Clinical, Genomic, and Pharmacological Study of MYCN-Amplified RB1 Wild-Type Metastatic Retinoblastoma

 , , , , , , , and add
Show full author list
, , , , , , , and add
Show full author list
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
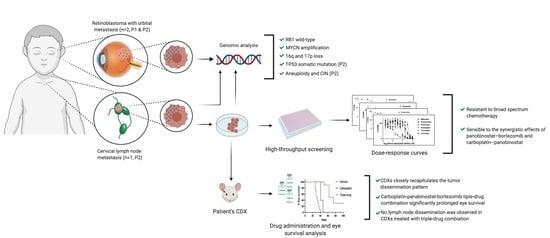
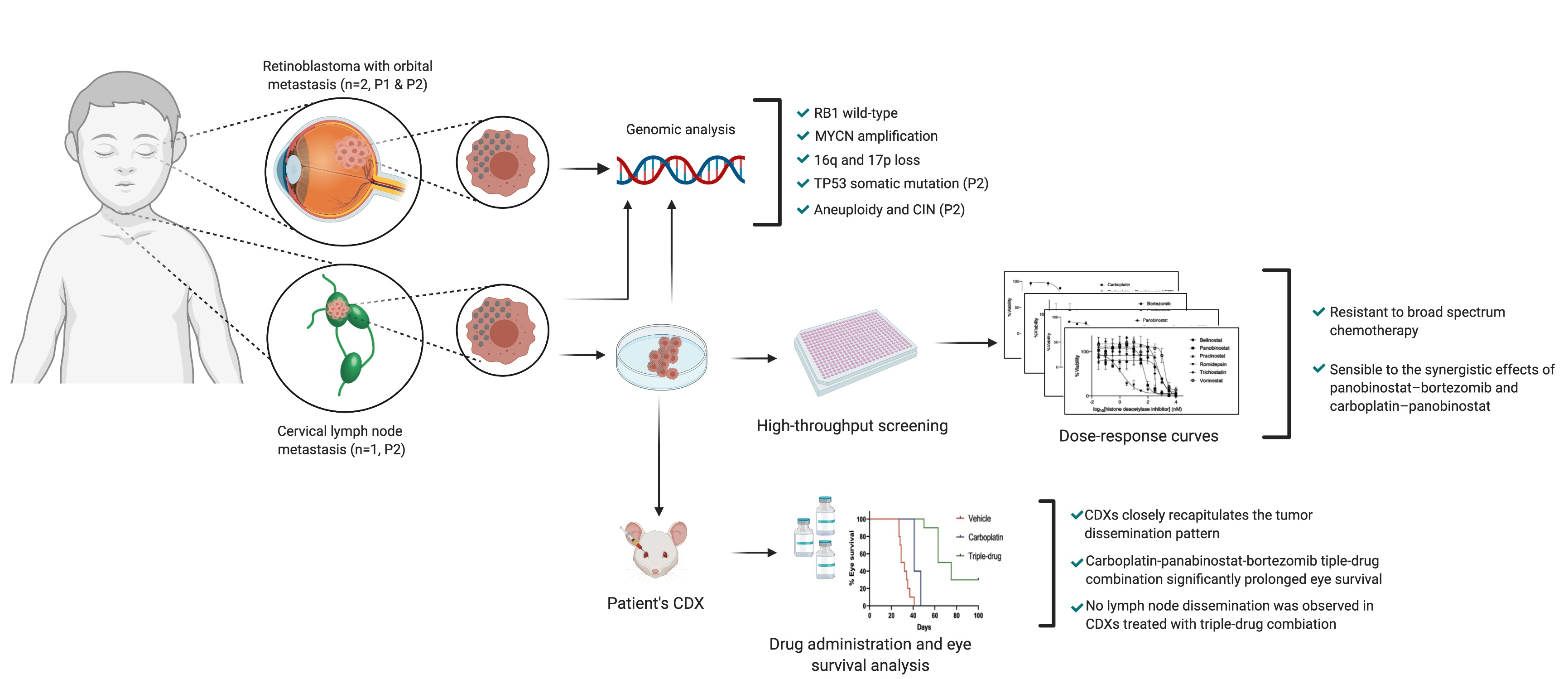
2. Results
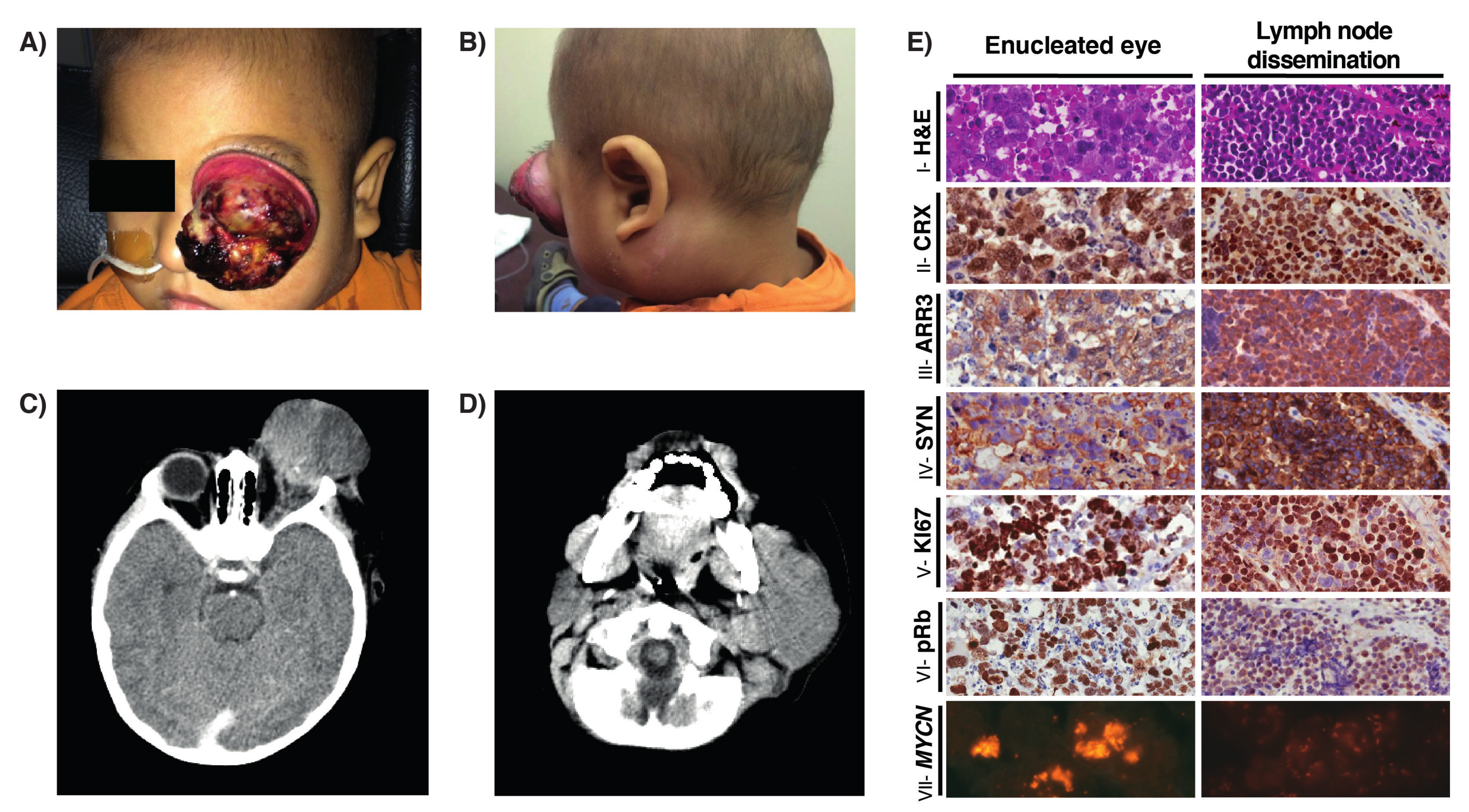
2.1. Report of Cases
2.1.1. Case 1
2.1.2. Case 2
2.2. Genomic Analysis
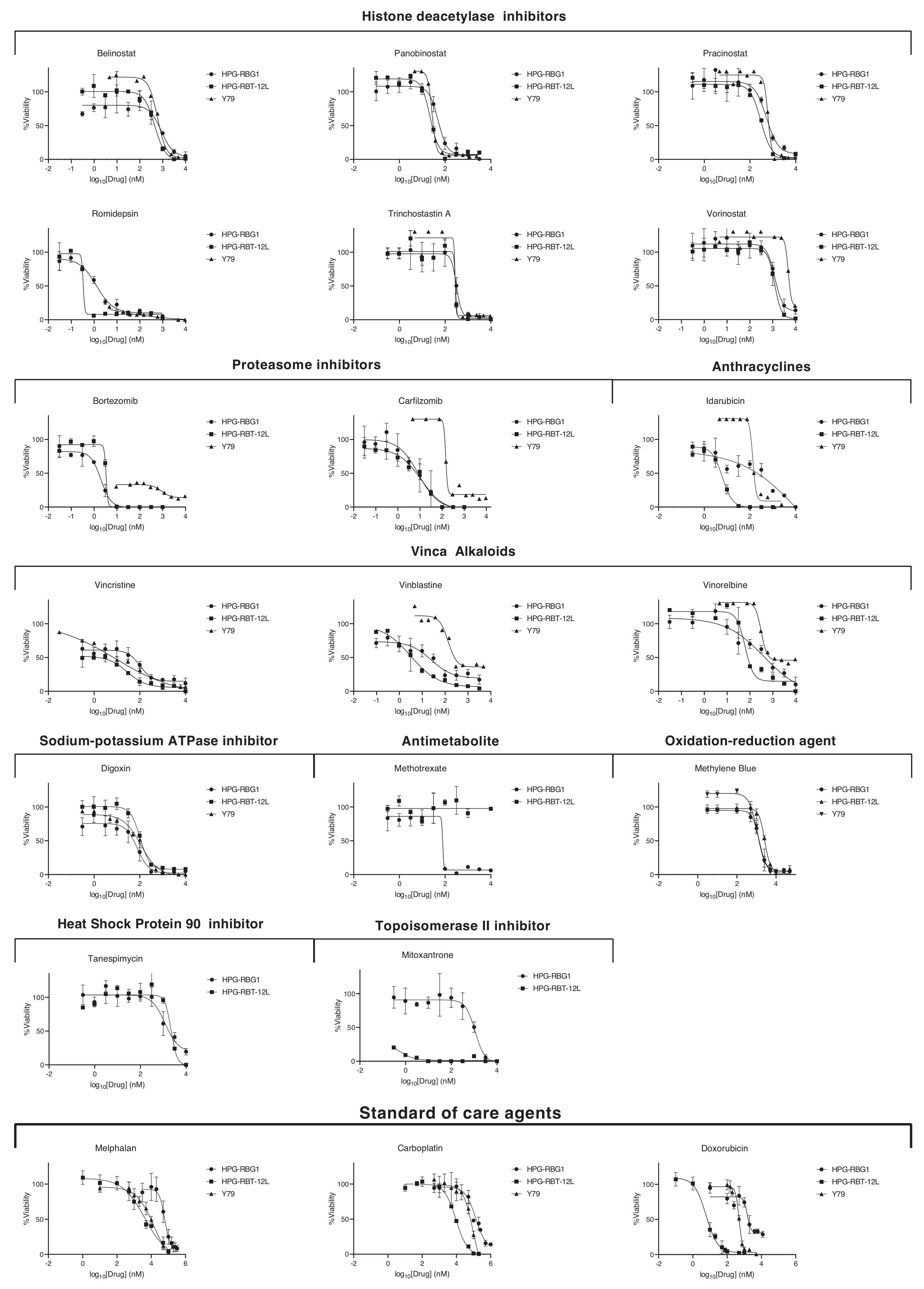
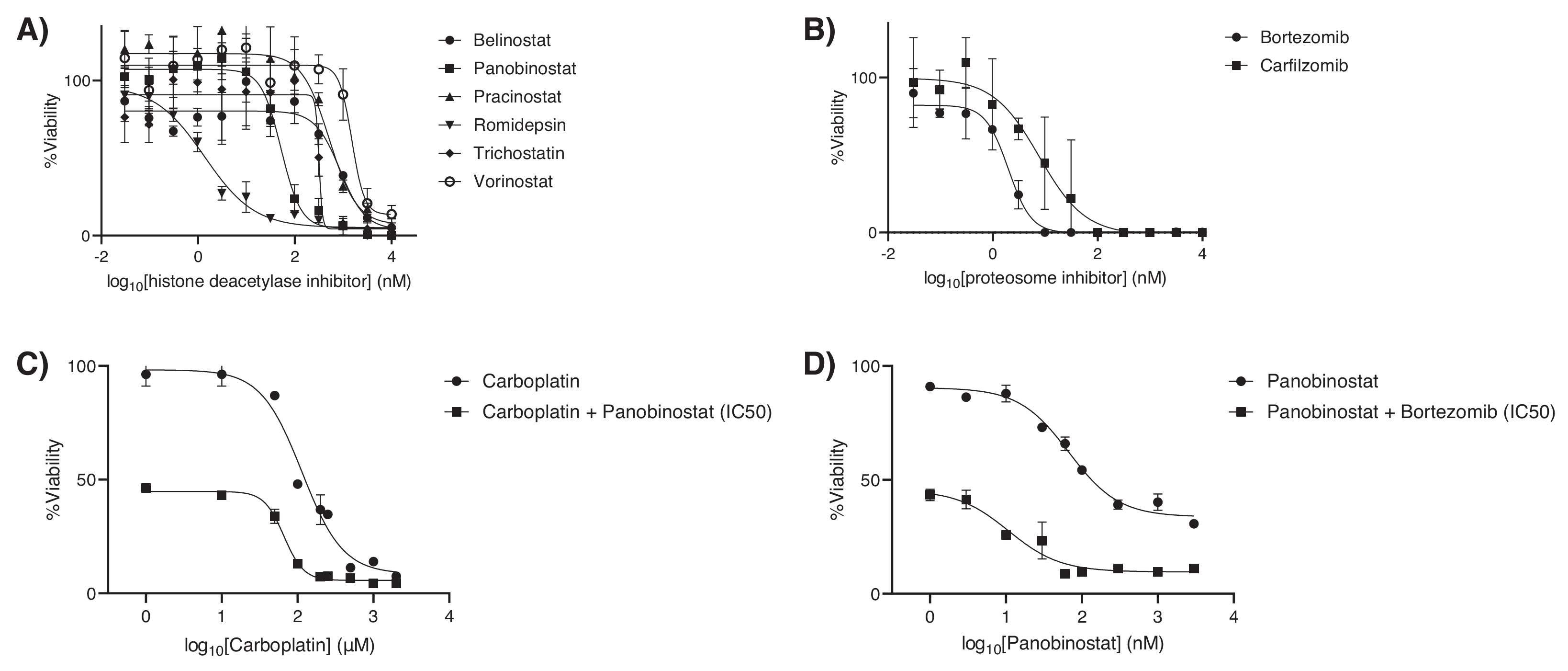
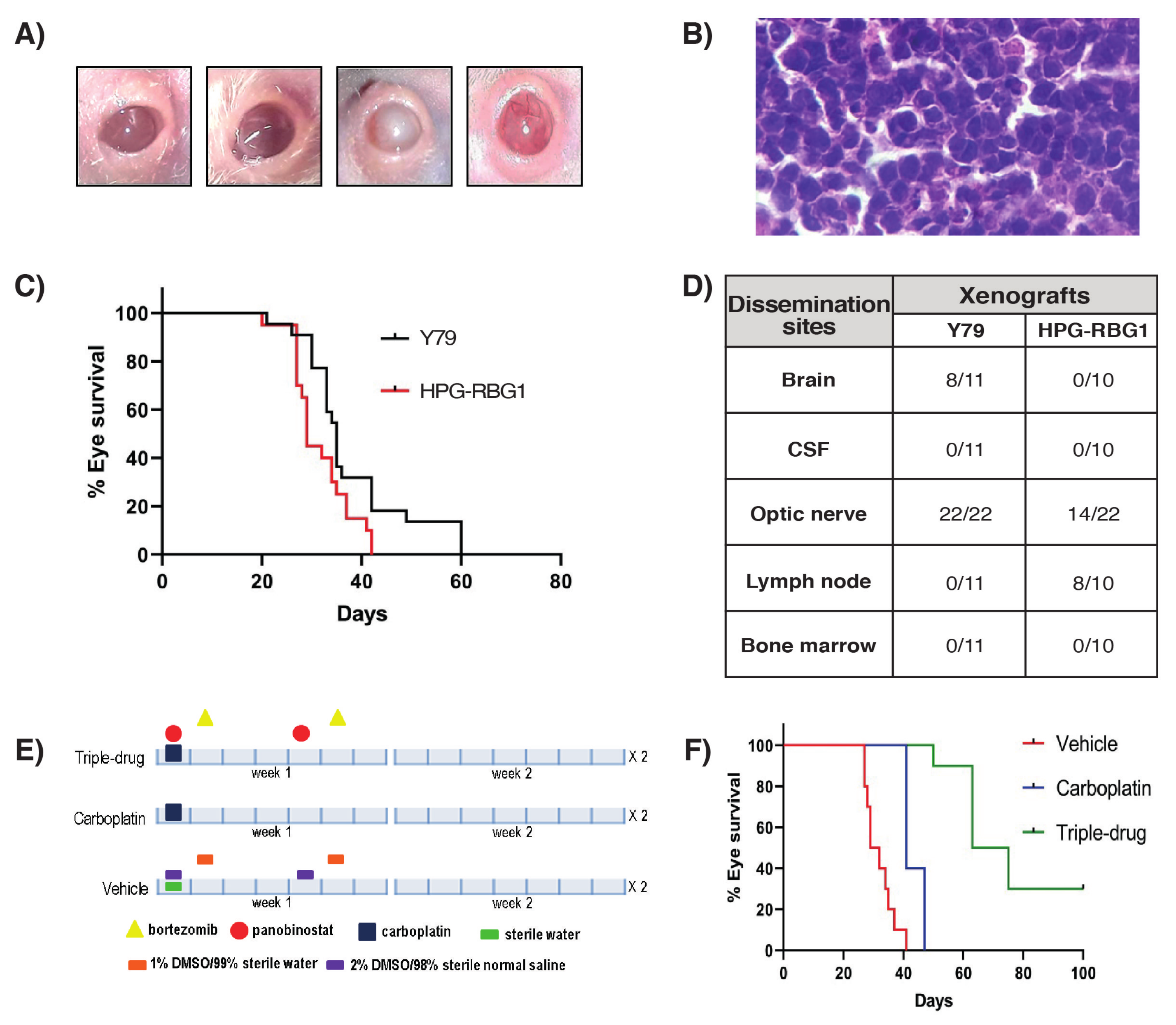
2.3. In Vitro and In Vivo Pharmacological Sensitivity
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Immunohistochemistry, Immunofluorescence, And Fluorescent In Situ Hybridization
4.3. Rb1 Alterations, Whole-Exome Sequencing and Oncoscan Copy Number Variation Analysis
4.4. Establishment and Characterization of Tumor Cell Line
4.5. Drug Screening, In Vitro and In Vivo Pharmacological Evaluations
4.6. Establishment, Characterization and Pharmacological Evaluation in a Patient-Derived Xenograft Model of MYCN RB1
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BAF | B-Allele frequency |
| CDX | Cell line derived xenograft |
| CNA | Copy number alteration |
| CNS | Central nervous system |
| COG-ARET0321 | Children’s Oncology Group trial (ARET0321) |
| CSF | Cerebrospinal fluid |
| CT | Computerized tomography |
| FISH | Fluorescence in situ hybridization |
| HPG-RBG1 | Lymph node metastasis-derived cell line |
| HTS | High throughput screening |
| LOH | Loss of heterozygosity |
| LRR | Log-ratio (Base-2 logarithmic ratio) |
| MLPA | Multiplex ligation-dependent probe amplification |
| MRI | Magnetic resonance imaging |
| MYCN | MYCN amplified |
| RB1 | wild-type RB1 |
| RB1 | aberrant RB1 |
| SEM | Standard error of the mean |
| STR | Short tandem repeat |
| WES | Whole exome sequencing |
| WGD | Whole genome doubling |
References
- Knudson, A.G. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corson, T.W.; Gallie, B.L. One hit, two hits, three hits, more? Genomic changes in the development of retinoblastoma Genes Chromosom. Cancer 2007, 46, 617–634. [Google Scholar] [CrossRef] [PubMed]
- Dimaras, H.; Khetan, V.; Halliday, W.; Orlic, M.; Prigoda, N.L.; Piovesan, B.; Marrano, P.; Corson, T.W.; Eagle, R.C.; Squire, J.A.; et al. Loss of RB1 induces non-proliferative retinoma: Increasing genomic instability correlates with progression to retinoblastoma. Hum. Mol. Genet. 2008, 17, 1363–1372. [Google Scholar] [CrossRef] [Green Version]
