial-mesenchymal transition and metastasis by TGF-β, P-bodies, and autophagy. Oncotarget 2017, 8, 103302–103314. [Google Scholar] [CrossRef] [Green Version]
- Shinde, A.; Hardy, S.D.; Kim, D.; Akhand, S.S.; Jolly, M.K.; Wang, W.H.; Anderson, J.C.; Khodadadi, R.B.; Brown, W.S.; George, J.T.; et al. Spleen tyrosine kinase–mediated autophagy is required for epithelial–mesenchymal plasticity and metastasis in breast cancer. Cancer Res. 2019, 79, 1831–1843. [Google Scholar] [CrossRef] [Green Version]
- Zeng, K.; He, B.; Yang, B.B.; Xu, T.; Chen, X.; Xu, M.; Liu, X.; Sun, H.; Pan, Y.; Wang, S. The pro-metastasis effect of circANKS1B in breast cancer 11 Medical and Health Sciences 1112 Oncology and Carcinogenesis. Mol. Cancer 2018, 17. [Google Scholar] [CrossRef]
- Lebret, S.C.; Newgreen, D.F.; Thompson, E.W.; Ackland, M.L. Induction of epithelial to mesenchymal transition in PMC42-LA human breast carcinoma cells by carcinoma-associated fibroblast secreted factors. Breast Cancer Res. 2007, 9. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Xiao, C.H.; Tan, L.D.; Wang, Q.S.; Li, X.Q.; Feng, Y.M. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-β signalling. Br. J. Cancer 2014, 110, 724–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giulianelli, S.; Cerliani, J.P.; Lamb, C.A.; Fabris, V.T.; Bottino, M.C.; Gorostiaga, M.A.; Novaro, V.; Góngora, A.; Baldi, A.; Molinolo, A.; et al. Carcinoma-associated fibroblasts activate progesterone receptors and induce hormone independent mammary tumor growth: A role for the FGF-2/FGFR-2 axis. Int. J. Cancer 2008, 123, 2518–2531. [Google Scholar] [CrossRef] [PubMed]
- Jedeszko, C.; Victor, B.C.; Podgorski, I.; Sloane, B.F. Fibroblast hepatocyte growth factor promotes invasion of human mammary ductal carcinoma in situ. Cancer Res. 2009, 69, 9148–9155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, X.C.; Zhang, W.R.; Cao, W.F.; Liu, B.W.; Zhang, F.; Zhao, H.M.; Meng, R.; Zhang, L.; Niu, R.F.; Hao, X.S.; et al. Aquaporin3 Is Required for FGF-2-Induced Migration of Human Breast Cancers. PLoS ONE 2013, 8, e56735. [Google Scholar] [CrossRef]
- Liu, Y.; Geng, Y.H.; Yang, H.; Yang, H.; Zhou, Y.T.; Zhang, H.Q.; Tian, X.X.; Fang, W.G. Extracellular ATP drives breast cancer cell migration and metastasis via S100A4 production by cancer cells and fibroblasts. Cancer Lett. 2018, 430, 1–10. [Google Scholar] [CrossRef]
- Sjöberg, E.; Meyrath, M.; Milde, L.; Herrera, M.; Lövrot, J.; Hägerstrand, D.; Frings, O.; Bartish, M.; Rolny, C.; Sonnhammer, E.; et al. A novel ACKR2-Dependent role of fibroblast-derived CXCL14 in epithelial-to-mesenchymal transition and metastasis of breast cancer. Clin. Cancer Res. 2019, 25, 3702–3717. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhang, J.; Sun, X.; Su, Q.; You, C. Down-regulation of miR-29b in carcinoma associated fibroblasts promotes cell growth and metastasis of breast cancer. Oncotarget 2017, 8, 39559–39570. [Google Scholar] [CrossRef] [Green Version]
- Tsuyada, A.; Chow, A.; Wu, J.; Somlo, G.; Chu, P.; Loera, S.; Luu, T.; Li, A.X.; Wu, X.; Ye, W.; et al. CCL2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells. Cancer Res. 2012, 72, 2768–2779. [Google Scholar] [CrossRef] [Green Version]
- Osuala, K.O.; Sameni, M.; Shah, S.; Aggarwal, N.; Simonait, M.L.; Franco, O.E.; Hong, Y.; Hayward, S.W.; Behbod, F.; Mattingly, R.R.; et al. Il-6 signaling between ductal carcinoma in situ cells and carcinoma-associated fibroblasts mediates tumor cell growth and migration. BMC Cancer 2015, 15, 584. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.; Tian, X.; Oh, S.Y.; Movassaghi, M.; Naber, S.P.; Kuperwasser, C.; Buchsbaum, R.J. The fibroblast Tiam1-osteopontin pathway modulates breast cancer invasion and metastasis. Breast Cancer Res. 2016, 18, 14. [Google Scholar] [CrossRef] [Green Version]
