Acquired Resistance to PD-1/PD-L1 Blockade in Lung Cancer: Mechanisms and Patterns of Failure

 ,
,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. PD-1/PD-L1 Axis in Normal Health and Tumorigenesis
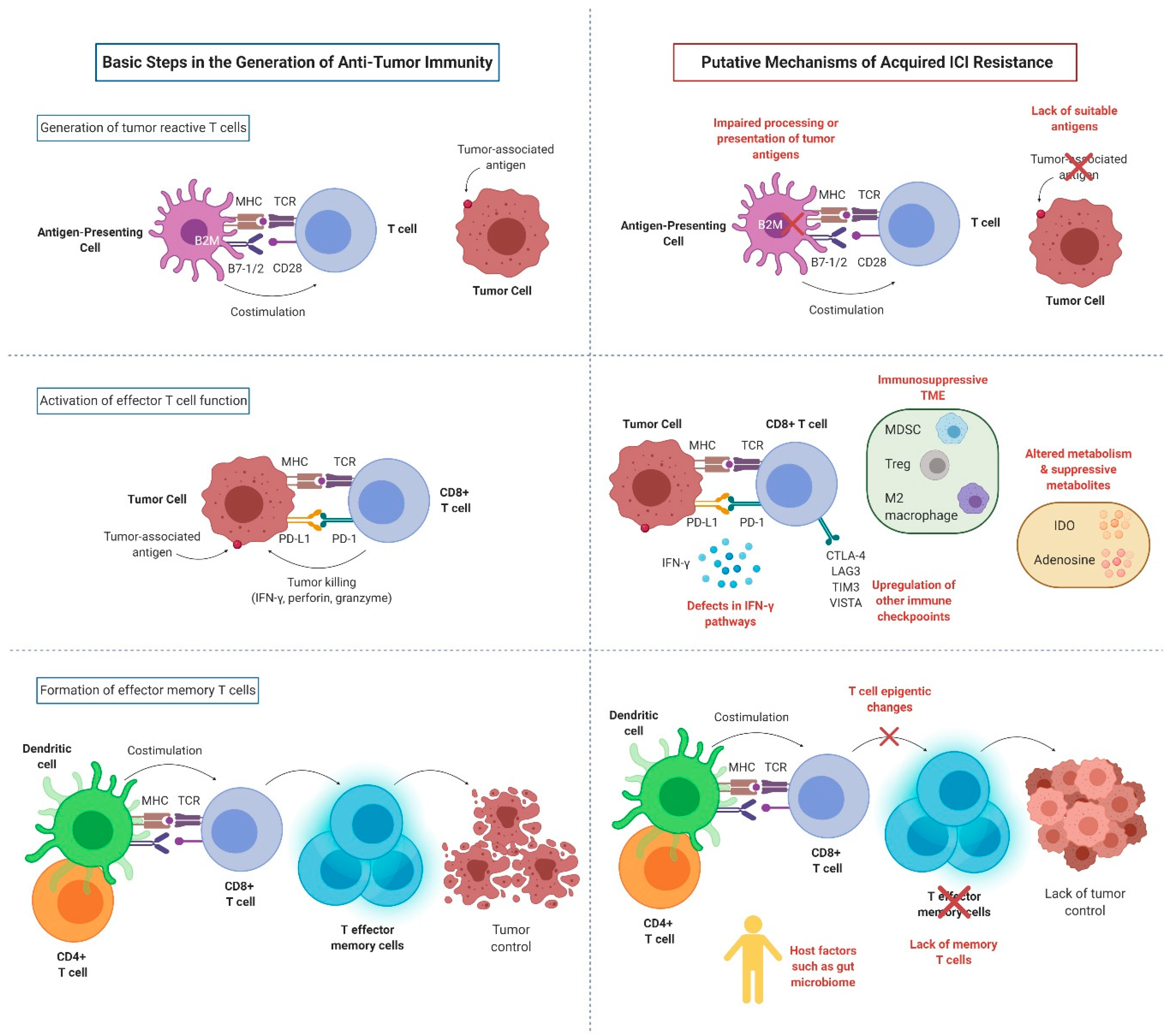
3. Mechanisms of Acquired ICI Resistance
3.1. Impaired T Cell Activation
3.1.1. Loss of Antigen Presentation
3.1.2. Fixed Epigenetic Modification and Lack of Memory T Cells
3.2. Impaired T Cell Effector Functions
3.2.1. Defects of Interferon Pathways
3.2.2. Upregulation of Other Immune Checkpoints
3.2.3. Altered Metabolism and Suppressive Metabolites
3.3. Other Local Immunosuppressive Factors within the TME
3.4. Role of Host Factors in ICI Resistance
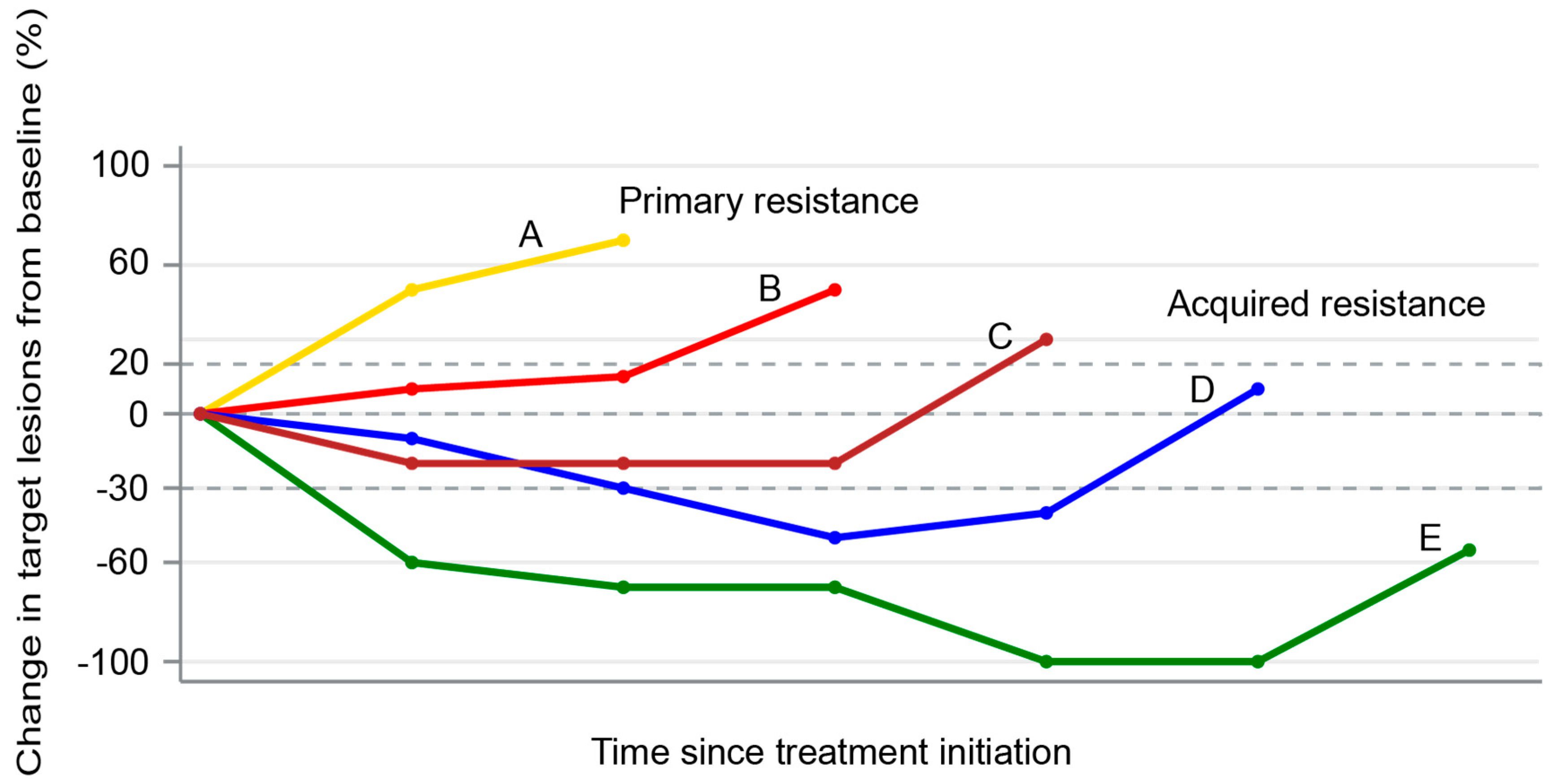
4. Patterns of Failures After PD-1/PD-L1 Blockade
5. Principles of Overcoming Acquired Resistance to PD-1/PD-L1 Blockade
6. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer. 2018, 118, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbour, K.; Shen, R.; Plodkowski, A.; Rizvi, H.; Ni, A.; Long, N.; Halpenny, D.; Sanchez-Vega, F.; Rudin, C.; Riely, G.; et al. MA19.09 Concurrent Mutations in STK11 and KEAP1 is Associated with Resistance to PD-(L)1 Blockade in Patients with NSCLC Despite High TMB. J. Thorac. Oncol. 2018, 13, S424. [Google Scholar] [CrossRef] [Green Version]
- Toyokawa, G.; Takada, K.; Tagawa, T.; Hamamoto, R.; Yamada, Y.; Shimokawa, M.; Oda, Y.; Maehara, Y. A Positive Correlation Between the EZH2 and PD-L1 Expression in Resected Lung Adenocarcinomas. Ann. Thorac. Surg. 2019, 107, 393–400. [Google Scholar] [CrossRef]
- Anagnostou, V.; Smith, K.N.; Forde, P.M.; Niknafs, N.; Bhattacharya, R.; White, J.; Zhang, T.; Adleff, V.; Phallen, J.; Wali, N.; et al. Evolution of Neoantigen Landscape during Immune Checkpoint Blockade in Non–Small Cell Lung Cancer. Cancer Discov. 2017, 7, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Gettinger, S.; Choi, J.; Hastings, K.; Truini, A.; Datar, I.; Sowell, R.; Wurtz, A.; Dong, W.; Cai, G.; Melnick, M.A.; et al. Impaired HLA Class I Antigen Processing and Presentation as a Mechanism of Acquired Resistance to Immune Checkpoint Inhibitors in Lung Cancer. Cancer Discov. 2017, 7, 1420–1435. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [Green Version]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, E.; Cheung, J.; Zhu, J.; Su, X.; Taylor, M.J.; Wallweber, H.A.; Sasmal, D.K.; Huang, J.; Kim, J.M.; Mellman, I.; et al. T cell costimulatory receptor CD28 is a primary target for PD-1–mediated inhibition. Science 2017, 355, 1428–1433. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, D.; Maruhashi, T.; Okazaki, I.-M.; Shimizu, K.; Maeda, T.K.; Takemoto, T.; Okazaki, T. Restriction of PD-1 function by cis-PD-L1/CD80 interactions is required for optimal T cell responses. Science 2019, 364, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer Genome Landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Laumont, C.M.; Vincent, K.; Hesnard, L.; Audemard, E.; Bonneil, É.; Laverdure, J.-P.; Gendron, P.; Courcelles, M.; Hardy, M.-P.; Côté, C.; et al. Noncoding regions are the main source of targetable tumor-specific antigens. Sci. Transl. Med. 2018, 10, eaau5516. [Google Scholar] [CrossRef] [Green Version]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.M.C.S.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198. [Google Scholar] [CrossRef]
- Hirano, F.; Kaneko, K.; Tamura, H.; Dong, H.; Wang, S.; Ichikawa, M.; Rietz, C.; Flies, D.B.; Lau, J.S.; Zhu, G.; et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 2005, 65, 1089–1096. [Google Scholar]
- Blank, C.; Brown, I.; Peterson, A.C.; Spiotto, M.; Iwai, Y.; Honjo, T.; Gajewski, T.F. PD-L1/B7H-1 Inhibits the Effector Phase of Tumor Rejection by T Cell Receptor (TCR) Transgenic CD8+T Cells. Cancer Res. 2004, 64, 1140–1145. [Google Scholar] [CrossRef] [Green Version]
- Miura, S.; Okudaira, K.; Hokari, R.; Tsuzuki, Y.; Okada, Y.; Komoto, S.; Watanabe, C.; Kurihara, C.; Kawaguchi, A.; Nagao, S.; et al. Blockade of B7-H1 or B7-DC induces an anti-tumor effect in a mouse pancreatic cancer model. Int. J. Oncol. 2009, 35, 741–749. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, T.; Honjo, T. PD-1 and PD-1 ligands: From discovery to clinical application. Int Immunol 2007, 19, 813–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Töpfer, K.; Kempe, S.; Müller, N.; Schmitz, M.; Bachmann, M.; Cartellieri, M.; Schackert, G.; Temme, A. Tumor Evasion from T Cell Surveillance. J. Biomed. Biotechnol. 2011, 2011, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGranahan, N.; Rosenthal, R.; Hiley, C.T.; Rowan, A.J.; Watkins, T.B.; Wilson, G.A.; Birkbak, N.J.; Veeriah, S.; Van Loo, P.; Herrero, J.; et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 2017, 171, 1259–1271.e11. [Google Scholar] [CrossRef] [PubMed]