- Singh, H.P.; Wang, S.; Stachelek, K.; Lee, S.; Reid, M.W.; Thornton, M.E.; Craft, C.M.; Grubbs, B.H.; Cobrinik, D. Developmental stage-specific proliferation and retinoblastoma genesis in RB-deficient human but not mouse cone precursors. Proc. Natl. Acad. Sci. USA 2018, 115, E9391–E9400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munier, F.L.; Beck-Popovic, M.; Chantada, G.L.; Cobrinik, D.; Kivelä, T.T.; Lohmann, D.; Maeder, P.; Moll, A.C.; Carcaboso, A.M.; Moulin, A.; et al. Conservative management of retinoblastoma: Challenging orthodoxy without compromising the state of metastatic grace. “Alive, with good vision and no comorbidity”. Prog. Retin. Eye Res. 2019, 73, 100764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rushlow, D.E.; Mol, B.M.; Kennett, J.Y.; Yee, S.; Pajovic, S.; Thériault, B.L.; Prigoda-Lee, N.L.; Spencer, C.; Dimaras, H.; Corson, T.W.; et al. Characterisation of retinoblastomas without RB1 mutations: Genomic, gene expression, and clinical studies. Lancet Oncol. 2013, 14, 327–334. [Google Scholar] [CrossRef] [Green Version]
- Torbidoni, A.V.; Laurent, V.E.; Sampor, C.; Ottaviani, D.; Vazquez, V.; Gabri, M.R.; Rossi, J.; de Dávila, M.T.; Alonso, C.; Alonso, D.F.; et al. Association of Cone-Rod Homeobox Transcription Factor Messenger RNA With Pediatric Metastatic Retinoblastoma. JAMA Ophthalmol. 2015, 133, 805–812. [Google Scholar] [CrossRef] [Green Version]
- Laurent, V.E.; Torbidoni, A.V.; Sampor, C.; Ottaviani, D.; Vazquez, V.; Gabri, M.R.; Garcia De Davila, M.T.; Ramirez-Ortiz, M.A.; Alonso, C.N.; Rossi, J.; et al. Minimal disseminated disease in no nMetastatic retinoblastoma with high-risk pathologic features and association with disease-free survival. JAMA Ophthalmol. 2016, 134, 1374–1379. [Google Scholar] [CrossRef]
- Laurent, V.E.; Sampor, C.; Solernou, V.; Rossi, J.; Gabri, M.; Eandi-Eberle, S.; De Davila, M.T.; Alonso, D.F.; Chantada, G.L. Detection of minimally disseminated disease in the cerebrospinal fluid of children with high-risk retinoblastoma by reverse transcriptase-polymerase chain reaction for GD2 synthase mRNA. Eur. J. Cancer 2013, 49, 2892–2899. [Google Scholar] [CrossRef]
- Chantada, G.L.; Rossi, J.; Casco, F.; Fandiño, A.; Scopinaro, M.; De Dávila, M.T.G.; Abramson, D.H. An aggressive bone marrow evaluation including immunocytology with GD2 for advanced retinoblastoma. J. Pediatr. Hematol. 2006, 28, 369–373. [Google Scholar] [CrossRef]
