Intercellular Mitochondrial Transfer in the Tumor Microenvironment

 , , , ,
, , , ,
Abstract
:1. Introduction
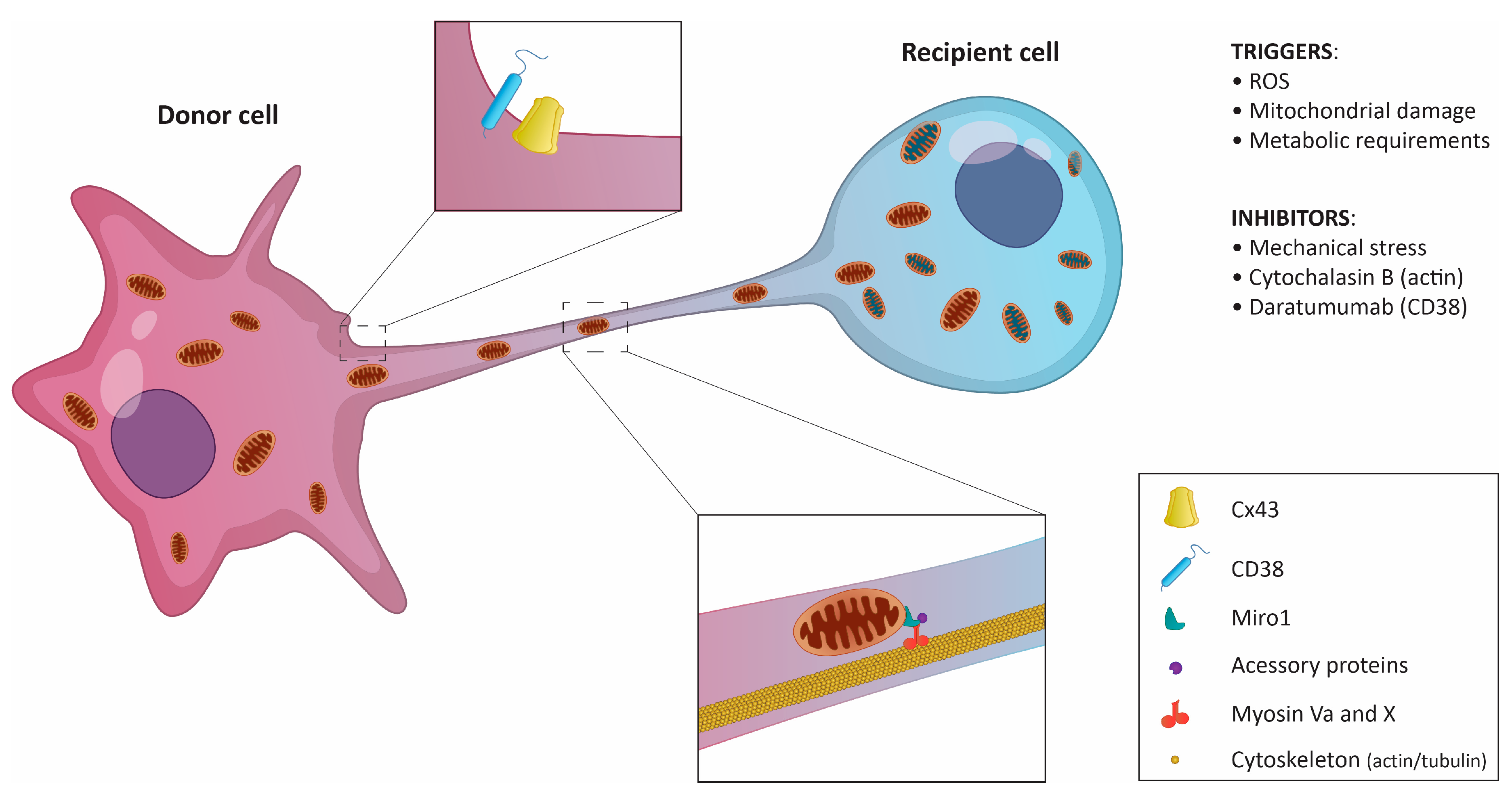
2. Means of Mitochondrial Transfer
3. Tunneling Nanotubes Are the Main Delivery Route for Mitochondria
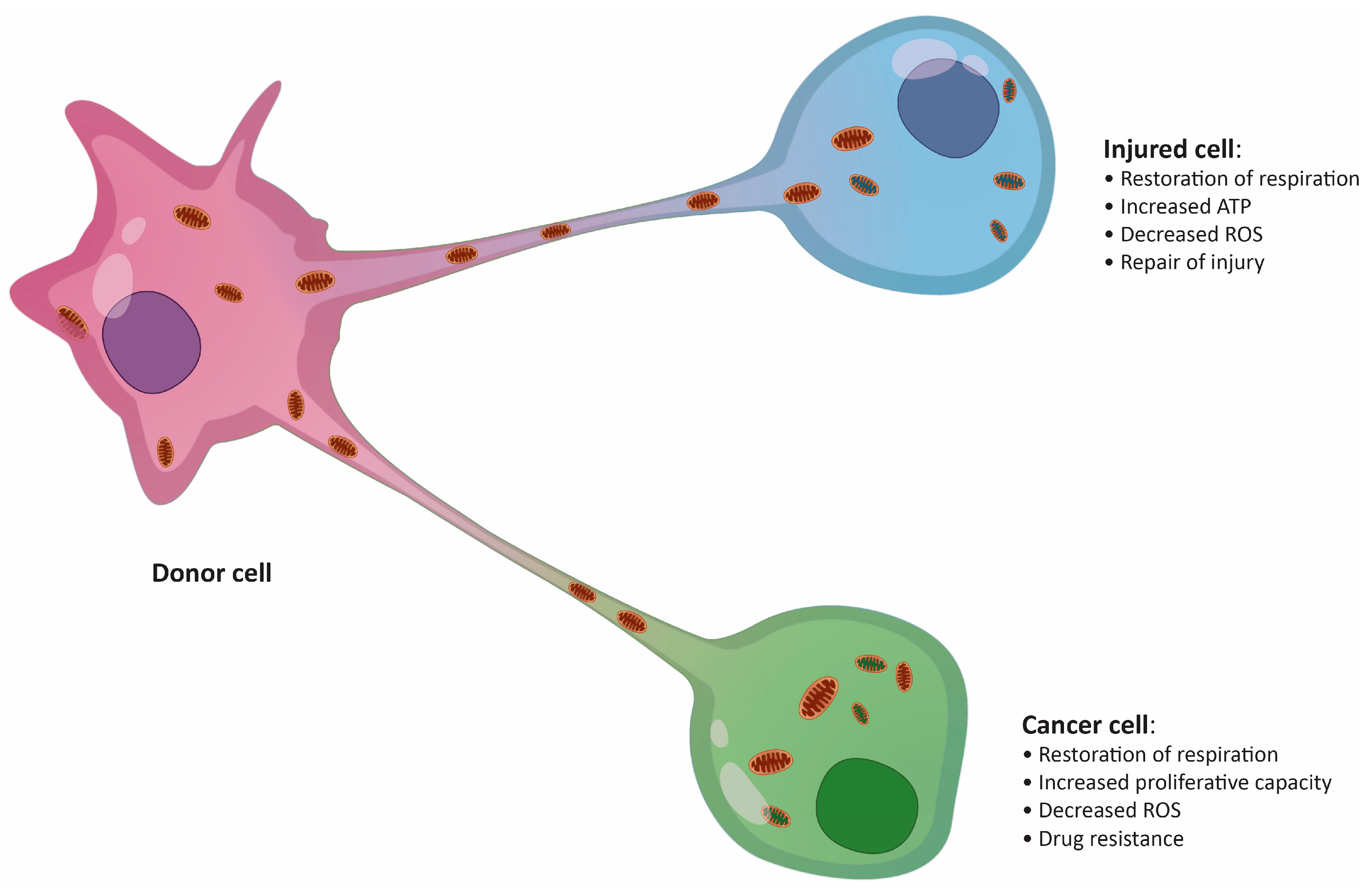
4. Mitochondrial Transfer in Solid Cancers
5. Mitochondrial Transfer in Hematological Malignancies
6. Mitochondrial Transfer in Acute Lymphoblastic Leukemia
7. Mitochondrial Transfer in Acute Myeloid Leukemia
8. Mitochondrial Transfer in Multiple Myeloma
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Warburg, O. On the Origin of Cancer Cells. Am. Assoc. Adv. Sci. 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Potter, M.; Newport, E.; Morten, K.J. The Warburg effect: 80 years on. Biochem. Soc. Trans. 2016, 44, 1499–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Sanchez, R.; Rodriguez-Enriquez, S.; Saavedra, E.; Marin-Hernandez, A.; Gallardo-Perez, J.C. The bioenergetics of cancer: Is glycolysis the main ATP supplier in all tumor cells? Biofactors 2009, 35, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Michelakis, E.D.; Webster, L.; Mackey, J.R. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br. J. Cancer 2008, 99, 989–994. [Google Scholar] [CrossRef] [Green Version]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Detappe, A.; Cai, K.; Keys, H.R.; Brune, Z.; Ying, W.; Thiru, P.; Reidy, M.; Kugener, G.; Rossen, J.; et al. Mitochondrial metabolism promotes adaptation to proteotoxic stress. Nat. Chem. Biol. 2019, 15, 681–689. [Google Scholar] [CrossRef]
- Kuntz, E.M.; Baquero, P.; Michie, A.M.; Dunn, K.; Tardito, S.; Holyoake, T.L.; Helgason, G.V.; Gottlieb, E. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat. Med. 2017, 23, 1234–1240. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Sanchez, R.; Rodriguez-Enriquez, S.; Marin-Hernandez, A.; Saavedra, E. Energy metabolism in tumor cells. FEBS J. 2007, 274, 1393–1418. [Google Scholar] [CrossRef]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2491. [Google Scholar] [CrossRef] [Green Version]
- Song, I.S.; Kim, H.K.; Lee, S.R.; Jeong, S.H.; Kim, N.; Ko, K.S.; Rhee, B.D.; Han, J. Mitochondrial modulation decreases the bortezomib-resistance in multiple myeloma cells. Int. J. Cancer 2013, 133, 1357–1367. [Google Scholar] [CrossRef]
- Chauncey, T.R. Drug resistance mechanisms in acute leukemia. Curr. Opin. Oncol. 2001, 13, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Zhan, J.; Gu, Y.; Chen, B. Mechanisms of drug resistance in acute myeloid leukemia. OncoTargets Ther. 2019, 12, 1937–1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roh, J.S.; Sohn, D.H. Damage-associated molecular patterns in inflammatory diseases. Immune Netw. 2018, 18, 1–14. [Google Scholar] [CrossRef]