- Marty, R.; Kaabinejadian, S.; Rossell, D.; Slifker, M.J.; Van De Haar, J.; Engin, H.B.; De Prisco, N.; Ideker, T.; Hildebrand, W.H.; Font-Burgada, J.; et al. MHC-I Genotype Restricts the Oncogenic Mutational Landscape. Cell 2017, 171, 1272–1283.e15. [Google Scholar] [CrossRef] [Green Version]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Philip, M.; Fairchild, L.; Sun, L.; Horste, E.L.; Camara, S.; Shakiba, M.; Scott, A.C.; Viale, A.; Lauer, P.; Merghoub, T.; et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 2017, 545, 452–456. [Google Scholar] [CrossRef]
- Pauken, K.E.; Sammons, M.A.; Odorizzi, P.M.; Manne, S.; Godec, J.; Khan, O.; Drake, A.M.; Chen, Z.; Sen, D.R.; Kurachi, M.; et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 2016, 354, 1160–1165. [Google Scholar] [CrossRef] [Green Version]
- Ribas, A.; Shin, D.S.; Zaretsky, J.; Frederiksen, J.; Cornish, A.; Avramis, E.; Seja, E.; Kivork, C.; Siebert, J.; Kaplan-Lefko, P.; et al. PD-1 Blockade Expands Intratumoral Memory T Cells. Cancer Immunol. Res. 2016, 4, 194–203. [Google Scholar] [CrossRef] [Green Version]
- Abiko, K.; Matsumura, N.; Hamanishi, J.; Horikawa, N.; Murakami, R.; Yamaguchi, K.; Yoshioka, Y.; Baba, T.; Konishi, I.; Mandai, M. IFN-γ from lymphocytes induces PD-L1 expression and promotes progression of ovarian cancer. Br. J. Cancer 2015, 112, 1501–1509. [Google Scholar] [CrossRef] [Green Version]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2016, 7, 188–201. [Google Scholar] [CrossRef] [Green Version]
- Manguso, R.T.; Pope, H.W.; Zimmer, M.D.; Brown, F.D.; Yates, K.B.; Miller, B.C.; Collins, N.B.; Bi, K.; LaFleur, M.W.; Juneja, V.R.; et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nat. Cell Biol. 2017, 547, 413–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, D.S.; Ribas, A. The evolution of checkpoint blockade as a cancer therapy: What’s here, what’s next? Curr. Opin. Immunol. 2015, 33, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Nixon, M.J.; Wang, Y.; Wang, D.Y.; Castellanos, E.; Estrada, M.V.; Ericsson-Gonzalez, P.I.; Cote, C.H.; Salgado, R.; Sanchez, V.; et al. Tumor-specific MHC-II expression drives a unique pattern of resistance to immunotherapy via LAG-3/FCRL6 engagement. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen–specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 2010, 207, 2175–2186. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef]
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; Van Der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [Green Version]