- McEvoy, J.; Nagahawatte, P.; Finkelstein, D.; Richards-Yutz, J.; Valentine, M.; Ma, J.; Mullighan, C.; Song, G.; Chen, X.; Wilson, M.; et al. RB1 gene inactivation by chromothripsis in human retinoblastoma. Oncotarget 2014, 5, 438–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef] [PubMed]
- Dimaras, H.; Corson, T.W.; Cobrinik, D.; White, A.; Zhao, J.; Munier, F.L.; Abramson, D.H.; Shields, C.L.; Chantada, G.L.; Njuguna, F.; et al. Retinoblastoma. Nat. Rev. Dis. Prim. 2015, 1. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Benavente, C.A.; McEvoy, J.; Flores-Otero, J.; Ding, L.; Chen, X.; Ulyanov, A.; Wu, G.; Wilson, M.; Wang, J.; et al. A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature 2012, 481, 329–334. [Google Scholar] [CrossRef]
- Winter, U.; Ganiewich, D.; Ottaviani, D.; Zugbi, S.; Aschero, R.; Sendoya, J.M.; Cafferata, E.G.; Mena, M.; Sgroi, M.; Sampor, C.; et al. Genomic and transcriptomic tumor heterogeneity in bilateral retinoblastoma. JAMA Ophthalmol. 2020. [Google Scholar] [CrossRef]
- Van Veggel, M.; Westerman, E.; Hamberg, P. Clinical Pharmacokinetics and Pharmacodynamics of Panobinostat. Clin. Pharmacokinet. 2018, 57, 21–29. [Google Scholar] [CrossRef]
- Wood, P.J.; Strong, R.; McArthur, G.A.; Michael, M.; Algar, E.; Muscat, A.; Rigby, L.; Ferguson, M.; Ashley, D.M. A phase I study of panobinostat in pediatric patients with refractory solid tumors, including CNS tumors. Cancer Chemother. Pharmacol. 2018, 82, 493–503. [Google Scholar] [CrossRef]
- Hennika, T.; Hu, G.; Olaciregui, N.G.; Barton, K.L.; Ehteda, A.; Chitranjan, A.; Chang, C.; Gifford, A.J.; Tsoli, M.; Ziegler, D.S.; et al. Pre-clinical study of panobinostat in xenograft and genetically engineered murine diffuse intrinsic pontine glioma models. PLoS ONE 2017, 12, 9485. [Google Scholar] [CrossRef] [Green Version]
- Stewart, E.; Federico, S.M.; Chen, X.; Shelat, A.A.; Bradley, C.; Gordon, B.; Karlstrom, A.; Twarog, N.R.; Clay, M.R.; Bahrami, A.; et al. Orthotopic patient-derived xenografts of paediatric solid tumours. Nature 2017, 549, 96–100. [Google Scholar] [CrossRef]
- Chawla, B.; Hasan, F.; Seth, R.; Pathy, S.; Pattebahadur, R.; Sharma, S.; Upadhyaya, A.; Azad, R. Multimodal Therapy for Stage III Retinoblastoma (International Retinoblastoma Staging System): A Prospective Comparative Study. Ophthalmology 2016, 123, 1933–1939. [Google Scholar] [CrossRef] [PubMed]