- Shinde, A.; Wilmanski, T.; Chen, H.; Teegarden, D.; Wendt, M.K. Pyruvate carboxylase supports the pulmonary tropism of metastatic breast cancer. Breast Cancer Res. 2018, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, S.; Hou, Y.; Fu, L.; Xi, L.; Yang, D.; Zhao, M.; Qin, Y.; Sun, K.; Teng, Y.; Liu, M. Cancer-associated fibroblast (CAF)-derived IL32 promotes breast cancer cell invasion and metastasis via integrin β3–p38 MAPK signalling. Cancer Lett. 2019, 442, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Hugo, H.J.; Lebret, S.; Tomaskovic-Crook, E.; Ahmed, N.; Blick, T.; Newgreen, D.F.; Thompson, E.W.; Ackland, M.L. Contribution of Fibroblast and Mast Cell (Afferent) and Tumor (Efferent) IL-6 Effects within the Tumor Microenvironment. Cancer Microenviron. 2012, 5, 83–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Studebaker, A.W.; Storci, G.; Werbeck, J.L.; Sansone, P.; Sasser, A.K.; Tavolari, S.; Huang, T.; Chan, M.W.Y.; Marini, F.C.; Rosol, T.J.; et al. Fibroblasts Isolated from Common Sites of Breast Cancer Metastasis Enhance Cancer Cell Growth Rates and Invasiveness in an Interleukin-6–Dependent Manner. Cancer Res. 2008, 68, 9087–9095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayatilaka, H.; Tyle, P.; Chen, J.J.; Kwak, M.; Ju, J.; Kim, H.J.; Lee, J.S.H.; Wu, P.-H.; Gilkes, D.M.; Fan, R.; et al. Synergistic IL-6 and IL-8 paracrine signalling pathway infers a strategy to inhibit tumour cell migration. Nat. Commun. 2017, 8, 15584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, W.; Zhang, W.; Strasner, A.; Grivennikov, S.; Cheng, J.Q.; Hoffman, R.M.; Karin, M. Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL–RANK signalling. Nature 2011, 470, 548–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Zeng, C.; Zhan, Y.; Wang, H.; Jiang, X.; Li, W. Aberrant low expression of p85α in stromal fibroblasts promotes breast cancer cell metastasis through exosome-mediated paracrine Wnt10b. Oncogene 2017, 36, 4692–4705. [Google Scholar] [CrossRef] [Green Version]
- Luga, V.; Zhang, L.; Viloria-Petit, A.M.; Ogunjimi, A.A.; Inanlou, M.R.; Chiu, E.; Buchanan, M.; Hosein, A.N.; Basik, M.; Wrana, J.L. Exosomes mediate stromal mobilization of autocrine Wnt-PCP signaling in breast cancer cell migration. Cell 2012, 151, 1542–1556. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, J.B.; El-Ashry, D.; Turley, E.A. Hyaluronan, cancer-associated fibroblasts and the tumor microenvironment in malignant progression. Front. Cell Dev. Biol. 2018, 6. [Google Scholar] [CrossRef]
- Farmaki, E.; Chatzistamou, I.; Kaza, V.; Kiaris, H. A CCL8 gradient drives breast cancer cell dissemination. Oncogene 2016, 35, 6309–6318. [Google Scholar] [CrossRef]
- Matsumura, Y.; Ito, Y.; Mezawa, Y.; Sulidan, K.; Daigo, Y.; Hiraga, T.; Mogushi, K.; Wali, N.; Suzuki, H.; Itoh, T.; et al. Stromal fibroblasts induce metastatic tumor cell clusters via epithelial–mesenchymal plasticity. Life Sci. Alliance 2019, 2, e201900425. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Boareto, M.; Debeb, B.G.; Aceto, N.; Farach-Carson, M.C.; Woodward, W.A.; Levine, H. Inflammatory breast cancer: A model for investigating cluster-based dissemination. NPJ Breast Cancer 2017, 3, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, B.; Esmaeili, A.A.; Gopalakrishna-Pillai, S.; Murad, J.P.; Andersen, E.S.; Reddy, N.K.; Srinivasan, G.; Armstrong, B.; Chu, C.; Kim, Y.; et al. Human brain metastatic stroma attracts breast cancer cells via chemokines CXCL16 and CXCL12. NPJ Breast Cancer 2017, 3, 6. [Google Scholar] [CrossRef] [Green Version]
- Hemalatha, S.K.; Sengodan, S.K.; Nadhan, R.; Dev, J.; Sushama, R.R.; Somasundaram, V.; Thankappan, R.; Rajan, A.; Latha, N.R.; Varghese, G.R.; et al. Brcal Defective Breast Cancer Cells Induce in vitro Transformation of Cancer Associated Fibroblasts (CAFs) to Metastasis Associated Fibroblasts (MAF). Sci. Rep. 2018, 8, 13903. [Google Scholar] [CrossRef] [PubMed]