- Rubartelli, A.; Lotze, M.T. Inside, outside, upside down: Damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol. 2007, 28, 429–436. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez, C.; Huebener, P.; Schwabe, R. Damage-associated molecular patterns in cancer: A double edged sword. Oncogene 2016, 35, 5931–5941. [Google Scholar] [CrossRef] [PubMed]
- Krysko, O.; Aaes, T.L.; Bachert, C.; Vandenabeele, P.; Krysko, D.V. Many faces of DAMPs in cancer therapy. Cell Death Dis. 2013, 4, e631. [Google Scholar] [CrossRef] [Green Version]
- Spees, J.L.; Olson, S.D.; Whitney, M.J.; Prockop, D.J. Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. USA 2006, 103, 1283–1288. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Zheng, X.; Li, F.; Yu, Y.; Chen, Z.; Liu, Z.; Xu, H.; Yang, W. Tunneling nanotubes promote intercellular mitochondria transfer followed by increased invasiveness in bladder cancer cells. Oncotarget 2017, 8, 15539–15552. [Google Scholar] [CrossRef] [Green Version]
- Pasquier, J.; Guerrouahen, B.S.; Al Thawadi, H.; Ghiabi, P.; Maleki, M.; Abu-Kaoud, N.; Jacob, A.; Mirshahi, M.; Galas, L.; Rafii, S.; et al. Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J. Transl. Med. 2013, 11, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burt, R.; Dey, A.; Aref, S.; Aguiar, M.; Akarca, A.; Bailey, K.; Day, W.; Hooper, S.; Kirkwood, A.; Kirschner, K.; et al. Activated stromal cells transfer mitochondria to rescue acute lymphoblastic leukemia cells from oxidative stress. Blood 2019, 134, 1415–1429. [Google Scholar] [CrossRef] [PubMed]
- Marlein, C.R.; Piddock, R.E.; Mistry, J.J.; Zaitseva, L.; Hellmich, C.; Horton, R.H.; Zhou, Z.; Auger, M.J.; Bowles, K.M.; Rushworth, S.A. CD38-driven mitochondrial trafficking promotes bioenergetic plasticity in multiple myeloma. Cancer Res. 2019, 79, 2285–2297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, L.F.; Kovarova, J.; Bajzikova, M.; Bezawork-Geleta, A.; Svec, D.; Endaya, B.; Sachaphibulkij, K.; Coelho, A.R.; Sebkova, N.; Ruzickova, A.; et al. Horizontal transfer of whole mitochondria restores tumorigenic potential in mitochondrial DNA-deficient cancer cells. eLife 2017, 6, e22187. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Gerdes, H.H. Transfer of mitochondria via tunneling nanotubes rescues apoptotic PC12 cells. Cell Death Differ. 2015, 22, 1181–1191. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Ji, K.; Guo, L.; Wu, W.; Lu, H.; Shan, P.; Yan, C. Mesenchymal stem cells rescue injured endothelial cells in an in vitro ischemia-reperfusion model via tunneling nanotube like structure-mediated mitochondrial transfer. Microvasc. Res. 2014, 92, 10–18. [Google Scholar] [CrossRef]
- Li, X.; Zhang, Y.; Yeung, S.C.; Liang, Y.; Liang, X.; Ding, Y.; Ip, M.S.M.; Tse, H.F.; Mak, J.C.W.; Lian, Q. Mitochondrial transfer of induced pluripotent stem cell-derived mesenchymal stem cells to airway epithelial cells attenuates cigarette smoke-induced damage. Am. J. Respir. Cell Mol. Biol. 2014, 51, 455–465. [Google Scholar] [CrossRef]