- Leal-Leal, C.A.; Rivera-Luna, R.; Flores-Rojo, M.; Juárez-Echenique, J.C.; Ordaz, J.C.; Amador-Zarco, J. Survival in extra-orbital metastatic retinoblastoma: Treatment results. Clin. Transl. Oncol. 2006, 8, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Canturk, S.; Qaddoumi, I.; Khetan, V.; Ma, Z.; Furmanchuk, A.; Antoneli, C.B.; Sultan, I.; Kebudi, R.; Sharma, T.; Rodriguez-Galindo, C.; et al. Survival of retinoblastoma in less-developed countries impact of socioeconomic and health-related indicators. Br. J. Ophthalmol. 2010, 94, 1432–1436. [Google Scholar] [CrossRef] [PubMed]
- Polski, A.; Xu, L.; Prabakar, R.K.; Gai, X.; Kim, J.W.; Shah, R.; Jubran, R.; Kuhn, P.; Cobrinik, D.; Hicks, J.; et al. Variability in retinoblastoma genome stability is driven by age and not heritability. Genes Chromosom. Cancer 2020. [Google Scholar] [CrossRef]
- Berry, J.L.; Xu, L.; Murphree, A.L.; Krishnan, S.; Stachelek, K.; Zolfaghari, E.; McGovern, K.; Lee, T.C.; Carlsson, A.; Kuhn, P.; et al. Potential of aqueous humor as a surrogate tumor biopsy for retinoblastoma. JAMA Ophthalmol. 2017, 135, 1221–1230. [Google Scholar] [CrossRef] [Green Version]
- Chevez-Barrios, P.; Hurwitz, M.Y.; Louie, K.; Marcus, K.T.; Holcombe, V.N.; Schafer, P.; Aguilar-Cordova, C.E.; Hurwitz, R.L. Metastatic and no nMetastatic models of retinoblastoma. Am. J. Pathol. 2000, 157, 1405–1412. [Google Scholar] [CrossRef] [Green Version]
- McClean, I.; Burnier, M.; Zimmerman, L.J.F. Tumors of the retina. Tumors of the eye and adnexa. Atlas of Tumor Pathology. Am. J. Surg. Pathol. Febr. 1994, 22, 100–135. [Google Scholar]
- Honavar, S.G.; Manjandavida, F.P.; Reddy, V.A.P. Orbital retinoblastoma: An update. Rev. Artic. 2017, 65, 435–442. [Google Scholar] [CrossRef]
- Pascual-Pasto, G.; Olaciregui, N.G.; Vila-Ubach, M.; Paco, S.; Monterrubio, C.; Rodriguez, E.; Winter, U.; Batalla-Vilacis, M.; Catala, J.; Salvador, H.; et al. Preclinical platform of retinoblastoma xenografts recapitulating human disease and molecular markers of dissemination. Cancer Lett. 2016, 380, 10–19. [Google Scholar] [CrossRef]
- MacPherson, D.; Conkrite, K.; Tam, M.; Mukai, S.; Mu, D.; Jacks, T. Murine bilateral retinoblastoma exhibiting rapid-onset, metastatic progression and N-myc gene amplification. EMBO J. 2007, 26, 784–794. [Google Scholar] [CrossRef] [Green Version]
- Donehower, L.A.; Soussi, T.; Korkut, A.; Liu, Y.; Schultz, A.; Cardenas, M.; Li, X.; Babur, O.; Hsu, T.K.; Lichtarge, O.; et al. Integrated Analysis of TP53 Gene and Pathway Alterations in The Cancer Genome Atlas. Cell Rep. 2019, 28, 1370–1384.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alejandro Sweet-Cordero, E.; Biegel, J.A. The genomic landscape of pediatric cancers: Implications for diagnosis and treatment. Science 2019, 363, 1170–1175. [Google Scholar] [CrossRef] [PubMed]