- Gui, Y.; Aguilar-Mahecha, A.; Krzemien, U.; Hosein, A.; Buchanan, M.; Lafleur, J.; Pollak, M.; Ferrario, C.; Basik, M. Metastatic breast carcinoma–associated fibroblasts have enhanced protumorigenic properties related to increased IGF2 expression. Clin. Cancer Res. 2019, 25, 7229–7242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daubriac, J.; Han, S.; Grahovac, J.; Smith, E.; Hosein, A.; Buchanan, M.; Basik, M.; Boucher, Y. The crosstalk between breast carcinoma-associated fibroblasts and cancer cells promotes RhoA-dependent invasion via IGF-1 and PAI-1. Oncotarget 2018, 9, 10375–10387. [Google Scholar] [CrossRef] [Green Version]
- Shani, O.; Raz, Y.; Megides, O.; Shacham, H.; Cohen, N.; Silverbush, D.; Monteran, L.; Sharan, R.; Tsarfaty, I.; Erez, N. Evolution of metastases-associated fibroblasts in the lung microenvironment is driven by stage-specific transcriptional plasticity. bioRxiv 2019, 778936. [Google Scholar] [CrossRef] [Green Version]
- Dumont, N.; Liu, B.; DeFilippis, R.A.; Chang, H.; Rabban, J.T.; Karnezis, A.N.; Tjoe, J.A.; Marx, J.; Parvin, B.; Tlsty, T.D. Breast Fibroblasts Modulate Early Dissemination, Tumorigenesis, and Metastasis through Alteration of Extracellular Matrix Characteristics. Neoplasia 2013, 15, 249–262. [Google Scholar] [CrossRef] [Green Version]
- Hüsemann, Y.; Geigl, J.B.; Schubert, F.; Musiani, P.; Meyer, M.; Burghart, E.; Forni, G.; Eils, R.; Fehm, T.; Riethmüller, G.; et al. Systemic Spread Is an Early Step in Breast Cancer. Cancer Cell 2008, 13, 58–68. [Google Scholar] [CrossRef] [Green Version]
- Trimboli, A.J.; Cantemir-Stone, C.Z.; Li, F.; Wallace, J.A.; Merchant, A.; Creasap, N.; Thompson, J.C.; Caserta, E.; Wang, H.; Chong, J.L.; et al. Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature 2009, 461, 1084–1091. [Google Scholar] [CrossRef] [Green Version]
- Provenzano, P.P.; Eliceiri, K.W.; Campbell, J.M.; Inman, D.R.; White, J.G.; Keely, P.J. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 2006, 4, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, C.E.; Hammer, A.M.; Cho, Y.; Sizemore, G.M.; Cukierman, E.; Yee, L.D.; Ghadiali, S.N.; Ostrowski, M.C.; Leight, J.L. Stromal PTEN Regulates Extracellular Matrix Organization in the Mammary Gland. Neoplasia 2019, 21, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Ying, H. The Rho kinase inhibitor fasudil inhibits tumor progression in human and rat tumor models. Mol. Cancer Ther. 2006, 5, 2158–2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conklin, M.W.; Eickhoff, J.C.; Riching, K.M.; Pehlke, C.A.; Eliceiri, K.W.; Provenzano, P.P.; Friedl, A.; Keely, P.J. Aligned Collagen Is a Prognostic Signature for Survival in Human Breast Carcinoma. Am. J. Pathol. 2011, 178, 1221–1232. [Google Scholar] [CrossRef]
- De Vincenzo, A.; Belli, S.; Franco, P.; Telesca, M.; Iaccarino, I.; Botti, G.; Carriero, M.V.; Ranson, M.; Stoppelli, M.P. Paracrine recruitment and activation of fibroblasts by c-Myc expressing breast epithelial cells through the IGFs/IGF-1R axis. Int. J. Cancer 2019, 145, 2827–2839. [Google Scholar] [CrossRef]
- Iams, W.T.; Lovly, C.M. Molecular Pathways: Clinical Applications and Future Direction of Insulin-like Growth Factor-1 Receptor Pathway Blockade. Clin. Cancer Res. 2015, 21, 4270–4277. [Google Scholar] [CrossRef] [Green Version]
- Brechbuhl, H.M.; Barrett, A.S.; Kopin, E.; Hagen, J.C.; Han, A.L.; Gillen, A.E.; Finlay-Schultz, J.; Cittelly, D.M.; Owens, P.; Horwitz, K.B.; et al. Fibroblast subtypes define a metastatic matrisome in breast cancer. JCI Insight 2020, 5, e130751. [Google Scholar] [CrossRef] [Green Version]
