Physical Interaction between HPV16E7 and the Actin-Binding Protein Gelsolin Regulates Epithelial-Mesenchymal Transition via HIPPO-YAP Axis

 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Line and Treatments
2.2. Plasmids and Cell Transfection
2.3. RNA Extraction and Quantitative RT-PCR
2.4. Cell Invasion Assay
2.5. RhoGTPases Activity
2.6. Immunoblot Analysis
2.7. Flow Cytometry
2.7.1. Quantitative Evaluation of Proteins
2.7.2. Fluorescence Resonance Energy Transfer (FRET)
2.8. Fluorescence Microscopy
2.9. Data Analyses and Statistics
3. Results
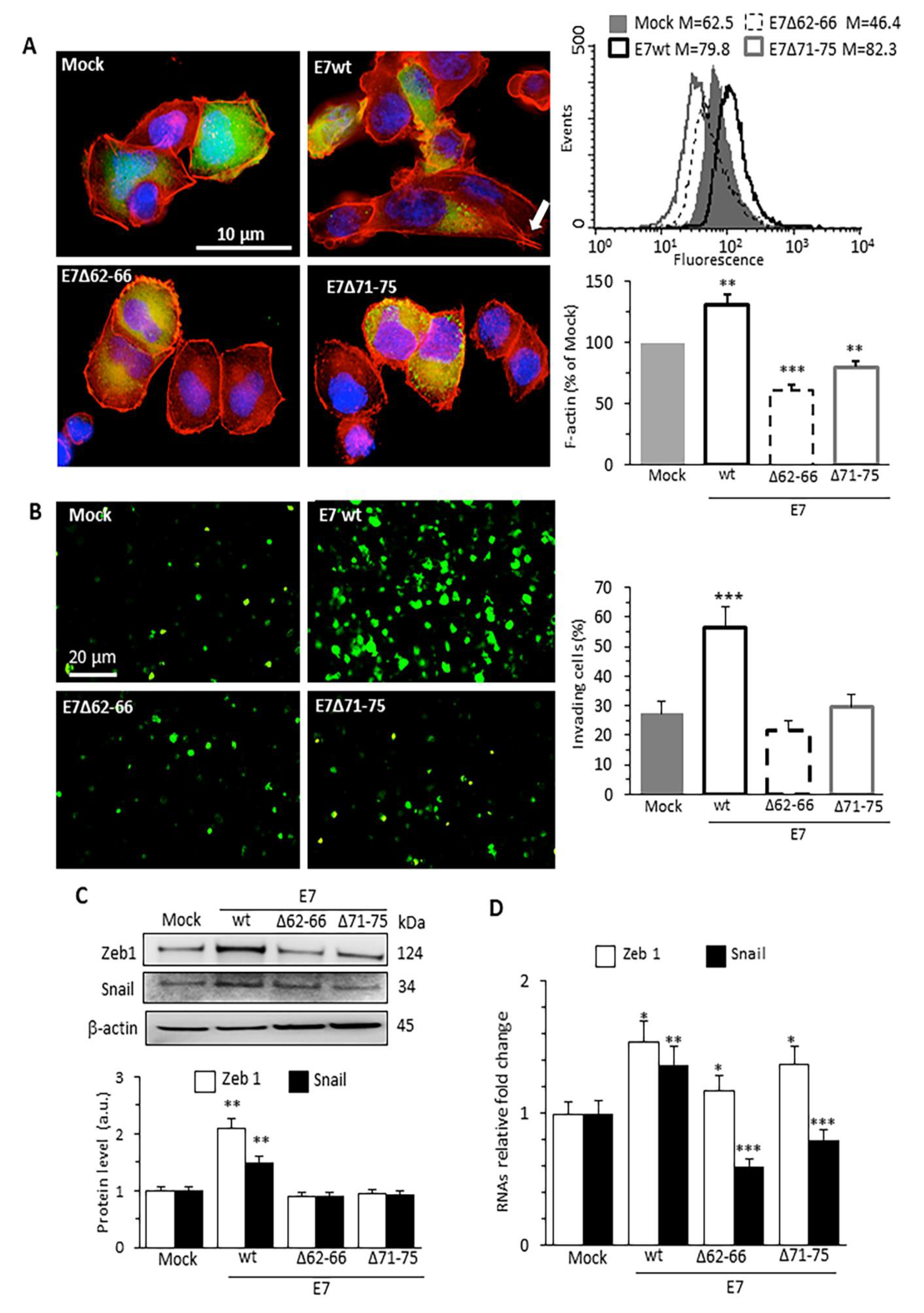
3.1. HPV16E7 Expression Promotes Actin Cytoskeleton Remodeling, Cell Invasion and EMT in Human Cervical Cancer Cells
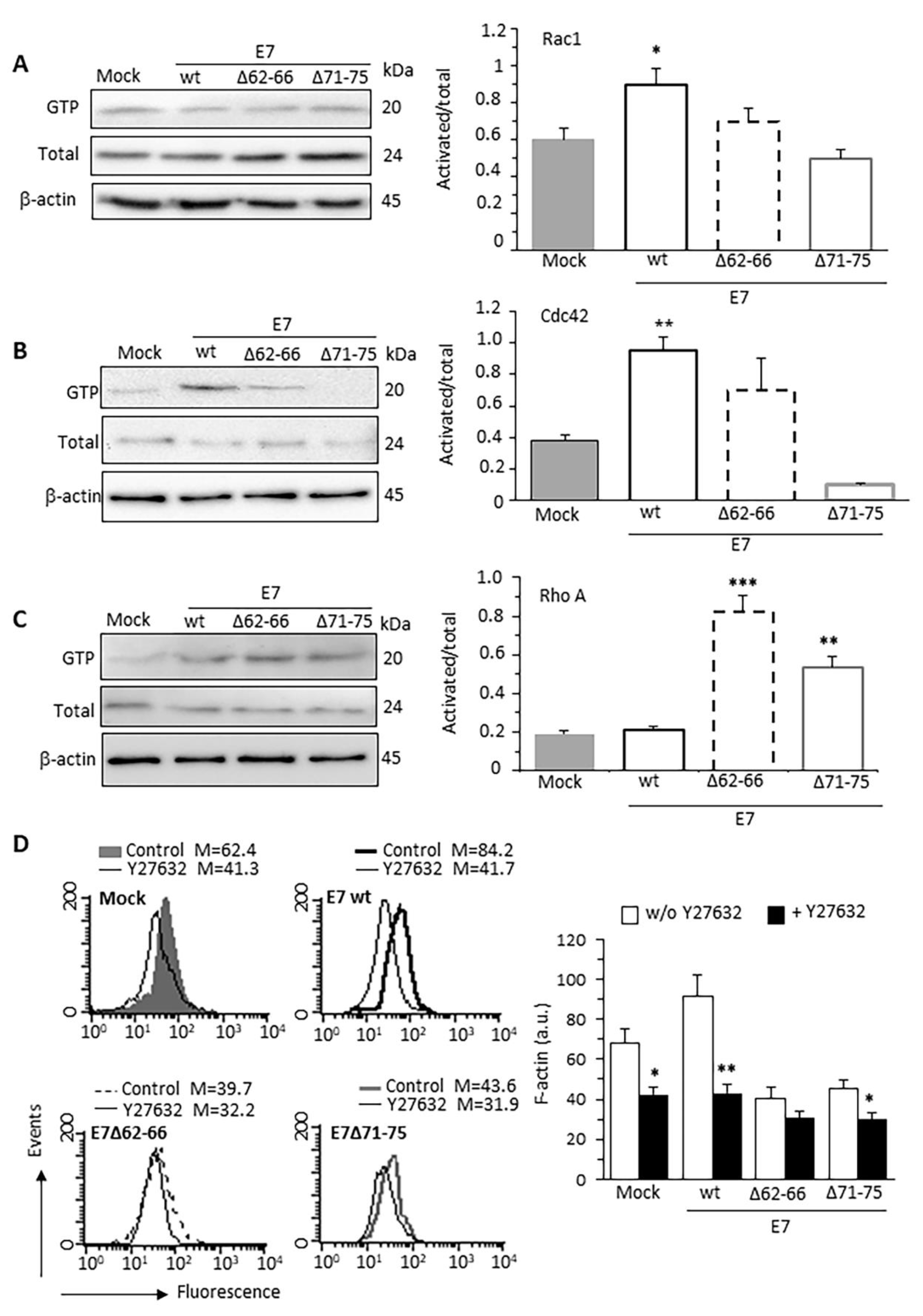
3.2. HPV16E7 Expression Induces Activation of Rac1 and Cdc42 RhoGTPases
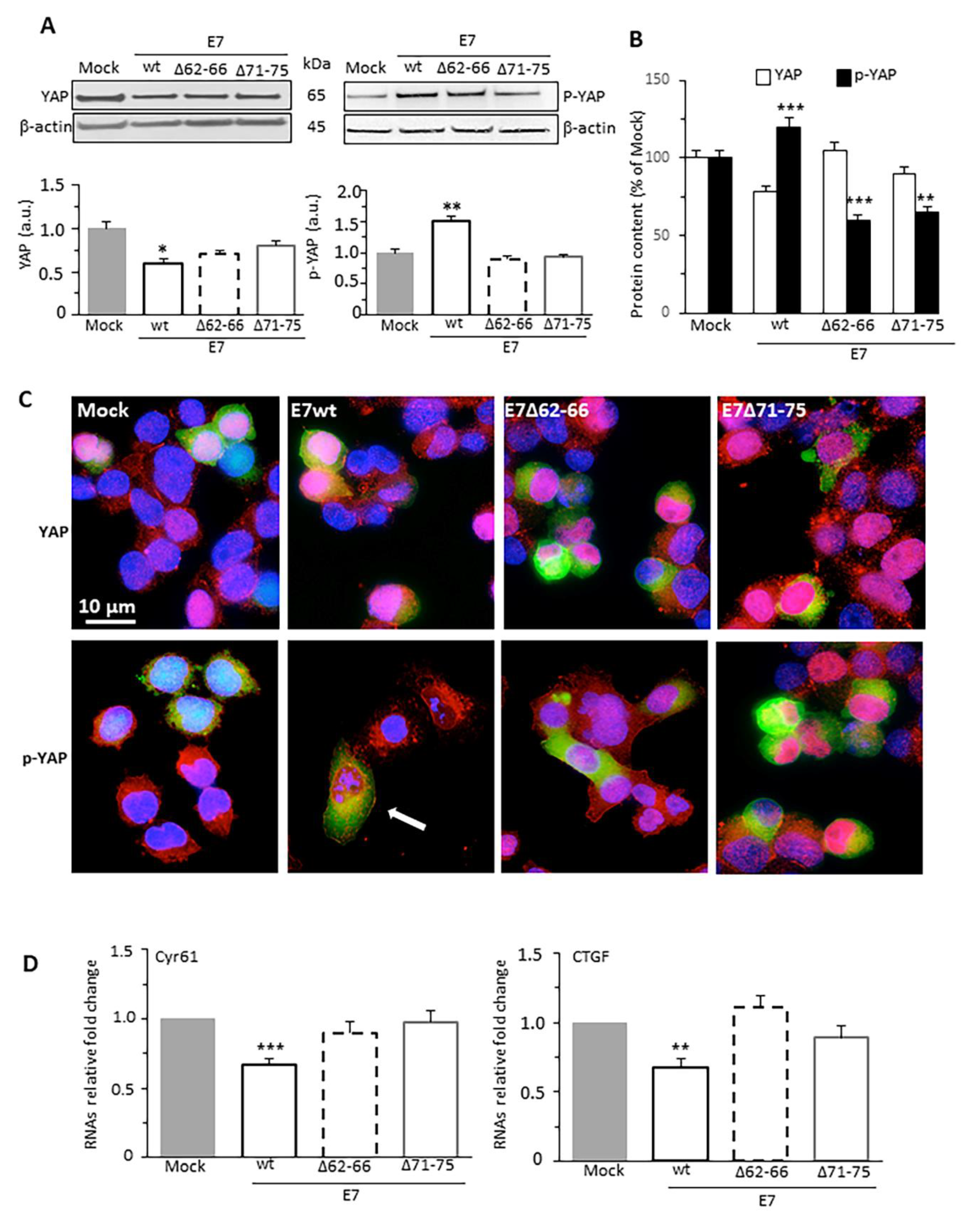
3.3. HPV16E7 Expression Promotes Phosphorylation of YAP
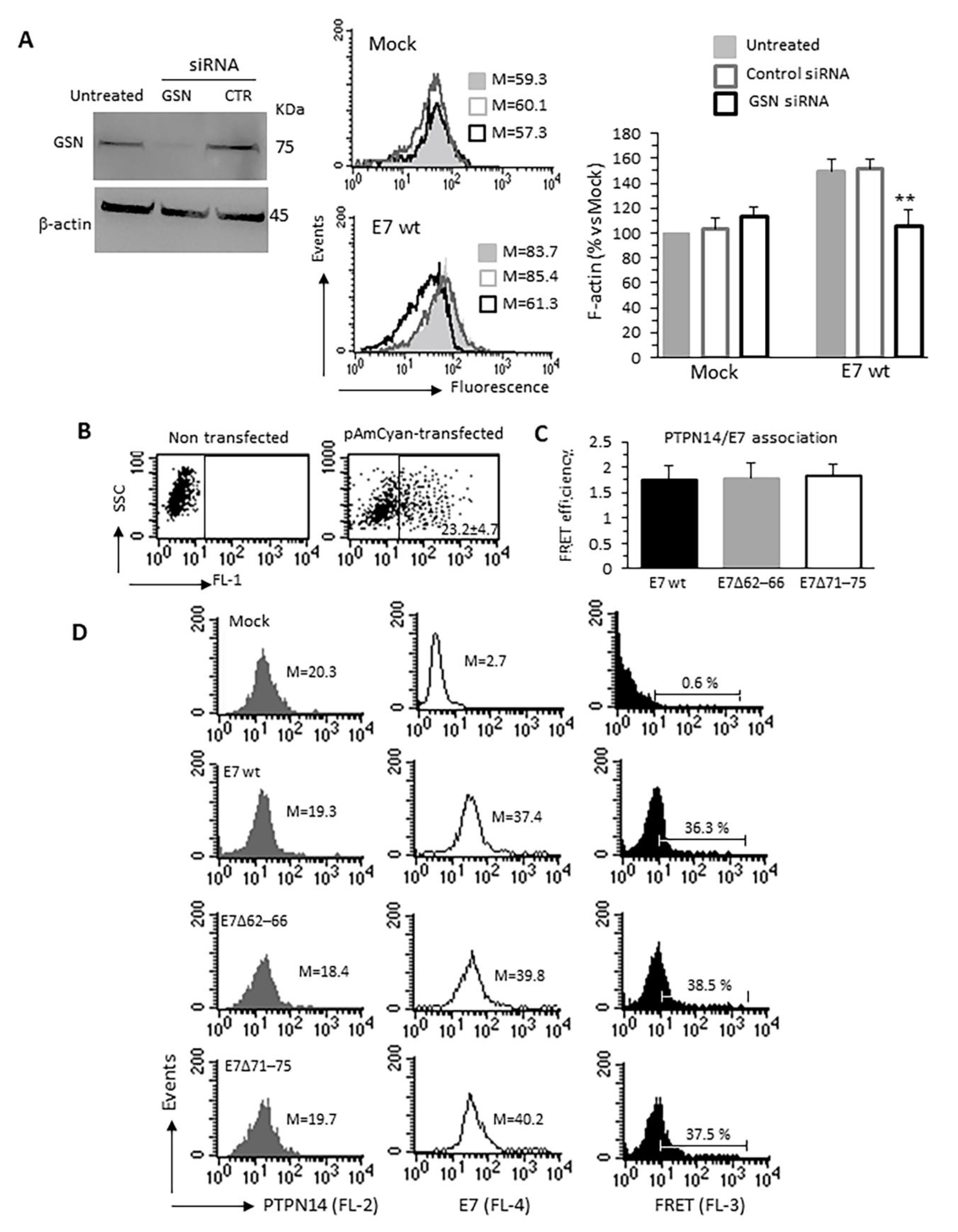
3.4. Specific Relevance of the E7–GSN Interaction
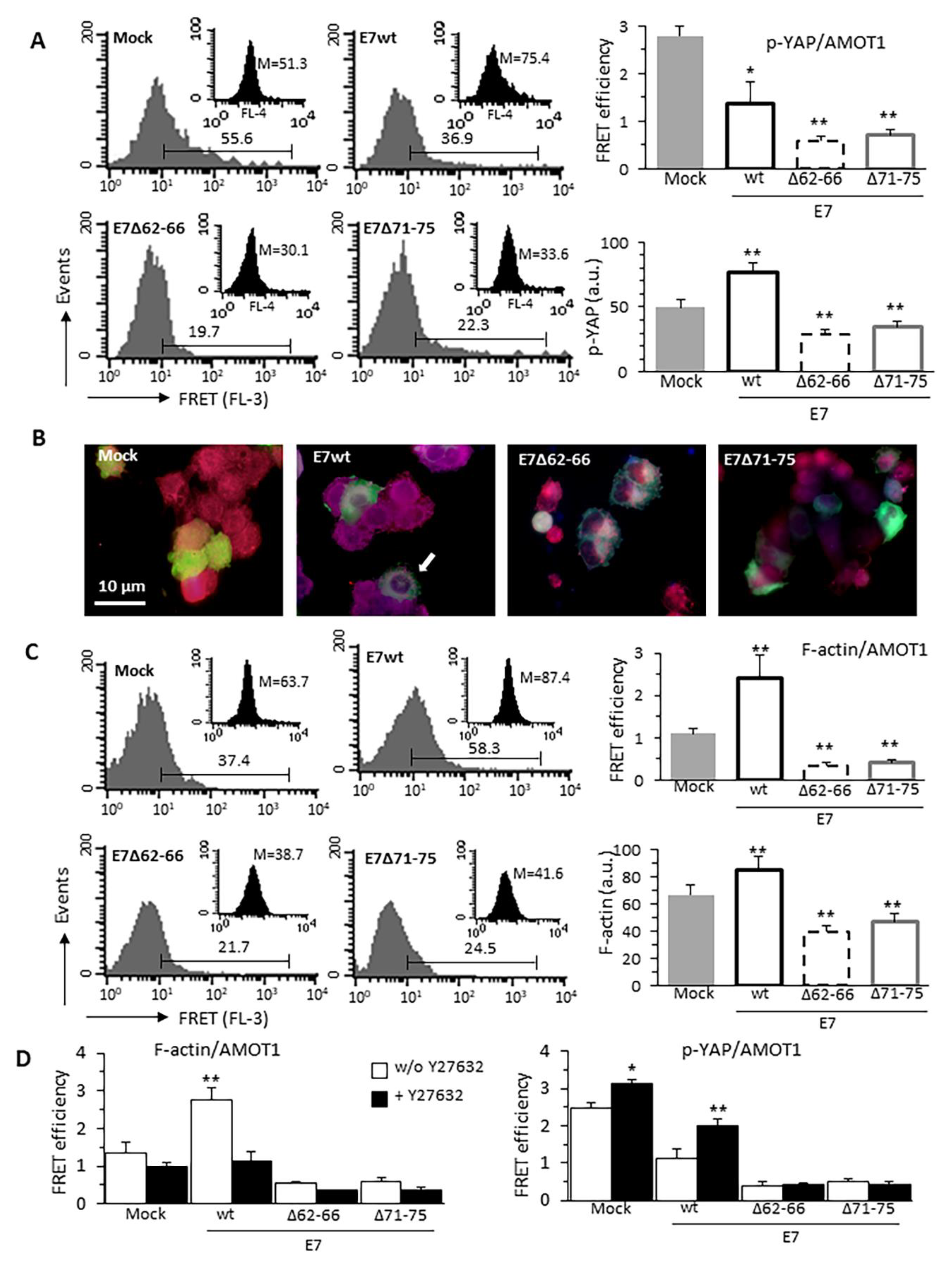
3.5. HPV16E7-Induced Cytoskeleton Alterations Interfere With p-YAP/AMOT1 Complex
3.6. Effect of Cytochalasin D on YAP Activity and EMT
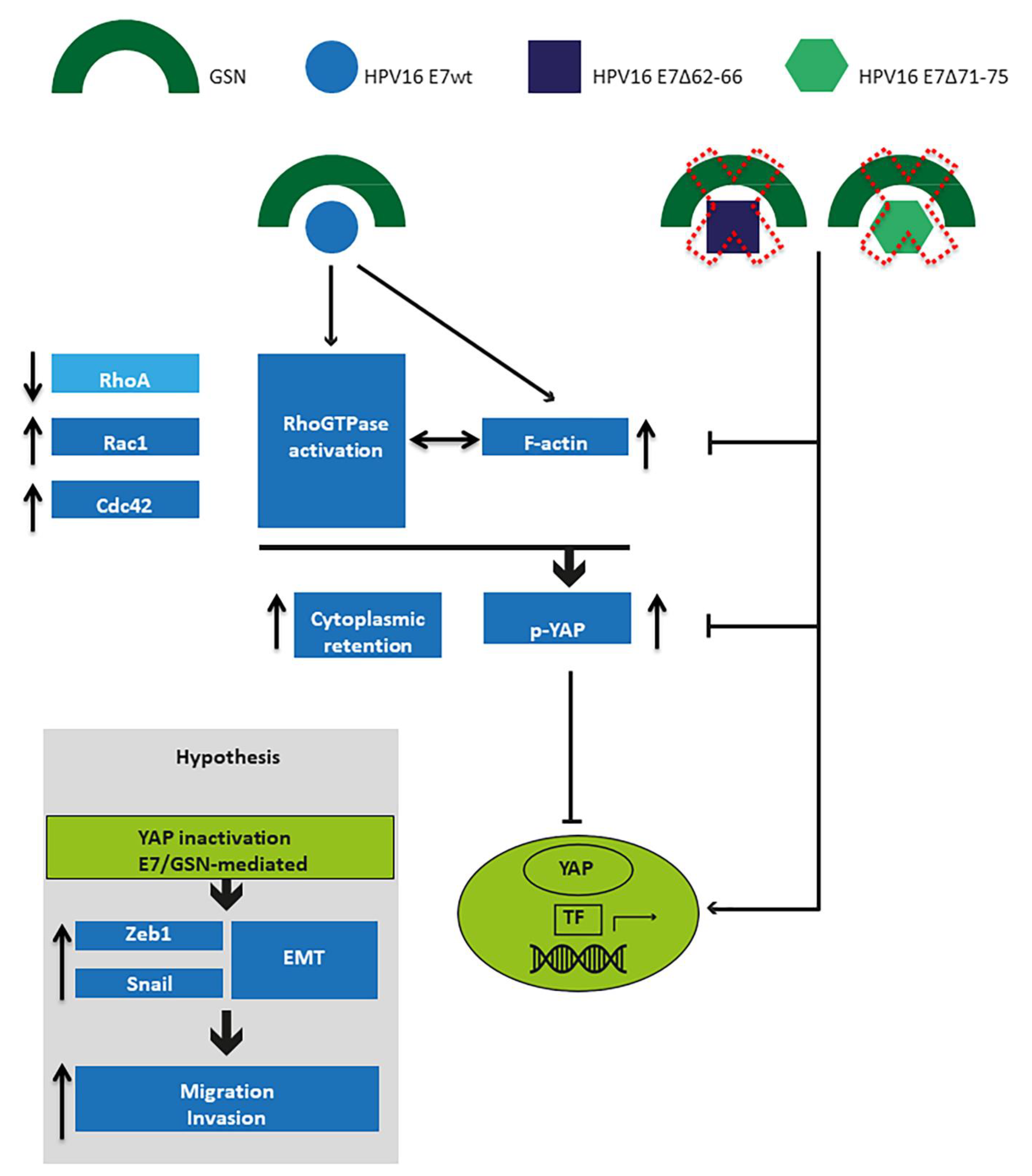
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hausen, H.Z. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Gheit, T. Mucosal and Cutaneous Human Papillomavirus Infections and Cancer Biology. Front. Oncol. 2019, 9, 355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clifford, G.M.; Smith, J.S.; Aguado, T.; Franceschi, S. Comparison of HPV type distribution in high-grade cervical lesions and cervical cancer: A meta-analysis. Br. J. Cancer 2003, 89, 101–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suarez, I.; Travé, G. Structural Insights in Multifunctional Papillomavirus Oncoproteins. Viruses 2018, 10, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo-Teh, N.S.L.; Ito, Y.; Jha, S. High-Risk Human Papillomaviral Oncogenes