- Oh, L.; Hafsi, H.; Hainaut, P.; Ariffin, H. P53, stem cell biology and childhood blastomas. Curr. Opin. Oncol. 2019, 31, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.R.; Ramkissoon, S.H.; Ross, J.; Weintraub, L. Tumor mutational burden and driver mutations: Characterizing the genomic landscape of pediatric brain tumors. Pediatr. Blood Cancer 2020, 67. [Google Scholar] [CrossRef]
- Kato, M.V.; Shimizu, T.; Ishizaki, K.; Kaneko, A.; Yandell, D.W.; Toguchida, J.; Sasaki, M.S. Loss of heterozygosity on chromosome 17 and mutation of the p53 gene in retinoblastoma. Cancer Lett. 1996, 106, 75–82. [Google Scholar] [CrossRef]
- Livide, G.; Epistolato, M.C.; Amenduni, M.; Disciglio, V.; Marozza, A.; Mencarelli, M.A.; Toti, P.; Lazzi, S.; Hadjistilianou, T.; De Francesco, S.; et al. Epigenetic and copy number variation analysis in retinoblastoma by MS-MLPA. Pathol. Oncol. Res. 2012, 18, 703–712. [Google Scholar] [CrossRef]
- Guo, Y.; Pajovic, S.; Gallie, B.L. Expression of p14ARF, MDM2, and MDM4 in human retinoblastoma. Biochem. Biophys. Res. Commun. 2008, 375, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Laurie, N.A.; Donovan, S.L.; Shih, C.S.; Zhang, J.; Mills, N.; Fuller, C.; Teunisse, A.; Lam, S.; Ramos, Y.; Mohan, A.; et al. Inactivation of the p53 pathway in retinoblastoma. Nature 2006, 444, 61–66. [Google Scholar] [CrossRef]
- Castéra, L.; Sabbagh, A.; Dehainault, C.; Michaux, D.; Mansuet-Lupo, A.; Patillon, B.; Lamar, E.; Aerts, I.; Lumbroso-Le Rouic, L.; Couturier, J.; et al. MDM2 as a modifier gene in retinoblastoma. J. Natl. Cancer Inst. 2010, 102, 1805–1808. [Google Scholar] [CrossRef]
- Conkrite, K.; Sundby, M.; Mu, D.; Mukai, S.; MacPherson, D. Cooperation between Rb and Arf in suppressing mouse retinoblastoma. J. Clin. Investig. 2012, 122, 1726–1733. [Google Scholar] [CrossRef] [Green Version]
- Benavente, C.A.; Dyer, M.A. Genetics and Epigenetics of Human Retinoblastoma. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 547–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, A.M.; Shih, J.; Ha, G.; Gao, G.F.; Zhang, X.; Berger, A.C.; Schumacher, S.E.; Wang, C.; Hu, H.; Liu, J.; et al. Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell 2018, 33, 676–689.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, J.; Valind, A.; Holmquist Mengelbier, L.; Bredin, S.; Cor nMark, L.; Jansson, C.; Wali, A.; Staaf, J.; Viklund, B.; Øra, I.; et al. Four evolutionary trajectories underlie genetic intratumoral variation in childhood cancer. Nat. Genet. 2018, 50, 944–950. [Google Scholar] [CrossRef]