- Vallabhaneni, K.C.; Haller, H.; Dumler, I. Vascular smooth muscle cells initiate proliferation of mesenchymal stem cells by mitochondrial transfer via tunneling nanotubes. Stem Cells Dev. 2012, 21, 3104–3113. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.N.; Das, S.R.; Emin, M.T.; Wei, M.; Sun, L.; Westphalen, K.; Rowlands, D.J.; Quadri, S.K.; Bhattacharya, S.; Bhattacharya, J. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 2012, 18, 759–765. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Gao, F.; Zhang, Y.; Wong, D.S.H.; Li, Q.; Tse, H.F.; Xu, G.; Yu, Z.; Lian, Q. Mitochondrial transfer of mesenchymal stem cells effectively protects corneal epithelial cells from mitochondrial damage. Cell Death Dis. 2016, 7, e2467. [Google Scholar] [CrossRef] [Green Version]
- Jackson, M.V.; Morrison, T.J.; Doherty, D.F.; McAuley, D.F.; Matthay, M.A.; Kissenpfennig, A.; O’Kane, C.M.; Krasnodembskaya, A.D. Mitochondrial Transfer via Tunneling Nanotubes is an Important Mechanism by Which Mesenchymal Stem Cells Enhance Macrophage Phagocytosis in the in vitro and in vivo Models of ARDS. Stem Cells 2016, 34, 2210–2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mistry, J.J.; Marlein, C.R.; Moore, J.A.; Hellmich, C.; Wojtowicz, E.E.; Smith, J.G.W.; Macaulay, I.; Sun, Y.; Morfakis, A.; Patterson, A.; et al. ROS-mediated PI3K activation drives mitochondrial transfer from stromal cells to hematopoietic stem cells in response to infection. Proc. Natl. Acad. Sci. USA 2019, 116, 24610–24619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marlein, C.R.; Zaitseva, L.; Piddock, R.E.; Robinson, S.D.; Edwards, D.R.; Shafat, M.S.; Zhou, Z.; Lawes, M.; Bowles, K.M.; Rushworth, S.A. NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood 2017, 130, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Polak, R.; Rooij, B.D.; Pieters, R.; Boer, M.L. Den B-cell precursor acute lymphoblastic leukemia cells use tunneling nanotubes to orchestrate their microenvironment. Blood 2015, 126, 2404–2414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, Y.M.; Kim, J.H.; Kim, M.; Park, S.J.; Koh, S.H.; Ahn, H.S.; Kang, G.H.; Lee, J.-B.; Park, K.S.; Lee, H.K. Mesenchymal stem cells transfer mitochondria to the cells with virtually no mitochondrial function but not with pathogenic mtDNA mutations. PLoS ONE 2012, 7, e32778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.; Liou, C.; Chen, S.; Hsu, T.; Chuang, J.; Wang, P.; Huang, S.; Tiao, M.; Chen, J.; Lin, T.; et al. Mitochondrial transfer from Wharton’s jelly-derived mesenchymal stem cells to mitochondria-defective cells recaptures impaired mitochondrial function. Mitochondrion 2015, 22, 31–34. [Google Scholar] [CrossRef]
- Moschoi, R.; Imbert, V.; Nebout, M.; Chiche, J.; Mary, D.; Prebet, T.; Saland, E.; Castellano, R.; Pouyet, L.; Collette, Y.; et al. Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood 2016, 128, 253–264. [Google Scholar] [CrossRef] [Green Version]