- Hancox, R.A.; Allen, M.D.; Holliday, D.L.; Edwards, D.R.; Pennington, C.J.; Guttery, D.S.; Shaw, J.A.; Walker, R.A.; Pringle, J.H.; Jones, J.L. Tumour-associated tenascin-C isoforms promote breast cancer cell invasion and growth by matrix metalloproteinase-dependent and independent mechanisms. Breast Cancer Res. 2009, 11, R24. [Google Scholar] [CrossRef] [Green Version]
- Samoszuk, M.K.; Nguyen, V.; Gluzman, I.; Pham, J.H. Occult Deposition of Eosinophil Peroxidase in a Subset of Human Breast Carcinomas. Am. J. Pathol. 1996, 148, 701–706. [Google Scholar]
- Zhang, N.; Francis, K.P.; Prakash, A.; Ansaldi, D. Enhanced detection of myeloperoxidase activity in deep tissues through luminescent excitation of near-infrared nanoparticles. Nat. Med. 2013, 19, 500–505. [Google Scholar] [CrossRef]
- Panagopoulos, V.; Leach, D.A.; Zinonos, I.; Ponomarev, V.; Licari, G.; Liapis, V.; Ingman, W.V.; Anderson, P.; DeNichilo, M.O.; Evdokiou, A. Inflammatory peroxidases promote breast cancer progression in mice via regulation of the tumour microenvironment. Int. J. Oncol. 2017, 50, 1191–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van’t Veer, L.J.; Dai, H.; van de Vijver, M.J.; He, Y.D.; Hart, A.A.M.; Mao, M.; Peterse, H.L.; van der Kooy, K.; Marton, M.J.; Witteveen, A.T.; et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002, 415, 530–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyengar, P.; Espina, V.; Williams, T.W.; Lin, Y.; Berry, D.; Jelicks, L.A.; Lee, H.; Temple, K.; Graves, R.; Pollard, J.; et al. Adipocyte-derived collagen VI affects early mammary tumor progression in vivo, demonstrating a critical interaction in the tumor/stroma microenvironment. J. Clin. Invest. 2005, 115, 1163–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, G.P.; Nguyen, D.X.; Chiang, A.C.; Bos, P.D.; Kim, J.Y.; Nadal, C.; Gomis, R.R.; Manova-Todorova, K.; Massagué, J. Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature 2007, 446, 765–770. [Google Scholar] [CrossRef]
- Eck, S.M.; Côté, A.L.; Winkelman, W.D.; Brinckerhoff, C.E. CXCR4 and matrix metalloproteinase-1 are elevated in breast carcinoma-associated fibroblasts and in normal mammary fibroblasts exposed to factors secreted by breast cancer cells. Mol. Cancer Res. 2009, 7, 1033–1044. [Google Scholar] [CrossRef] [Green Version]
- Holliday, D.L.; Hughes, S.; Shaw, J.A.; Walker, R.A.; Jones, J.L. Intrinsic genetic characteristics determine tumor-modifying capacity of fibroblasts: Matrix metalloproteinase-3 5A/5A genotype enhances breast cancer cell invasion. Breast Cancer Res. 2007, 9, R67. [Google Scholar] [CrossRef] [Green Version]
- Radisky, E.S.; Radisky, D.C. Matrix metalloproteinases as breast cancer drivers and therapeutic targets. Front. Biosci. Landmark 2015, 20, 1144–1163. [Google Scholar] [CrossRef]
- Nabeshima, K.; Inoue, T.; Shimao, Y.; Sameshima, T. Matrix metalloproteinases in tumor invasion: Role for cell migration. Pathol. Int. 2002, 52, 255–264. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix Metalloproteinases: Regulators of the Tumor Microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [Green Version]
- Del Casar, J.M.; González, L.O.; Alvarez, E.; Junquera, S.; Marín, L.; González, L.; Bongera, M.; Vázquez, J.; Vizoso, F.J. Comparative analysis and clinical value of the expression of metalloproteases and their inhibitors by intratumor stromal fibroblasts and those at the invasive front of breast carcinomas. Breast Cancer Res. Treat. 2009, 116, 39–52. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.N.; Albo, D.; Tuszynski, G.P. Fibroblasts Promote Breast Cancer Cell Invasion by Upregulating Tumor Matrix metalloproteinase-9 Production. Surgery 2002, 132. [Google Scholar] [CrossRef] [PubMed]