- Ben-David, U.; Amon, A. Context is everything: Aneuploidy in cancer. Nat. Rev. Genet. 2020. [Google Scholar] [CrossRef]
- Hill, R.M.; Kuijper, S.; Lindsey, J.C.; Petrie, K.; Schwalbe, E.C.; Barker, K.; Boult, J.K.; Williamson, D.; Ahmad, Z.; Hallsworth, A.; et al. Combined MYC and P53 defects emerge at medulloblastoma relapse and define rapidly progressive, therapeutically targetable disease. Cancer Cell 2015, 27, 72–84. [Google Scholar] [CrossRef]
- Alexandrova, E.M.; Mirza, S.A.; Xu, S.; Schulz-Heddergott, R.; Marchenko, N.D.; Moll, U.M. P53 loss-of-heterozygosity is a necessary prerequisite for mutant p53 stabilization and gain-of-function in vivo. Cell Death Dis. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Andersson, N.; Bakker, B.; Karlsson, J.; Valind, A.; Mengelbier, L.H.; Spierings, D.C.; Foijer, F.; Gisselsson, D. Extensive clonal branching shapes the evolutionary history of high-risk pediatric cancers. Cancer Res. 2020, 80, 1512–1523. [Google Scholar] [CrossRef]
- Glubrecht, D.D.; Kim, J.H.; Russell, L.; Bamforth, J.S.; Godbout, R. Differential CRX and OTX2 expression in human retina and retinoblastoma. J. Neurochem. 2009, 111, 250–263. [Google Scholar] [CrossRef] [Green Version]
- Deng, C.; Dai, R.; Li, X.; Liu, F. Genetic variation frequencies in Wilms’ tumor: A meta-analysis and systematic review. Cancer Sci. 2016, 107, 690–699. [Google Scholar] [CrossRef]
- Schneiderman, J.; London, W.B.; Brodeur, G.M.; Castleberry, R.P.; Look, A.T.; Cohn, S.L. Clinical significance of MYCN amplification and ploidy in favorable-stage neuroblastoma: A report from the Children’s Oncology Group. J. Clin. Oncol. 2008, 26, 913–918. [Google Scholar] [CrossRef]
- Schleiermacher, G.; Janoueix-Lerosey, I.; Ribeiro, A.; Klijanienko, J.; Couturier, J.; Pierron, G.; Mosseri, V.; Valent, A.; Auger, N.; Plantaz, D.; et al. Accumulation of segmental alterations determines progression in neuroblastoma. J. Clin. Oncol. 2010, 28, 3122–3130. [Google Scholar] [CrossRef]