- Gide, T.N.; Quek, C.; Menzies, A.M.; Tasker, A.T.; Shang, P.; Holst, J.; Madore, J.; Lim, S.Y.; Velickovic, R.; Wongchenko, M.; et al. Distinct Immune Cell Populations Define Response to Anti-PD-1 Monotherapy and Anti-PD-1/Anti-CTLA-4 Combined Therapy. Cancer Cell 2019, 35, 238–255.e6. [Google Scholar] [CrossRef] [Green Version]
- Spranger, S.; Spaapen, R.M.; Zha, Y.; Williams, J.; Meng, Y.; Ha, T.T.; Gajewski, T.F. Up-Regulation of PD-L1, IDO, and Tregs in the Melanoma Tumor Microenvironment Is Driven by CD8+ T Cells. Sci. Transl. Med. 2013, 5, 200ra116. [Google Scholar] [CrossRef] [Green Version]
- Ricciuti, B.; Leonardi, G.C.; Puccetti, P.; Fallarino, F.; Bianconi, V.; Sahebkar, A.; Baglivo, S.; Chiari, R.; Pirro, M. Targeting indoleamine-2,3-dioxygenase in cancer: Scientific rationale and clinical evidence. Pharmacol. Ther. 2019, 196, 105–116. [Google Scholar] [CrossRef]
- Botticelli, A.; Cerbelli, B.; Lionetto, L.; Zizzari, I.G.; Salati, M.; Pisano, A.; Mazzuca, F.; Simmaco, M.; Nuti, M.; Marchetti, P. Can IDO activity predict primary resistance to anti-PD-1 treatment in NSCLC? J. Transl. Med. 2018, 16, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Sek, K.; Mølck, C.; Stewart, G.D.; Kats, L.M.; Darcy, P.K.; Beavis, P.A. Targeting Adenosine Receptor Signaling in Cancer Immunotherapy. Int. J. Mol. Sci. 2018, 19, 3837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, A.; Ngiow, S.F.; Gao, Y.; Patch, A.-M.; Barkauskas, D.; Messaoudene, M.; Lin, G.; Coudert, J.D.; Stannard, K.A.; Zitvogel, L.; et al. A2AR Adenosine Signaling Suppresses Natural Killer Cell Maturation in the Tumor Microenvironment. Cancer Res. 2018, 78, 1003–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohta, A.; Esitkovsky, M. Extracellular Adenosine-Mediated Modulation of Regulatory T Cells. Front. Immunol. 2014, 5, 304. [Google Scholar] [CrossRef] [PubMed]
- Horenstein, A.L.; Chillemi, A.; Zaccarello, G.; Bruzzone, S.; Quarona, V.; Zito, A.; Serra, S.; Malavasi, F. A CD38/CD203a/CD73 ectoenzymatic pathway independent of CD39 drives a novel adenosinergic loop in human T lymphocytes. OncoImmunology 2013, 2, e26246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beavis, P.A.; Milenkovski, N.; Henderson, M.A.; John, L.B.; Allard, B.; Loi, S.; Kershaw, M.H.; Stagg, J.; Darcy, P.K. Adenosine Receptor 2A Blockade Increases the Efficacy of Anti–PD-1 through Enhanced Antitumor T-cell Responses. Cancer Immunol. Res. 2015, 3, 506–517. [Google Scholar] [CrossRef] [Green Version]