- Krishnamachary, B.; Stasinopoulos, I.; Kakkad, S.; Penet, M.-F.; Jacob, D.; Wildes, F.; Mironchik, Y.; Pathak, A.P.; Solaiyappan, M.; Bhujwalla, Z.M. Breast cancer cell cyclooxygenase-2 expression alters extracellular matrix structure and function and numbers of cancer associated fibroblasts. Oncotarget 2017, 8, 17981–17994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, F.; Li, M.; Zhang, C.; Cui, J.; Liu, J.; Li, J.; Jiang, H. Clinicopathological and prognostic significance of COX-2 immunohistochemical expression in breast cancer: A metaanalysis. Oncotarget 2017, 8, 6003–6012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, K.; Mazo, I.; Leung, H.; Engelke, K.; Von Andrian, U.H.; Deryugina, E.I.; Strongin, A.Y.; Bröcker, E.B.; Friedl, P. Compensation mechanism in tumor cell migration: Mesenchymal-amoeboid transition after blocking of pericellular proteolysis. J. Cell Biol. 2003, 160, 267–277. [Google Scholar] [CrossRef] [Green Version]
- Provenzano, P.P.; Inman, D.R.; Eliceiri, K.W.; Knittel, J.G.; Yan, L.; Rueden, C.T.; White, J.G.; Keely, P.J. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Kakkad, S.M.; Solaiyappan, M.; Argani, P.; Sukumar, S.; Jacobs, L.K.; Leibfritz, D.; Bhujwalla, Z.M.; Glunde, K. Collagen I fiber density increases in lymph node positive breast cancers: Pilot study. J. Biomed. Opt. 2012, 17, 116017. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Peluffo, G.; Chen, H.; Gelman, R.; Schnitt, S.; Polyak, K. Role of COX-2 in epithelial-stromal cell interactions and progression of ductal carcinoma in situ of the breast. Proc. Natl. Acad. Sci. USA 2009, 106, 3372–3377. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.Y.; Li, C.F.; Kuo, C.C.; Tsai, K.K.; Hou, M.F.; Hung, W.C. Cancer/stroma interplay via cyclooxygenase-2 and indoleamine 2,3-dioxygenase promotes breast cancer progression. Breast Cancer Res. 2014, 16, 410. [Google Scholar] [CrossRef] [Green Version]
- Pereg, D.; Lishner, M. Non-steroidal anti-inflammatory drugs for the prevention and treatment of cancer. J. Intern. Med. 2005, 258, 115–123. [Google Scholar] [CrossRef]
- Albano, F.; Arcucci, A.; Granato, G.; Romano, S.; Montagnani, S.; De Vendittis, E.; Ruocco, M.R. Markers of mitochondrial dysfunction during the diclofenac-induced apoptosis in melanoma cell lines. Biochimie 2013, 95, 934–945. [Google Scholar] [CrossRef]
- Harris, R.E.; Alshafie, G.A.; Abou-Issa, H.; Seibert, K. Chemoprevention of Breast Cancer in Rats by Celecoxib, a Cyclooxygenase 2 Inhibitor. Cancer Res. 2000, 60, 2101–2103. [Google Scholar]
- Ulrich, C.M.; Bigler, J.; Potter, J.D. Non-steroidal anti-inflammatory drugs for cancer prevention: Promise, perils and pharmacogenetics. Nat. Rev. Cancer 2006, 6, 130–140. [Google Scholar] [CrossRef]
- Egan, K.M.; Stampfer, M.J.; Giovannucci, E.; Rosner, B.A.; Colditz, G.A. Prospective Study of Regular Aspirin Use and the Risk of Breast Cancer. J. Natl. Cancer Inst. 1996, 88, 988–993. [Google Scholar] [CrossRef] [Green Version]
- Arun, B.; Goss, P. The role of COX-2 inhibition in breast cancer treatment and prevention. Semin. Oncol. 2004, 31, 22–29. [Google Scholar] [CrossRef]
- Granato, G.; Ruocco, M.R.; Iaccarino, A.; Masone, S.; Calì, G.; Avagliano, A.; Russo, V.; Bellevicine, C.; Di Spigna, G.; Fiume, G.; et al. Generation and analysis of spheroids from human primary skin myofibroblasts: An experimental system to study myofibroblasts deactivation. Cell Death Discov. 2017, 3, 17038. [Google Scholar] [CrossRef] [Green Version]
- Avagliano, A.; Ruocco, M.R.; Nasso, R.; Aliotta, F.; Sanità, G.; Iaccarino, A.; Bellevicine, C.; Calì, G.; Fiume, G.; Masone, S.; et al. Development of a Stromal Microenvironment Experimental Model Containing Proto-Myofibroblast Like Cells and Analysis of Its Crosstalk with Melanoma Cells: A New Tool to Potentiate and Stabilize Tumor Suppressor Phenotype of Dermal Myofibroblasts. Cells 2019, 8, 1435. [Google Scholar] [CrossRef] [Green Version]