- Hegarty, S.V.; Togher, K.L.; O’Leary, E.; Solger, F.; Sullivan, A.M.; O’Keeffe, G.W. Romidepsin induces caspase-dependent cell death in human neuroblastoma cells. Neurosci. Lett. 2017, 653, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Weiss, W.A. Neuroblastoma and MYCN. Cold Spring Harb. Perspect. Med. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Shahbazi, J.; Liu, P.Y.; Atmadibrata, B.; Bradner, J.E.; Marshall, G.M.; Lock, R.B.; Liu, T. The bromodomain inhibitor jq1 and the histone deacetylase inhibitor panobinostat synergistically reduce n-myc expression and induce anticancer effects. Clin. Cancer Res. 2016, 22, 2534–2544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Jiang, J.; Chen, H.; Wang, L.; Guo, H.; Yang, L.; Xiao, D.; Qing, G.; Liu, H. FDA-approved drug screen identifies proteasome as a synthetic lethal target in MYC-driven neuroblastoma. Oncogene 2019, 38, 6737–6751. [Google Scholar] [CrossRef] [PubMed]
- Boccadoro, M.; Morgan, G.; Cavenagh, J. Preclinical evaluation of the proteasome inhbitor bortezomib in cancer therapy. Cancer Cell Int. 2005, 5, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, J.; Regan, P.L.; Edo, R.; Leonhardt, P.; Jeng, E.I.; Rappaport, E.F.; Ikegaki, N.; Tang, X.X. Biological effects of induced MYCN hyper-expression in MYCN-amplified neuroblastomas. Int. J. Oncol. 2010, 37. [Google Scholar] [CrossRef]
- Ferrario, A.; Luna, M.; Rucker, N.; Wong, S.; Lederman, A.; Kim, J.; Gomer, C. Targeting survivin enhances chemosensitivity in retinoblastoma cells and orthotopic tumors. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [Green Version]
- Henssen, A.; Thor, T.; Odersky, A.; Heukamp, L.; El-Hindy, N.; Beckers, A.; Speleman, F.; Althoff, K.; Schäfers, S.; Schramm, A.; et al. BET bromodomain protein inhibition is a therapeutic option for medulloblastoma. Oncotarget 2013, 4, 2080–2095. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Carbonero, R.; Carnero, A.; Paz-Ares, L. Inhibition of HSP90 molecular chaperones: Moving into the clinic. Lancet Oncol. 2013, 14, e358–e369. [Google Scholar] [CrossRef]
- Wu, W.C.; Wu, M.H.; Chang, Y.C.; Hsieh, M.C.; Wu, H.J.; Cheng, K.C.; Lai, Y.H.; Kao, Y.H. Geldanamycin and its analog induce cytotoxicity in cultured human retinal pigment epithelial cells. Exp. Eye Res. 2010, 91, 211–219. [Google Scholar] [CrossRef]
- Lorenzon, I.; Pellarin, I.; Pellizzari, I.; D’Andrea, S.; Belletti, B.; Sonego, M.; Baldassarre, G.; Schiappacassi, M. Identification and Characterization of a New Platinum-Induced TP53 Mutation in MDAH Ovarian Cancer Cells. Cells 2019, 9, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyault, S.; Drouet, Y.; Navarro, C.; Bachelot, T.; Lasset, C.; Treilleux, I.; Tabone, E.; Puisieux, A.; Wang, Q. Mutational characterization of individual breast tumors: TP53 and PI3K pathway genes are frequently and distinctively mutated in different subtypes. Breast Cancer Res. Treat. 