- Leone, R.D.; Sun, I.-M.; Oh, M.-H.; Wen, J.; Englert, J.; Powell, J.D. Inhibition of the adenosine A2a receptor modulates expression of T cell coinhibitory receptors and improves effector function for enhanced checkpoint blockade and ACT in murine cancer models. Cancer Immunol. Immunother. 2018, 67, 1271–1284. [Google Scholar] [CrossRef]
- Arlauckas, S.P.; Garris, C.S.; Kohler, R.H.; Kitaoka, M.; Cuccarese, M.F.; Yang, K.; Miller, M.A.; Carlson, J.C.; Freeman, G.J.; Anthony, R.M.; et al. In vivo imaging reveals a tumor-associated macrophage–mediated resistance pathway in anti–PD-1 therapy. Sci. Transl. Med. 2017, 9, eaal3604. [Google Scholar] [CrossRef] [Green Version]
- Arina, A.; Bronte, V. Myeloid-derived suppressor cell impact on endogenous and adoptively transferred T cells. Curr. Opin. Immunol. 2015, 33, 120–125. [Google Scholar] [CrossRef]
- Hou, A.; Hou, K.; Huang, Q.; Lei, Y.; Chen, W. Targeting Myeloid-Derived Suppressor Cell, a Promising Strategy to Overcome Resistance to Immune Checkpoint Inhibitors. Front. Immunol. 2020, 11, 783. [Google Scholar] [CrossRef]
- Saleh, R.A.; Elkord, E. Treg-mediated acquired resistance to immune checkpoint inhibitors. Cancer Lett. 2019, 457, 168–179. [Google Scholar] [CrossRef]
- Oweida, A.; Hararah, M.K.; Phan, A.V.; Binder, D.C.; Bhatia, S.; Lennon, S.; Bukkapatnam, S.; Van Court, B.; Uyanga, N.; Darragh, L.; et al. Resistance to Radiotherapy and PD-L1 Blockade Is Mediated by TIM-3 Upregulation and Regulatory T-Cell Infiltration. Clin. Cancer Res. 2018, 24, 5368–5380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fares, C.M.; Van Allen, E.M.; Drake, C.G.; Allison, J.P.; Hu-Lieskovan, S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Datta, M.; Coussens, L.M.; Nishikawa, H.; Hodi, F.S.; Jain, R.K. Reprogramming the Tumor Microenvironment to Improve Immunotherapy: Emerging Strategies and Combination Therapies. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.-L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science. 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iida, N.; Dzutsev, A.; Stewart, C.A.; Smith, L.; Bouladoux, N.; Weingarten, R.A.; Molina, D.A.; Salcedo, R.; Back, T.; Cramer, S.; et al. Commensal Bacteria Control Cancer Response to Therapy by Modulating the Tumor Microenvironment. Science 2013, 342, 967–970. [Google Scholar] [CrossRef] [PubMed]