E6 and E7 Target Key Cellular Pathways to Achieve Oncogenesis. Int. J. Mol. Sci. 2018, 19, 1706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Bode, A.M.; Dong, Z.; Cao, Y. The epithelial–mesenchymal transition (EMT) is regulated by oncoviruses in cancer. FASEB J. 2016, 30, 3001–3010. [Google Scholar] [CrossRef] [Green Version]
- Matarrese, P.; Abbruzzese, C.; Mileo, A.M.; Vona, R.; Ascione, B.; Visca, P.; Rollo, F.; Benevolo, M.; Malorni, W.; Paggi, M.G. Interaction between the human papillomavirus 16 E7 oncoprotein and gelsolin ignites cancer cell motility and invasiveness. Oncotarget 2016, 7, 50972–50985. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, R.; Holmes, K.C. Actin Structure and Function. Annu. Rev. Biophys. 2011, 40, 169–186. [Google Scholar] [CrossRef] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.-S.; Kato, I.; Kim, H.-R.C. A novel function of HPV16-E6/E7 in epithelial–mesenchymal transition. Biochem. Biophys. Res. Commun. 2013, 435, 339–344. [Google Scholar] [CrossRef]
- Mileo, A.M.; Abbruzzese, C.; Vico, C.; Bellacchio, E.; Matarrese, P.; Ascione, B.; Federico, A.; Della Bianca, S.; Mattarocci, S.; Malorni, W.; et al. The human papillomavirus-16 E7 oncoprotein exerts antiapoptotic effects via its physical interaction with the actin-binding protein gelsolin. Carcinogenesis 2013, 34, 2424–2433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, H.T.; Machesky, L.M. Actin cytoskeletal control during epithelial to mesenchymal transition: Focus on the pancreas and intestinal tract. Br. J. Cancer 2015, 112, 613–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, T.R.; Stephenson, R.E.; Miller, A.L. Rho GTPases and actomyosin: Partners in regulating epithelial cell-cell junction structure and function. Exp. Cell Res. 2017, 358, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Irvine, K.D. Cellular Organization and Cytoskeletal Regulation of the Hippo Signaling Network. Trends Cell Biol. 2016, 26, 694–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nardone, G.; La Cruz, J.O.