2012, 132, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, J.; Pan, H.; Tai, M.C.; Maher, M.H.; Jia, R.; Ge, S.; Lu, L. Dinutuximab synergistically enhances the cytotoxicity of natural killer cells to retinoblastoma through the perforin-granzyme B pathway. Oncotargets Ther. 2020, 13, 3903–3920. [Google Scholar] [CrossRef] [PubMed]
- Parma, D.; Ferrer, M.; Luce, L.; Giliberto, F.; Szijan, I. RB1 gene mutations in Argentine retinoblastoma patients. Implications for genetic counseling. PLoS ONE 2017, 12. [Google Scholar] [CrossRef]
- Boeva, V.; Popova, T.; Bleakley, K.; Chiche, P.; Cappo, J.; Schleiermacher, G.; Janoueix-Lerosey, I.; Delattre, O.; Barillot, E. Control-FREEC: A tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics 2012, 28, 423–425. [Google Scholar] [CrossRef]
- Sherry, S.T.; Ward, M.; Sirotkin, K. dbSNP - database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res. 1999, 9, 677–679. [Google Scholar] [CrossRef]
- Winter, U.; Aschero, R.; Fuentes, F.; Buontempo, F.; Zugbi, S.; Sgroi, M.; Sampor, C.; Abramson, D.H.; Carcaboso, A.M.; Schaiquevich, P. Tridimensional Retinoblastoma Cultures as Vitreous Seeds Models for Live-Cell Imaging of Chemotherapy Penetration. Int. J. Mol. Sci. 2019, 20, 1077. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zugbi, S.; Ganiewich, D.; Bhattacharyya, A.; Aschero, R.; Ottaviani, D.; Sampor, C.; Cafferata, E.G.; Mena, M.; Sgroi, M.; Winter, U.; et al. Clinical, Genomic, and Pharmacological Study of MYCN-Amplified RB1 Wild-Type Metastatic Retinoblastoma. Cancers 2020, 12, 2714. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12092714
Zugbi S, Ganiewich D, Bhattacharyya A, Aschero R, Ottaviani D, Sampor C, Cafferata EG, Mena M, Sgroi M, Winter U, et al. Clinical, Genomic, and Pharmacological Study of MYCN-Amplified RB1 Wild-Type Metastatic Retinoblastoma. Cancers. 2020; 12(9):2714. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12092714
Chicago/Turabian StyleZugbi, Santiago, Daiana Ganiewich, Arpita Bhattacharyya, Rosario Aschero, Daniela Ottaviani, Claudia Sampor, Eduardo G. Cafferata, Marcela Mena, Mariana Sgroi, Ursula Winter, and et al. 2020. "Clinical, Genomic, and Pharmacological Study of MYCN-Amplified RB1 Wild-Type Metastatic Retinoblastoma" Cancers 12, no. 9: 2714. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12092714