- Routy, B.; Le Chatelier, E.; DeRosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Elkrief, A.; DeRosa, L.; Kroemer, G.; Zitvogel, L.; Routy, B. The negative impact of antibiotics on outcomes in cancer patients treated with immunotherapy: A new independent prognostic factor? Ann. Oncol. 2019, 30, 1572–1579. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.-Z.; Gao, P.; Song, Y.-X.; Xu, Y.; Sun, J.-X.; Chen, X.-W.; Zhao, J.-H.; Wang, Z.-N. Antibiotic use and the efficacy of immune checkpoint inhibitors in cancer patients: A pooled analysis of 2740 cancer patients. OncoImmunology 2019, 8, e1665973. [Google Scholar] [CrossRef] [Green Version]
- DeRosa, L.; Hellmann, M.; Spaziano, M.; Halpenny, D.; Fidelle, M.; Rizvi, H.; Long, N.; Plodkowski, A.; Arbour, K.; Chaft, J.; et al. Negative association of antibiotics on clinical activity of immune checkpoint inhibitors in patients with advanced renal cell and non-small-cell lung cancer. Ann. Oncol. 2018, 29, 1437–1444. [Google Scholar] [CrossRef]
- Pinato, D.J.; Howlett, S.; Ottaviani, D.; Urus, H.; Patel, A.; Mineo, T.; Brock, C.; Power, D.; Hatcher, O.; Falconer, A.; et al. Association of Prior Antibiotic Treatment With Survival and Response to Immune Checkpoint Inhibitor Therapy in Patients With Cancer. JAMA Oncol. 2019, 5, 1774–1778. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.E.; Coughlin, L.A.; Kim, J.; Froehlich, T.W.; Xie, Y.; Frenkel, E.P.; Koh, A.Y. Metagenomic Shotgun Sequencing and Unbiased Metabolomic Profiling Identify Specific Human Gut Microbiota and Metabolites Associated with Immune Checkpoint Therapy Efficacy in Melanoma Patients. Neoplasia 2017, 19, 848–855. [Google Scholar] [CrossRef] [PubMed]
- Arbour, K.C.; Mezquita, L.; Long, N.; Rizvi, H.; Auclin, E.; Ni, A.; Martínez-Bernal, G.; Ferrara, R.; Lai, W.V.; Hendriks, L.E.L.; et al. Impact of Baseline Steroids on Efficacy of Programmed Cell Death-1 and Programmed Death-Ligand 1 Blockade in Patients With Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 2872–2878. [Google Scholar] [CrossRef] [PubMed]
- Ricciuti, B.; Dahlberg, S.E.; Adeni, A.; Sholl, L.M.; Nishino, M.; Awad, M.M. Immune Checkpoint Inhibitor Outcomes for Patients With Non–Small-Cell Lung Cancer Receiving Baseline Corticosteroids for Palliative Versus Nonpalliative Indications. J. Clin. Oncol. 2019, 37, 1927–1934. [Google Scholar] [CrossRef]
- Gettinger, S.; Wurtz, A.; Goldberg, S.B.; Rimm, D.; Schalper, K.; Kaech, S.; Kavathas, P.; Chiang, A.; Lilenbaum, R.; Zelterman, D.; et al. Clinical Features and Management of Acquired Resistance to PD-1 Axis Inhibitors in 26 Patients With Advanced Non–Small Cell Lung Cancer. J. Thorac. Oncol. 2018, 13, 831–839. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.; Wood, K.; Labadie, B.; Won, B.; Brisson, R.; Karrison, T.; Hensing, T.; Kozloff, M.; Bao, R.; Patel, J.D.; et al. Clinical and molecular features of innate and acquired resistance to anti-PD-1/PD-L1 therapy in lung cancer. Oncotarget 2017, 9, 4375–4384. [Google Scholar] [CrossRef] [Green Version]