-D.; Vrbsky, J.; Martini, C.; Pribyl, J.; Skládal, P.; Pešl, M.; Caluori, G.; Pagliari, S.; Martino, F.; et al. YAP regulates cell mechanics by controlling focal adhesion assembly. Nat. Commun. 2017, 8, 15321. [Google Scholar] [CrossRef]
- Halder, G.; Dupont, S.; Piccolo, S. Transduction of mechanical and cytoskeletal cues by YAP and TAZ. Nat. Rev. Mol. Cell Biol. 2012, 13, 591–600. [Google Scholar] [CrossRef]
- Yu, F.-X.; Zhao, B.; Guan, K.L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef] [Green Version]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The Biology of YAP/TAZ: Hippo Signaling and Beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nat. Cell Biol. 2011, 474, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Barry, E.R.; Morikawa, T.; Butler, B.L.; Shrestha, K.; De La Rosa, R.; Yan, K.S.; Fuchs, C.S.; Magness, S.T.; Smits, R.; Ogino, S.; et al. Restriction of intestinal stem cell expansion and the regenerative response by YAP. Nat. Cell Biol. 2013, 493, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Mello, S.S.; Valente, L.J.; Raj, N.; Seoane, J.A.; Flowers, B.M.; McClendon, J.; Bieging-Rolett, K.T.; Lee, J.; Ivanochko, D.; Kozak, M.M.; et al. A p53 Super-tumor Suppressor Reveals a Tumor Suppressive p53-Ptpn14-Yap Axis in Pancreatic Cancer. Cancer Cell 2017, 32, 460–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2–ΔΔCT. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Matarrese, P.; Garofalo, T.; Manganelli, V.; Gambardella, L.; Marconi, M.; Grasso, M.; Tinari, A.; Misasi, R.; Malorni, W.; Sorice, M. Evidence for the involvement of GD3 ganglioside in autophagosome formation and maturation. Autophagy 2014, 10, 750–765. [Google Scholar] [CrossRef]
- Riemann, D.; Tcherkes, A.; Hansen, G.H.; Wulfaenger, J.; Blosz, T.; Danielsen, E.M. Functional co-localization of monocytic aminopeptidase N/CD13 with the Fc gamma receptors CD32 and CD64. Biochem. Biophys. Res. Commun. 2005, 331, 1408–1412. [Google Scholar] [CrossRef]