- Marlein, C.R.; Zaitseva, L.; Piddok, R.; Schafat, M.; Collins, A.; Bowles, K.; Rushworth, S. PGC1α Driven Mitochondrial Biogenesis within the Bone Marrow Stromal Cells of the Acute Myeloid Leukemia Micro-Environment Is a Pre-Requisite for Mitochondrial Transfer to Leukemic Blasts. Blood 2017, 130 (Suppl. 1), 3927. [Google Scholar]
- Torralba, D.; Baixauli, F.; Sánchez-Madrid, F. Mitochondria know no boundaries: Mechanisms and functions of intercellular mitochondrial transfer. Front. Cell Dev. Biol. 2016, 4, 107. [Google Scholar] [CrossRef] [Green Version]
- Rustom, A.; Saffrich, R.; Markovic, I.; Walther, P.; Gerdes, H.H. Nanotubular Highways for Intercellular Organelle Transport. Science 2004, 303, 1007–1010. [Google Scholar] [CrossRef] [Green Version]
- Schapman, D.; Lebon, A.; Monterroso, B.; Bellenger, M.; Foll, F.L.; Pasquier, J.; Vaudry, H.; Vaudry, D.; Galas, L. Structural and functional analysis of tunneling nanotubes (TnTs) using g CW STED and g confocal approaches. Biol. Cell 2015, 107, 419–425. [Google Scholar] [CrossRef]
- Gousset, K.; Marzo, L.; Commere, P.; Zurzolo, C. Myo10 is a key regulator of TNT formation in neuronal cells. J. Cell Sci. 2013, 126, 4424–4435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tardivel, M.; Bégard, S.; Bousset, L.; Dujardin, S.; Coens, A.; Melki, R. Tunneling nanotube (TNT)-mediated neuron-to neuron transfer of pathological Tau protein assemblies. Acta Neuropathol. Commun. 2016, 4, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Yu, Z.; Jiang, D.; Liang, X.; Liao, S.; Zhang, Z.; Yue, W.; Li, X.; Chiu, S.M.; Chai, Y.H.; et al. iPSC-MSCs with High Intrinsic MIRO1 and Sensitivity to TNF-α Yield Efficacious Mitochondrial Transfer to Rescue Anthracycline-Induced Cardiomyopathy. Stem Cell Rep. 2016, 7, 749–763. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, T.; Mukherjee, S.; Pattnaik, B.; Kumar, M.; Singh, S.; Rehman, R.; Tiwari, B.K.; Jha, K.A.; Barhanpurkar, A.P.; Wani, M.R.; et al. Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J. 2014, 33, 994–1010. [Google Scholar] [CrossRef]
- López-Doménech, G.; Covill-Cooke, C.; Ivankovic, D.; Halff, E.F.; Sheehan, D.F.; Norkett, R.; Birsa, N.; Kittler, J.T. Miro proteins coordinate microtubule- and actin-dependent mitochondrial transport and distribution. EMBO J. 2018, 37, 321–336. [Google Scholar] [CrossRef]
- Zhang, Y. Tunneling-nanotube. Commun. Integr. Biol. 2011, 4, 324–325. [Google Scholar] [CrossRef]
- Hase, K.; Kimura, S.; Takatsu, H.; Ohmae, M.; Kawano, S.; Kitamura, H.; Ito, M.; Watarai, H.; Hazelett, C.; Yeaman, C.; et al. M-Sec promotes membrane nanotube formation by interacting with Ral and the exocyst complex. Nat. Cell Biol. 2009, 12, 1427–1432. [Google Scholar] [CrossRef]
- Ohno, H.; Hase, K.; Kimura, S. M-Sec: Emerging secrets of tunneling nanotube formation. Commun. Integr. Biol. 2010, 3, 231–233. [Google Scholar] [CrossRef] [Green Version]