- Trimmer, C.; Sotgia, F.; Whitaker-Menezes, D.; Balliet, R.M.; Eaton, G.; Martinez-Outschoorn, U.E.; Pavlides, S.; Howell, A.; Iozzo, R.V.; Pestell, R.G.; et al. Caveolin-1 and mitochondrial SOD2 (MnSOD) function as tumor suppressors in the stromal microenvironment: A new genetically tractable model for human cancer associated fibroblasts. Cancer Biol. Ther. 2011, 11, 383–394. [Google Scholar] [CrossRef]
- Pavlides, S.; Tsirigos, A.; Vera, I.; Flomenberg, N.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Pestell, R.G.; Martinez-Outschoorn, U.E.; Howell, A.; et al. Transcriptional evidence for the “Reverse Warburg Effect” in human breast cancer tumor stroma and metastasis: Similarities with oxidative stress, inflammation, Alzheimer’s disease, and “Neuron-Glia Metabolic Coupling”. Aging 2010, 2, 185–199. [Google Scholar] [CrossRef] [Green Version]
- Weaver, V.M.; Petersen, O.W.; Wang, F.; Larabell, C.A.; Briand, P.; Damsky, C.; Bissell, M.J. Reversion of the Malignant Phenotype of Human Breast Cells in Three-Dimensional Culture and In Vivo by Integrin Blocking Antibodies. J. Cell Biol. 1997, 137, 231–245. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Drabsch, Y.; Pujuguet, P.; Ren, J.; van Laar, T.; Zhang, L.; van Dam, H.; Clément-Lacroix, P.; ten Dijke, P. Genetic depletion and pharmacological targeting of αv integrin in breast cancer cells impairs metastasis in zebrafish and mouse xenograft models. Breast Cancer Res. 2015, 17, 28. [Google Scholar] [CrossRef]
- Sayyad, M.R.; Puchalapalli, M.; Vergara, N.G.; Wangensteen, S.M.; Moore, M.; Mu, L.; Edwards, C.; Anderson, A.; Kall, S.; Sullivan, M.; et al. Syndecan-1 facilitates breast cancer metastasis to the brain. Breast Cancer Res. Treat. 2019, 178, 35–49. [Google Scholar] [CrossRef]
- Dedes, P.G.; Gialeli, C.; Tsonis, A.I.; Kanakis, I.; Theocharis, A.D.; Kletsas, D.; Tzanakakis, G.N.; Karamanos, N.K. Expression of Matrix Macromolecules and Functional Properties of Breast Cancer Cells Are Modulated by the Bisphosphonate Zoledronic Acid. Biochim. Biophys. Acta 2012, 1820, 1926–1939. [Google Scholar] [CrossRef]
- Rousseau, C.; Ruellan, A.L.; Bernardeau, K.; Kraeber-Bodéré, F.; Gouard, S.; Loussouarn, D.; Saï-Maurel, C.; Faivre-Chauvet, A.; Wijdenes, J.; Barbet, J.; et al. Syndecan-1 antigen, a promising new target for triple-negative breast cancer immuno-PET and radioimmunotherapy. A preclinical study on MDA-MB-468 xenograft tumors. EJNMMI Res. 2011, 1, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.P.; Hielscher, A. Fibronectin: How its aberrant expression in tumors may improve therapeutic targeting. J. Cancer 2017, 8, 674–682. [Google Scholar] [CrossRef] [Green Version]
- Hielscher, A.; Ellis, K.; Qiu, C.; Porterfield, J.; Gerecht, S. Fibronectin Deposition Participates in Extracellular Matrix Assembly and Vascular Morphogenesis. PLoS ONE 2016, 11, e0147600. [Google Scholar] [CrossRef] [Green Version]
- Tabariès, S.; Dong, Z.; Annis, M.G.; Omeroglu, A.; Pepin, F.; Ouellet, V.; Russo, C.; Hassanain, M.; Metrakos, P.; Diaz, Z.; et al. Claudin-2 is selectively enriched in and promotes the formation of breast cancer liver metastases through engagement of integrin complexes. Oncogene 2011, 30, 1318–1328. [Google Scholar] [CrossRef] [Green Version]
- Takai, K.; Le, A.; Weaver, V.M.; Werb, Z. Targeting the cancer-associated fibroblasts as a treatment in triple-negative breast cancer. Oncotarget 2016, 7, 82889–82901. [Google Scholar] [CrossRef] [Green Version]