- Mack, N.A.; Whalley, H.J.; Castillo-Lluva, S.; Malliri, A. The diverse roles of Rac signaling in tumorigenesis. Cell Cycle 2011, 10, 1571–1581. [Google Scholar] [CrossRef] [Green Version]
- Stengel, K.; Zheng, Y. Cdc42 in oncogenic transformation, invasion, and tumorigenesis. Cell. Signal. 2011, 23, 1415–1423. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.H.; Kim, J.; Hong, H.; Lee, S.; Lee, J.; Jung, E.; Kim, J. Actin remodeling confers BRAF inhibitor resistance to melanoma cells through YAP/TAZ activation. EMBO J. 2016, 35, 462–478. [Google Scholar] [CrossRef]
- van Soldt, B.J.; Qian, J.; Li, J.; Tang, N.; Lu, J.; Cardoso, W.V. Yap and its subcellular localization have distinct compart-ment-specific roles in the developing lung. Development 2019. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.; Kim, J. Regulation of Hippo signaling by actin remodeling. BMB Rep. 2018, 51, 151–156. [Google Scholar] [CrossRef]
- Wada, K.-I.; Itoga, K.; Okano, T.; Yonemura, S.; Sasaki, H. Hippo pathway regulation by cell morphology and stress fibers. Development 2011, 138, 3907–3914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mason, D.E.; Collins, J.M.; Dawahare, J.H.; Nguyen, T.D.; Lin, Y.; Voytik-Harbin, S.L.; Zorlutuna, P.; Yoder, M.C.; Boerckel, J.D. YAP and TAZ limit cytoskeletal and focal adhesion maturation to enable persistent cell motility. J. Cell Biol. 2019, 218, 1369–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007, 21, 2747–2761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, Y.; Chun, A.; Cheung, K.; Rashidi, B.; Yang, X. Tumor Suppressor LATS1 Is a Negative Regulator of OncogeneYAP. J. Biol. Chem. 2008, 283, 5496–5509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatterschide, J.; Bohidar, A.E.; Grace, M.; Nulton, T.J.; Kim, H.W.; Windle, B.; Morgan, I.M.; Munger, K.; White, E.A. PTPN14 degradation by high-risk human papillomavirus E7 limits keratinocyte differentiation and contributes to HPV-mediated on-cogenesis. Proc. Natl. Acad. Sci. USA 2019, 116, 7033–7042. [Google Scholar] [CrossRef] [Green Version]