- Aarhus, R.; Graeff, R.M.; Dickey, D.M.; Walseth, T.F.; Lee, H.C. ADP-ribosyl cyclase and CD38 catalyze the synthesis of a calcium-mobilizing metabolite from NADP. J. Biol. Chem. 1995, 270, 30327–30333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.C. Physiologival functions of cyclic ADP-ribose and NAADP as calcium messangers. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 317–345. [Google Scholar] [CrossRef] [PubMed]
- Bruzzone, S.; Moreschi, I.; Guida, L.; Usai, C.; Zocchi, E.; De Flora, A. Extracellular NAD+ regulates intracellular calcium levels and induces activation of human granulocytes. Biochem. J. 2006, 393, 697–704. [Google Scholar] [CrossRef]
- Ariazi, J.; Benowitz, A.; De Biasi, V.; Den Boer, M.L.; Cherqui, S.; Cui, H.; Douillet, N.; Eugenin, E.A.; Favre, D.; Goodman, S.; et al. Tunneling Nanotubes and Gap Junctions—Their Role in Long-Range Intercellular Communication during Development, Health, and Disease Conditions. Front. Mol. Neurosci. 2017, 10, 333. [Google Scholar] [CrossRef] [PubMed]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain tumour cells interconnect to a functional and resistant network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef]
- Wang, X.; Veruki, M.L.; Bukoreshtliev, N.V.; Hartveit, E.; Gerdes, H. Animal cells connected by nanotubes can be electrically coupled through interposed gap-junction channels. Proc. Natl. Acad. Sci. USA 2010, 107, 17194–17199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, K.; Shi, X.; Zhang, X.; Dang, S.; Ma, X.; Liu, F.; Xu, M.; Lv, Z.; Han, D.; Fang, X.; et al. Long-distance intercellular connectivity between cardiomyocytes and cardiofibroblasts mediated by membrane nanotubes. Cardiovasc. Res. 2011, 92, 39–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eugenin, E.A. Role of Connexin/Pannexin containing channels in infectious diseases. FEBS Lett. 2014, 588, 1389–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aasen, T. Connexins: Junctional and non-junctional modulators of proliferation. Cell Tissue Res. 2015, 360, 685–699. [Google Scholar] [CrossRef]
- Gerdes, H.H.; Carvalho, R.N. Intercellular transfer mediated by tunneling nanotubes. Curr. Opin. Cell Biol. 2008, 20, 470–475. [Google Scholar] [CrossRef]
- Gerdes, H.H.; Rustom, A.; Wang, X. Tunneling nanotubes, an emerging intercellular communication route in development. Mech. Dev. 2013, 130, 381–387. [Google Scholar] [CrossRef]
- Whiteside, T. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, T.; Dash, C.; Khiste, S.; Sengupta, S. A novel mechanism of immunosuppression via nanotube mediated mitochondrial trafficking between cancer cell and immune cell. Cancer Res. 2019. [Google Scholar] [CrossRef]
- Ippolito, L.; Morandi, A.; Taddei, M.L.; Parri, M.; Comito, G.; Iscaro, A.; Raspollini, M.R.; Magherini, F.; Rapizzi, E.; Masquelier, J.; et al. Cancer-associated fibroblasts promote prostate cancer malignancy via metabolic rewiring and mitochondrial transfer. Oncogene 2019, 38, 5339–5355. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, X.; Qiu, Y.; Shi, Y.; Cai, J.; Wang, B.; Wei, X.; Ke, Q.; Sui, X.; Wang, Y.; et al. Cell adhesion-mediated mitochondria transfer contributes to mesenchymal stem cell-induced chemoresistance on T cell acute lymphoblastic leukemia cells. J. Hematol. Oncol. 2018, 11, 11. [Google Scholar] [CrossRef]