- Seymour, L.; Bogaerts, J.; Perrone, A.; Ford, R.; Schwartz, L.H.; Mandrekar, S.; Lin, N.U.; Litière, S.; Dancey, J.; Chen, A.; et al. iRECIST: Guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol. 2017, 18, e143–e152. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, S.B.; Narayan, A.; Kole, A.J.; Decker, R.H.; Teysir, J.; Carriero, N.J.; Lee, A.; Nemati, R.; Nath, S.K.; Mane, S.M.; et al. Early Assessment of Lung Cancer Immunotherapy Response via Circulating Tumor DNA. Clin. Cancer Res. 2018, 24, 1872–1880. [Google Scholar] [CrossRef] [Green Version]
- Hogan, S.; Courtier, A.; Cheng, P.F.; Jaberg-Bentele, N.F.; Goldinger, S.M.; Manuel, M.; Perez, S.; Plantier, N.; Mouret, J.-F.; Nguyen-Kim, T.D.L.; et al. Peripheral Blood TCR Repertoire Profiling May Facilitate Patient Stratification for Immunotherapy against Melanoma. Cancer Immunol. Res. 2019, 7, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Auslander, N.; Zhang, G.; Lee, J.S.; Frederick, D.T.; Miao, B.; Moll, T.; Tian, T.; Wei, Z.; Madan, S.; Sullivan, R.J.; et al. Robust prediction of response to immune checkpoint blockade therapy in metastatic melanoma. Nat. Med. 2018, 24, 1545–1549. [Google Scholar] [CrossRef]
- Bensch, F.; Van Der Veen, E.L.; Lub-de Hooge, M.N.; Jorritsma-Smit, A.; Boellaard, R.; Kok, I.C.; Oosting, S.F.; Schröder, C.P.; Hiltermann, T.J.N.; Van Der Wekken, A.J.; et al. 89Zr-atezolizumab imaging as a non-invasive approach to assess clinical response to PD-L1 blockade in cancer. Nat. Med. 2018, 24, 1852–1858. [Google Scholar] [CrossRef] [PubMed]
- Klemen, N.D.; Wang, M.; Feingold, P.L.; Cooper, K.; Pavri, S.N.; Han, D.; Detterbeck, F.C.; Boffa, D.J.; Khan, S.A.; Olino, K.; et al. Patterns of failure after immunotherapy with checkpoint inhibitors predict durable progression-free survival after local therapy for metastatic melanoma. J. Immunother. Cancer 2019, 7, 196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, I.C.; Long, G.V.; Quek, C.; Gonzalez, M.; Carlino, M.S.; Long, G.V.; Menzies, A.M. Site-specific response patterns, pseudoprogression, and acquired resistance in patients with melanoma treated with ipilimumab combined with anti–PD-1 therapy. Cancer 2020, 126, 86–97. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.B.; Li, S.; Higgs, E.F.; Cabanov, A.; Wang, X.; Huang, H.; Gajewski, T. Tumor heterogeneity and clonal cooperation influence the immune selection of IFN-γ-signaling mutant cancer cells. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popel, A.S. Immunoactivating the tumor microenvironment enhances immunotherapy as predicted by integrative computational model. Proc. Natl. Acad. Sci. USA 2020, 117, 4447–4449. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Mechanisms of Acquired ICI Resistance | Potential Therapeutic Approaches |
|---|---|
| Impaired processing or presentation of tumor-associated antigens | Chemotherapy, radiotherapy, cancer vaccines, oncolytic viruses, TLR agonists |
| Fixed epigenetic modification of T cells | Epigenetic modulators (hypomethylating agents, histone deacetylase inhibitors) |
| Defects in IFN-γ pathways | STING agonists, c-di-GMP, JAK inhibitors |
| Upregulation of other immune checkpoints | Combination immune checkpoint inhibitors (such as CTLA-4, TIGIT, LAG-3, TIM-3, VISTA, BTLA), immune stimulatory agents (such as OX40, CD40, GITR, ICOS, 4-1BB) |
| Immunosuppressive cells/inhibitory metabolites in the tumor microenvironment | Macrophage inhibitors (such as CSF1R inhibitors), cytokine/chemokine inhibitors, targeted therapies targeting canonical pathways (such as against VEGF, PI3K), IDO inhibitors, TGF-β inhibitors, CXCR2 inhibitors, CXCR4 inhibitors, A2AR inhibitors/anti-CD73 |
| Gut microbiome | Fecal microbiota transplantation, probiotics, diet interventions |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pathak, R.; Pharaon, R.R.; Mohanty, A.; Villaflor, V.M.; Salgia, R.; Massarelli, E. Acquired Resistance to PD-1/PD-L1 Blockade in Lung Cancer: Mechanisms and Patterns of Failure. Cancers 2020, 12, 3851. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12123851
Pathak R, Pharaon RR, Mohanty A, Villaflor VM, Salgia R, Massarelli E. Acquired Resistance to PD-1/PD-L1 Blockade in Lung Cancer: Mechanisms and Patterns of Failure. Cancers. 2020; 12(12):3851. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12123851
Chicago/Turabian StylePathak, Ranjan, Rebecca R. Pharaon, Atish Mohanty, Victoria M. Villaflor, Ravi Salgia, and Erminia Massarelli. 2020. "Acquired Resistance to PD-1/PD-L1 Blockade in Lung Cancer: Mechanisms and Patterns of Failure" Cancers 12, no. 12: 3851. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12123851