- Mana-Capelli, S.; Paramasivam, M.; Dutta, S.; Mccollum, D. Angiomotins link F-actin architecture to Hippo pathway signaling. Mol. Biol. Cell 2014, 25, 1676–1685. [Google Scholar] [CrossRef]
- Lammers, B.; Zhao, Y.; Hoekstra, M.; Hildebrand, R.B.; Ye, D.; Meurs, I.; Van Berkel, T.J.; Van Eck, M. Augmented athero-genesis in LDL receptor deficient mice lacking both macrophage ABCA1 and ApoE. PLoS ONE 2011, 6, e26095. [Google Scholar] [CrossRef] [Green Version]
- Moleirinho, S.; Hoxha, S.; Mandati, V.; Curtale, G.; Troutman, S.; Ehmer, U.; Kissil, J.L. Regulation of localization and function of the transcriptional co-activator YAP by angiomotin. eLife 2017, 6, e23966. [Google Scholar] [CrossRef]
- Martino, F.; Perestrelo, A.R.; Vinarský, V.; Pagliari, S.; Forte, G. Cellular Mechanotransduction: From Tension to Function. Front. Physiol. 2018, 9, 824. [Google Scholar] [CrossRef]
- Zemke, N.R.; Gou, D.; Berk, A.J. Dedifferentiation by adenovirus E1A due to inactivation of Hippo pathway effectors YAP and TAZ. Genes Dev. 2019, 33, 828–843. [Google Scholar] [CrossRef] [Green Version]
- Felsani, A.; Mileo, A.M.; Paggi, M.G. Retinoblastoma family proteins as key targets of the small DNA virus oncoproteins. Oncogene 2006, 25, 5277–5285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chellappan, S.; Kraus, V.B.; Kroger, B.; Munger, K.; Howley, P.M.; Phelps, W.C.; Nevins, J.R. Adenovirus E1A, simian virus 40 tumor antigen, and human papillomavirus E7 protein share the capacity to disrupt the interaction between transcription factor E2F and the retinoblastoma gene product. Proc. Natl. Acad. Sci. USA 1992, 89, 4549–4553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helt, A.M.; Galloway, D.A. Mechanisms by which DNA tumor virus oncoproteins target the Rb family of pocket proteins. Carcinogenesis 2003, 24, 159–169. [Google Scholar] [CrossRef] [PubMed]