- Poburko, D.; Santo-Domingo, J.; Demaurex, N. Dynamic regulation of the mitochondrial proton gradient during cytosolic calcium elevations. J. Biol. Chem. 2011, 286, 11672–11684. [Google Scholar] [CrossRef] [Green Version]
- Basak, N.P.; Banerjee, S. Mitochondrial dependency in progression of acute myeloid leukemia. Mitochondrion 2015, 21, 41–48. [Google Scholar] [CrossRef]
- Putten, W.V.; Sc, M.; Schouten, H.C.; Graux, C.; Ferrant, A.; Sonneveld, P.; Biemond, B.J.; Gratwohl, A.; Greef, G.E.D.; Verdonck, L.F.; et al. Cytarabine Dose for Acute Myeloid Leukemia. N. Engl. J. Med. 2011, 1027–1036. [Google Scholar]
- Greenberg, P.L.; Lee, S.J.; Advani, R.; Tallman, M.S.; Sikic, B.I.; Letendre, L.; Dugan, K.; Lum, B.; Chin, D.L.; Dewald, G.; et al. Mitoxantrone, Etoposide, and Cytarabine with or without Valspodar in Patients with Relapsed or Refractory Acute Myeloid Leukemia and High-Risk Myelodysplastic Syndrome: A Phase III Trial (E2995). J. Clin. Oncol. 2004, 22, 1078. [Google Scholar] [CrossRef]
- Teuffel, O.; Leibundgut, K.; Lehrnbecher, T.; Alonzo, T.A.; Beyene, J.; Sung, L. Anthracyclines during induction therapy in acute myeloid leukaemia: A systematic review and meta-analysis. Br. J. Haematol. 2013, 161, 192–203. [Google Scholar] [CrossRef] [Green Version]
- Hole, P.S.; Zabkiewicz, J.; Munje, C.; Newton, Z.; Pearn, L.; White, P.; Marquez, N.; Hills, R.K.; Burnett, A.K.; Tonks, A.; et al. Overproduction of NOX-derived ROS in AML promotes proliferation and is associated with defective oxidative stress signaling. Blood 2013, 122, 3322–3330. [Google Scholar] [CrossRef]
- Naik, J.; Themeli, M.; de Jong-Korlaar, R.; Ruiter, R.W.J.; Poddighe, P.J.; Yuan, H.; de Bruijin, J.D.; Ossenkoppele, G.J.; Zweegman, S.; Smit, L.; et al. CD38 as a therapeutic target for adult acute myeloid leukemia and T-cell acute lymphoblastic leukemia Acute. Haematologica 2019, 14, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, N.; Kumar, S.K. Daratumumab in untreated newly diagnosed multiple myeloma. Ther. Adv. Hematol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Mateos, M.; Spencer, A.; Nooka, A.K.; Pour, L.; Weisel, K.; Cavo, M.; Laubach, J.P.; Cook, G. Daratumumab-based regimens are highly effective and well tolerated in relapsed or refractory multiple myeloma regardless of patient age: Subgroup analysis of the phase 3 CASTOR and POLLUX studies. Haematologica 2020, 105, 468–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mistry, J.; Hellmich, C.; Moore, J.A.; Marlein, C.; Pillinger, G.; Collings, A.; Bowles, K.; Rushworth, S. Daratumumab Inhibits AML Metabolic Capacity and Tumor Growth through Inhibition of CD38 Mediated Mitochondrial Transfer from Bone Marrow Stromal Cells to Blasts in the Leukemic Microenvironment. Blood 2019. [Google Scholar] [CrossRef]