- Diop-Frimpong, B.; Chauhan, V.P.; Krane, S.; Boucher, Y.; Jain, R.K. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc. Natl. Acad. Sci. USA 2011, 108, 2909–2914. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Liu, X.; Ran, W.; Meng, J.; Zhai, Y.; Zhang, P.; Yin, Q.; Yu, H.; Zhang, Z.; Li, Y. Regulating Cancer Associated Fibroblasts With Losartan-Loaded Injectable Peptide Hydrogel to Potentiate Chemotherapy in Inhibiting Growth and Lung Metastasis of Triple Negative Breast Cancer. Biomaterials 2017, 144, 60–72. [Google Scholar] [CrossRef]
- Gioni, V.; Karampinas, T.; Voutsinas, G.; Roussidis, A.E.; Papadopoulos, S.; Karamanos, N.K.; Kletsas, D. Imatinib Mesylate Inhibits Proliferation and Exerts an Antifibrotic Effect in Human Breast Stroma Fibroblasts. Mol. Cancer Res. 2008, 6, 706–714. [Google Scholar] [CrossRef] [Green Version]
- Kanapathipillai, M.; Mammoto, A.; Mammoto, T.; Kang, J.H.; Jiang, E.; Ghosh, K.; Korin, N.; Gibbs, A.; Mannix, R.; Ingber, D.E. Inhibition of mammary tumor growth using lysyl oxidase-targeting nanoparticles to modify extracellular matrix. Nano Lett. 2012, 12, 3213–3217. [Google Scholar] [CrossRef]
- Van Den Eynden, G.G.; Van Der Auwera, I.; Van Laere, S.J.; Colpaert, C.G.; Turley, H.; Harris, A.L.; Van Dam, P.; Dirix, L.Y.; Vermeulen, P.B.; Van Marck, E.A. Angiogenesis and hypoxia in lymph node metastases is predicted by the angiogenesis and hypoxia in the primary tumour in patients with breast cancer. Br. J. Cancer 2005, 93, 1128–1136. [Google Scholar] [CrossRef] [Green Version]
- Longatto Filho, A.; Lopes, J.M.; Schmitt, F.C. Angiogenesis and breast cancer. J. Oncol. 2010, 2010, 576384. [Google Scholar] [CrossRef]
- Ren, J.; Guo, H.; Wu, H.; Tian, T.; Dong, D.; Zhang, Y.; Sui, Y.; Zhang, Y.; Zhao, D.; Wang, S.; et al. GPER in CAFs regulates hypoxia-driven breast cancer invasion in a CTGF-dependent manner. Oncol. Rep. 2015, 33, 1929–1937. [Google Scholar] [CrossRef] [Green Version]
- De Francesco, E.M.; Lappano, R.; Santolla, M.F.; Marsico, S.; Caruso, A.; Maggiolini, M. HIF-1α/GPER signaling mediates the expression of VEGF induced by hypoxia in breast cancer associated fibroblasts (CAFs). Breast Cancer Res. 2013, 15. [Google Scholar] [CrossRef] [Green Version]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 10. [Google Scholar] [CrossRef]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef]
- Ershaid, N.; Sharon, Y.; Doron, H.; Raz, Y.; Shani, O.; Cohen, N.; Monteran, L.; Leider-Trejo, L.; Ben-Shmuel, A.; Yassin, M.; et al. NLRP3 inflammasome in fibroblasts links tissue damage with inflammation in breast cancer progression and metastasis. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, D.; Zhao, J. The Role of Chemokine Receptor CXCR4 in Breast Cancer Metastasis. Am. J. Cancer Res. 2013, 3, 46–57. [Google Scholar]
- Gründker, C.; Bauerschmitz, G.; Knapp, J.; Schmidt, E.; Olbrich, T.; Emons, G. Inhibition of SDF-1/CXCR4-induced epithelial–mesenchymal transition by kisspeptin-10. Breast Cancer Res. Treat. 2015, 152, 41–50. [Google Scholar] [CrossRef]
- Tamamura, H.; Hori, A.; Kanzaki, N.; Hiramatsu, K.; Mizumoto, M.; Nakashima, H.; Yamamoto, N.; Otaka, A.; Fujii, N. T140 analogs as CXCR4 antagonists identified as anti-metastatic agents in the treatment of breast cancer. FEBS Lett. 2003, 550, 79–83. [Google Scholar] [CrossRef]
- Liang, Z.; Wu, T.; Lou, H.; Yu, X.; Taichman, R.S.; Lau, S.K.; Nie, S.; Umbreit, J.; Shim, H. Inhibition of breast cancer metastasis by selective synthetic polypeptide against CXCR4. Cancer Res. 2004, 64, 4302–4308. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.C.P.; Luker, K.E.; Garbow, J.R.; Prior, J.L.; Jackson, E.; Piwnica-Worms, D.; Luker, G.D. CXCR4 regulates growth of both primary and metastatic breast cancer. Cancer Res. 2004, 64, 8604–8612. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.; Park, B. Baohuoside I suppresses invasion of cervical and breast cancer cells through the downregulation of CXCR4 chemokine receptor expression. Biochemistry 2014, 53, 7562–7569. [Google Scholar] [CrossRef]