- White, E.A. Manipulation of Epithelial Differentiation by HPV Oncoproteins. Viruses 2019, 11, 369. [Google Scholar] [CrossRef] [Green Version]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [Green Version]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef]
- Zheng, Y.; Pan, D. The Hippo Signaling Pathway in Development and Disease. Dev. Cell 2019, 50, 264–282. [Google Scholar] [CrossRef]
- Oka, T.; Sudol, M. Nuclear localization and pro-apoptotic signaling of YAP2 require intact PDZ-binding motif. Genes Cells 2009, 14, 607–615. [Google Scholar] [CrossRef]
- Levy, D.; Adamovich, Y.; Reuven, N.; Shaul, Y. Yap1 Phosphorylation by c-Abl Is a Critical Step in Selective Activation of Proapoptotic Genes in Response to DNA Damage. Mol. Cell 2008, 29, 350–361. [Google Scholar] [CrossRef]
- Raj, N.; Bam, R. Reciprocal Crosstalk Between YAP1/Hippo Pathway and the p53 Family Proteins: Mechanisms and Outcomes in Cancer. Front. Cell Dev. Biol. 2019, 7, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, J.H.; Fernando, A.T.P.; Faure, N.; Andrabi, S.; Hahn, W.C.; Schaffhausen, B.S.; Roberts, T.M. Polyomavirus Small t Antigen Interacts with Yes-Associated Protein to Regulate Cell Survival and Differentiation. J. Virol. 2014, 88, 12055–12064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, H.; Wu, L.; Zheng, H.; Huili, W.; Wan, H.; Liang, G.; Zhao, Y.; Liang, J. Expression of Yes-Associated Protein in Cervical Squamous Epithelium Lesions. Int. J. Gynecol. Cancer 2014, 24, 1575–1582. [Google Scholar] [CrossRef]
- He, C.; Mao, D.; Hua, G.; Lv, X.; Chen, X.; Angeletti, P.C.; Dong, J.; Remmenga, S.W.; Rodabaugh, K.J.; Zhou, J.; et al. The Hippo/YAP pathway interacts with EGFR signaling and HPV oncoproteins to regulate cervical cancer progression. EMBO Mol. Med. 2015, 7, 1426–1449. [Google Scholar] [CrossRef] [PubMed]