- Farber, M.; Arnold, L.; Chen, Y.; Mollmann, M.; Duehrsen, U.; Hanoun, M. Inhibition of CD38 Shows Anti-Leukemic Activity in Acute Myeloid Leukemia. Blood 2018, 132, 1456. [Google Scholar] [CrossRef]
- Saba, F.; Soleimani, M.; Abroun, S. New role of hypoxia in pathophysiology of multiple myeloma through miR-210. EXCLI J. 2018, 17, 647–662. [Google Scholar] [CrossRef]
- Lipchick, B.; Fink, E.; Nikiforoc, M. Oxidative Stress and Proteasome Inhibitors in Multiple Myeloma. Pharmacol. Res. 2016, 105, 210–215. [Google Scholar] [CrossRef] [Green Version]
- Krejcik, J.; Casneuf, T.; Nijhof, I.S.; Verbist, B.; Bald, J.; Plesner, T.; Syed, K.; Liu, K.; van de Donk, N.W.C.J.; Weiss, B.M.; et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood 2016, 128, 384–394. [Google Scholar] [CrossRef] [Green Version]
- Moreno, A.L.; Perez, C.; Zabaleta, A.; Manrique, I.; Garate, S.; Jelinek, T.; Segura, V.; Moreno, C. The Mechanism of Action of The Anti-CD38 Monoclonal Antibody Isatuximab In Multiple Myeloma. Clin. Cancer Res. 2019, 25, 3176–3187. [Google Scholar] [CrossRef] [Green Version]
- Martin, T.; Strickland, S.; Glenn, M.; Charpentier, E.; Guillemin, H.; Hsu, K.; Mikhael, J. Phase I trial of isatuximab monotherapy in the treatment of refractory multiple myeloma. Blood Cancer J. 2019, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Spencer, A.; Lentzsch, S.; Weisel, K.; Avet-Loiseau, H.; Mark, T.M.; Spicka, I.; Masszi, T.; Lauri, B.; Levin, M.D.; Bosi, A.; et al. Daratumumab plus bortezomib and dexamethasone versus bortezomib and dexamethasone in relapsed or refractory multiple myeloma: Updated analysis of CASTOR. Haematologica 2018, 103, 2079–2087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Read, M.A.; Neish, A.S.; Luscinskas, F.W.; Palombella, V.J.; Yaniatis, T.; Collins, T. The Proteasome Pathway is Required for Cytokine-Induced Endothelial-Leukocyte Adhesion Molecule Expression. Immunity 1995, 2, 493–506. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, D.; Uchiyama, H.; Akbarali, Y.; Urashima, M.; Yamamoto, K.I.; Libermann, T.A.; Anderson, K.C. Multiple myeloma cell adhesion-induced interleukin-6 expression in bone marrow stromal cells involves activation of NF-κB. Blood 1996, 87, 1104–1112. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Donor Cells | Recipient Cells | Mechanism of Transport | Triggers | Cellular Effect | Reference |
|---|---|---|---|---|---|
| NON-TUMOR CELLS | |||||
| cardio-myocytes | cardio-fibroblasts | TNTs | ND | Transfer in both directions | [56] |
| MSCs | vascular smooth muscle cells | TNTs | ND | Stimulation of MSCs proliferation | [28] |
| BMSCs | alveolar epithelium | microvesicles | LPS-induced lung injury | Protection against acute lung injury | [29] |
| MSCs | HUVEC | TNTs | Hypoxia | Rescue of injured endothelial cells | [26] |
| MSCs | Epithelial cells | TNTs | Miro1 overexpression | ND | [45] |