- Chua, A.W.L.; Hay, H.S.; Rajendran, P.; Shanmugam, M.K.; Li, F.; Bist, P.; Koay, E.S.C.; Lim, L.H.K.; Kumar, A.P.; Sethi, G. Butein downregulates chemokine receptor CXCR4 expression and function through suppression of NF-κB activation in breast and pancreatic tumor cells. Biochem. Pharmacol. 2010, 80, 1553–1562. [Google Scholar] [CrossRef]
- Zhan, W.; Liang, Z.; Zhu, A.; Kurtkaya, S.; Shim, H.; Snyder, J.P.; Liotta, D.C. Discovery of small molecule CXCR4 antagonists. J. Med. Chem. 2007, 50, 5655–5664. [Google Scholar] [CrossRef]
- Yang, Q.; Zhang, F.; Ding, Y.; Huang, J.; Chen, S.; Wu, Q.; Wang, Z.; Chen, C. Antitumour activity of the recombination polypeptide GST-NT21MP is mediated by inhibition of CXCR4 pathway in breast cancer. Br. J. Cancer 2014, 110, 1288–1297. [Google Scholar] [CrossRef] [Green Version]
- Suchanski, J.; Tejchman, A.; Zacharski, M.; Piotrowska, A.; Grzegrzolka, J.; Chodaczek, G.; Nowinska, K.; Rys, J.; Dziegiel, P.; Kieda, C.; et al. Podoplanin increases the migration of human fibroblasts and affects the endothelial cell network formation: A possible role for cancer-associated fibroblasts in breast cancer progression. PLoS ONE 2017, 12, e0184970. [Google Scholar] [CrossRef] [Green Version]
- Niemiec, J.; Adamczyk, A.; Harazin-Lechowska, A.; Ambicka, A.; Grela-Wojewoda, A.; Majchrzyk, K.; Kruczak, A.; Sas-Korczyńska, B.; Ryś, J. Podoplanin-positive Cancer-associated stromal fibroblasts in primary tumor and synchronous lymph node metastases of HER2-overexpressing breast carcinomas. Anticancer Res. 2018, 38, 1957–1965. [Google Scholar]
- Bernfield, M.; Götte, M.; Park, P.W.; Reizes, O.; Fitzgerald, M.L.; Lincecum, J.; Zako, M. Functions of Cell Surface Heparan Sulfate Proteoglycans. Annu. Rev. Biochem. 1999, 68, 729–777. [Google Scholar] [CrossRef]
- Derksen, P.W.B.; Keehnen, R.M.J.; Evers, L.M.; Van Oers, M.H.J.; Spaargaren, M.; Pals, S.T. Cell surface proteoglycan syndecan-1 mediates hepatocyte growth factor binding and promotes Met signaling in multiple myeloma. Blood 2002, 99, 1405–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purushothaman, A.; Uyama, T.; Kobayashi, F.; Yamada, S.; Sugahara, K.; Rapraeger, A.C.; Sanderson, R.D. Heparanase-enhanced shedding of syndecan-1 by myeloma cells promotes endothelial invasion and angiogenesis. Blood 2010, 115, 2449–2457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szatmári, T.; Ötvös, R.; Hjerpe, A.; Dobra, K. Syndecan-1 in Cancer: Implications for Cell Signaling, Differentiation, and Prognostication. Dis. Markers 2015, 2015, 796052. [Google Scholar] [CrossRef] [PubMed] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Avagliano, A.; Fiume, G.; Ruocco, M.R.; Martucci, N.; Vecchio, E.; Insabato, L.; Russo, D.; Accurso, A.; Masone, S.; Montagnani, S.; et al. Influence of Fibroblasts on Mammary Gland Development, Breast Cancer Microenvironment Remodeling, and Cancer Cell Dissemination. Cancers 2020, 12, 1697. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061697
Avagliano A, Fiume G, Ruocco MR, Martucci N, Vecchio E, Insabato L, Russo D, Accurso A, Masone S, Montagnani S, et al. Influence of Fibroblasts on Mammary Gland Development, Breast Cancer Microenvironment Remodeling, and Cancer Cell Dissemination. Cancers. 2020; 12(6):1697. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061697
Chicago/Turabian StyleAvagliano, Angelica, Giuseppe Fiume, Maria Rosaria Ruocco, Nunzia Martucci, Eleonora Vecchio, Luigi Insabato, Daniela Russo, Antonello Accurso, Stefania Masone, Stefania Montagnani, and et al. 2020. "Influence of Fibroblasts on Mammary Gland Development, Breast Cancer Microenvironment Remodeling, and Cancer Cell Dissemination" Cancers 12, no. 6: 1697. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061697