- Olmedo-Nieva, L.; Muñoz-Bello, J.O.; Manzo-Merino, J.; Lizano, M. New insights in Hippo signalling alteration in human papillomavirus-related cancers. Cell. Signal. 2020, 76, 109815. [Google Scholar] [CrossRef]
- He, C.; Lv, X.; Huang, C.; Angeletti, P.C.; Hua, G.; Dong, J.; Zhou, J.; Wang, Z.; Ma, B.; Chen, X.; et al. A Human Papilloma-virus-Independent Cervical Cancer Animal Model Reveals Unconventional Mechanisms of Cervical Carcinogenesis. Cell Rep. 2019, 26, 2636–2650.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanz-Moreno, V.; Gadea, G.; Ahn, J.; Paterson, H.; Marra, P.; Pinner, S.; Sahai, E.; Marshall, C.J. Rac Activation and Inactivation Control Plasticity of Tumor Cell Movement. Cell 2008, 135, 510–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.W.; Sun, M.S.; Liao, M.Y.; Chung, C.H.; Chi, Y.H.; Chiou, L.T.; Yu, J.; Lou, K.L.; Wu, H.C. Podocalyxin-like 1 promotes invadopodia formation and metastasis through activation of Rac1/Cdc42/cortactin signaling in breast cancer cells. Carcinogenesis 2014, 35, 2425–2435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adler, J.J.; Johnson, D.E.; Heller, B.L.; Bringman, L.R.; Ranahan, W.P.; Conwell, M.D.; Sun, Y.; Hudmon, A.; Wells, C.D. Serum deprivation inhibits the transcriptional co-activator YAP and cell growth via phosphorylation of the 130-kDa isoform of An-giomotin by the LATS1/2 protein kinases. Proc. Natl. Acad. Sci. USA 2013, 110, 17368–17373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, Y.S.; Li, S.; Wang, X.; Zhu, J.; Zhuo, S.; Han, Y.; Yue, T.; Yang, Y.; Jiang, J. CDK7 regulates organ size and tumor growth by safeguarding the Hippo pathway effector Yki/Yap/Taz in the nucleus. Genes Dev. 2020, 34, 53–71. [Google Scholar] [CrossRef]
- Leung, C.Y.; Zernicka-Goetz, M. Angiomotin prevents pluripotent lineage differentiation in mouse embryos via Hippo path-way-dependent and -independent mechanisms. Nat. Commun. 2013, 4, 2251. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, B.G.; Gaspar, P.; Brás-Pereira, C.; Jezowska, B.; Rebelo, S.R.; Janody, F. Actin-Capping Protein and the Hippo pathway regulate F-actin and tissue growth in Drosophila. Development 2011, 138, 2337–2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, E.A.; Münger, K.; Howley, P.M. High-Risk Human Papillomavirus E7 Proteins Target PTPN14 for Degradation. mBio 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, E.A.; Sowa, M.E.; Tan, M.J.A.; Jeudy, S.; Hayes, S.D.; Santha, S.; Münger, K.; Harper, J.W.; Howley, P.M. Systematic identification of interactions between host cell proteins and E7 oncoproteins from diverse human papillomaviruses. Proc. Natl. Acad. Sci. USA 2012, 109, E260–E267. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Mock | E7wt | E7Δ62–66 | E7Δ71–75 | ||||
|---|---|---|---|---|---|---|---|---|
| No CytoD | CytoD | No CytoD | CytoD | No CytoD | CytoD | No CytoD | CytoD | |
| p-YAP | 51.4 ± 7.3 | 53.2 ± 6.1 | 73.6 ± 7.2 | 50.4 ± 5.7 | 39.4 ± 4.5 | 27.9 ± 3.6 | 40.0 ± 5.2 | 30.3 ± 4,0 |
| AMOT1 | 25.6 ± 4.7 | 22.6 ± 3.5 | 23.7 ± 3.9 | 19.2 ± 2.3 | 25.1 ± 3.3 | 20.4 ± 3.1 | 23.5 ± 3.7 | 21.3 ± 3.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matarrese, P.; Vona, R.; Ascione, B.; Paggi, M.G.; Mileo, A.M. Physical Interaction between HPV16E7 and the Actin-Binding Protein Gelsolin Regulates Epithelial-Mesenchymal Transition via HIPPO-YAP Axis. Cancers 2021, 13, 353. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13020353
Matarrese P, Vona R, Ascione B, Paggi MG, Mileo AM. Physical Interaction between HPV16E7 and the Actin-Binding Protein Gelsolin Regulates Epithelial-Mesenchymal Transition via HIPPO-YAP Axis. Cancers. 2021; 13(2):353. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13020353
Chicago/Turabian StyleMatarrese, Paola, Rosa Vona, Barbara Ascione, Marco G. Paggi, and Anna Maria Mileo. 2021. "Physical Interaction between HPV16E7 and the Actin-Binding Protein Gelsolin Regulates Epithelial-Mesenchymal Transition via HIPPO-YAP Axis" Cancers 13, no. 2: 353. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13020353