| iPSC-MSCs | epithelial cells | TNTs | Cigarette smoke | Repair of damaged cells | [27] |
| PC12 cells | PC12 cells | TNTs | Damaged mitochondria in receiver cells | Rescue from apoptosis | [25] |
| astrocytes | neurons | microvesicles | Damage by stroke | Neuroprotection/recovery | [19] |
| MSCs | corneal epithelial cells | TNTs | OXPHOS inhibition | Protection from oxidative damage | [30] |
| BM-MSCs | macrophage | TNTs | Acute respiratory distress syndrome | Enhanced phagocytosis | [31] |
| iPSC-MSCs/BM-MSCs | cardio-myocytes | TNTs | Anthracycline | Increased mitochondrial transfer | [44] |
| BMSCs | hematopoietic stem cells | not specified | Bacterial infection-induced ROS | Granulocytes activation | [32] |
| SOLID TUMORS | |||||
| BMSCs | A549 cells | not specified | Non-functional mitochondria | Rescue of aerobic respiration | [18] |
| BMSCs | 143B cells | not specified | Restrictive media | Rescue of mitochondria functions | [35] |
| MSCs/ epithelial cells | ovarian and breast cancer cells | TNTs | ND | Specific selection of donor cells | [21] |
| MSCs | lung adeno-carcinoma cells | TNTs | Miro1 increased mitochondrial donor capacity | ND | [45] |
| Wharton’s jelly-derived MSCs | 143B | not specified | Absence of mitochondria | Rescue of mitochondria functions | [36] |
| mouse tissue | melanoma cells | not specified | Absence of mitochondria | Rescue of mitochondria functions and tumor formation | [24] |
| Prostate cancer-associated fibroblasts | prostate cancer cell | TNTs | ND | Enhanced lactate metabolism and mitochondria motility | [63] |
| NKT cells | breast cancer cells | TNTs | ND | ND | [62] |
| HEMATOLOGICAL TUMORS | |||||
| BMSCs | AML | endocytosis | Chemotherapy agents | Increased viability | [37] |
| BMSCs | AML | TNTs | NOX2-derived ROS | ND | [33] |
| BMSCs | AML | TNTs | ND | ND | [38] |
| BMSCs | T-ALL | TNTs | ND | Chemoresistance | [64] |
| MSCs | ALL | TNTs | ROS | rescue from chemotheraphy | [22] |
| BMSCs | MM | TNTs | ND | Metabolic switch | [23] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sahinbegovic, H.; Jelinek, T.; Hrdinka, M.; Bago, J.R.; Turi, M.; Sevcikova, T.; Kurtovic-Kozaric, A.; Hajek, R.; Simicek, M. Intercellular Mitochondrial Transfer in the Tumor Microenvironment. Cancers 2020, 12, 1787. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12071787
Sahinbegovic H, Jelinek T, Hrdinka M, Bago JR, Turi M, Sevcikova T, Kurtovic-Kozaric A, Hajek R, Simicek M. Intercellular Mitochondrial Transfer in the Tumor Microenvironment. Cancers. 2020; 12(7):1787. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12071787
Chicago/Turabian StyleSahinbegovic, Hana, Tomas Jelinek, Matous Hrdinka, Juli R. Bago, Marcello Turi, Tereza Sevcikova, Amina Kurtovic-Kozaric, Roman Hajek, and Michal Simicek. 2020. "Intercellular Mitochondrial Transfer in the Tumor Microenvironment" Cancers 12, no. 7: 1787. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12071787