
Small Proline-Rich Protein 2A and 2D Are Regulated by the RBM38-p73 Axis and Associated with p73-Dependent Suppression of Chronic Inflammation

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. MEF Isolation
2.3. Cell Culture, Cell Line Generation, and Differentiation
2.4. Western Blot Analysis
2.5. RNA Isolation, RT-PCR
2.6. Chromatin Immunoprecipitation Assay
2.7. Histological Analysis
2.8. Statistical Analysis
3. Results
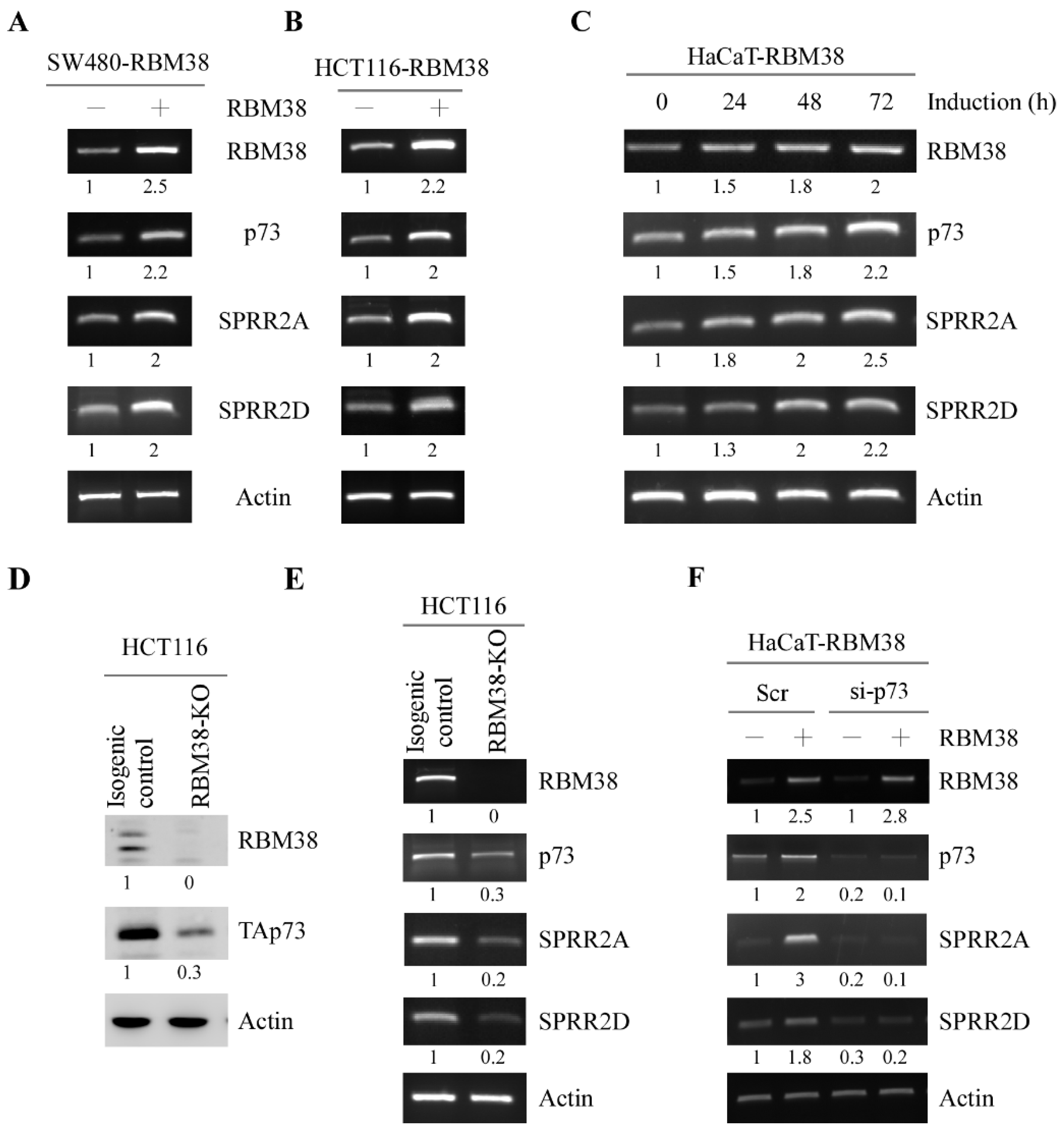
3.1. RBM38 Regulates SPRR2A/2D Expression via p73
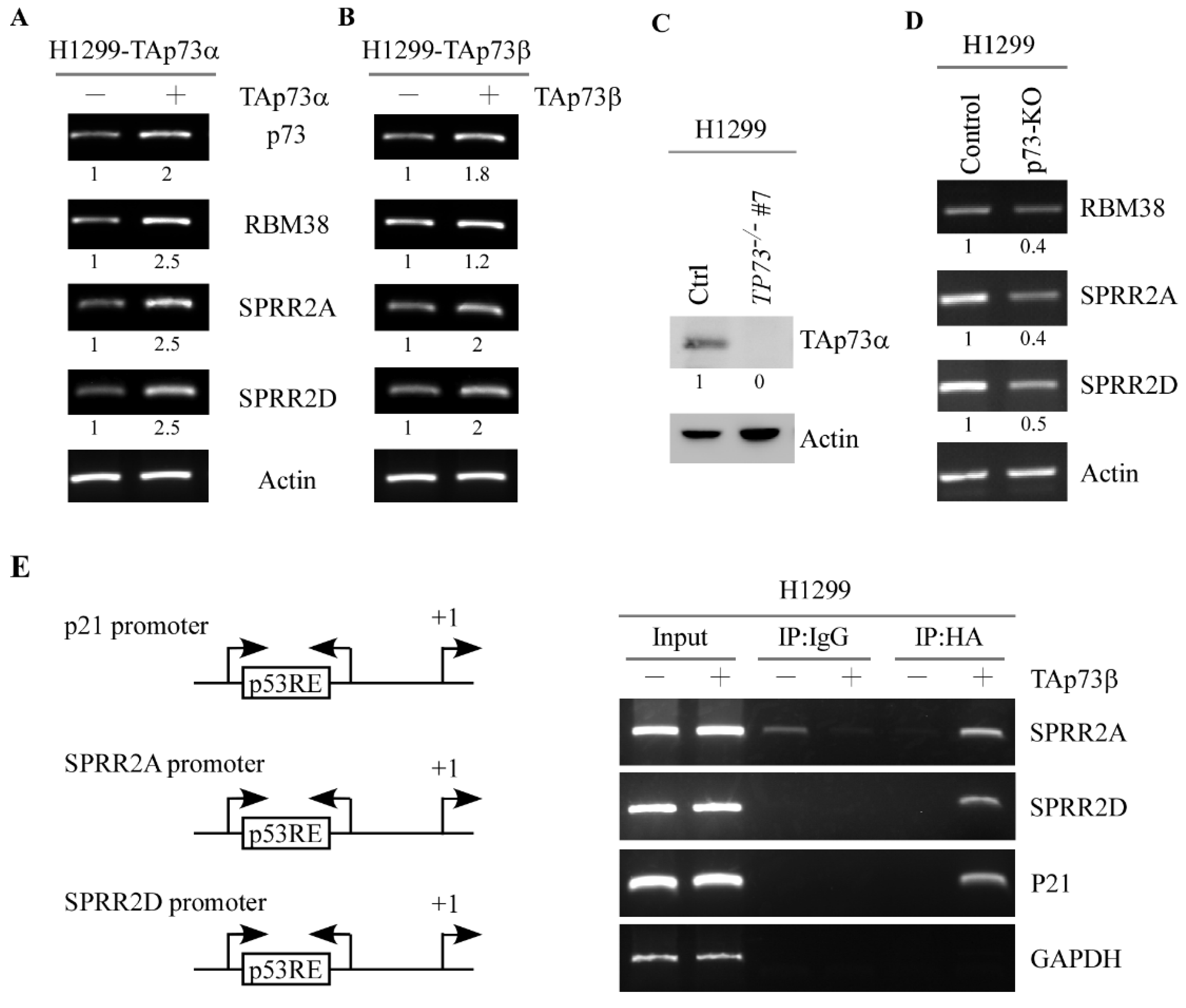
3.2. p73 Directly Induces SPRR2A and SPRR2D Expression via Binding to Their Promoters
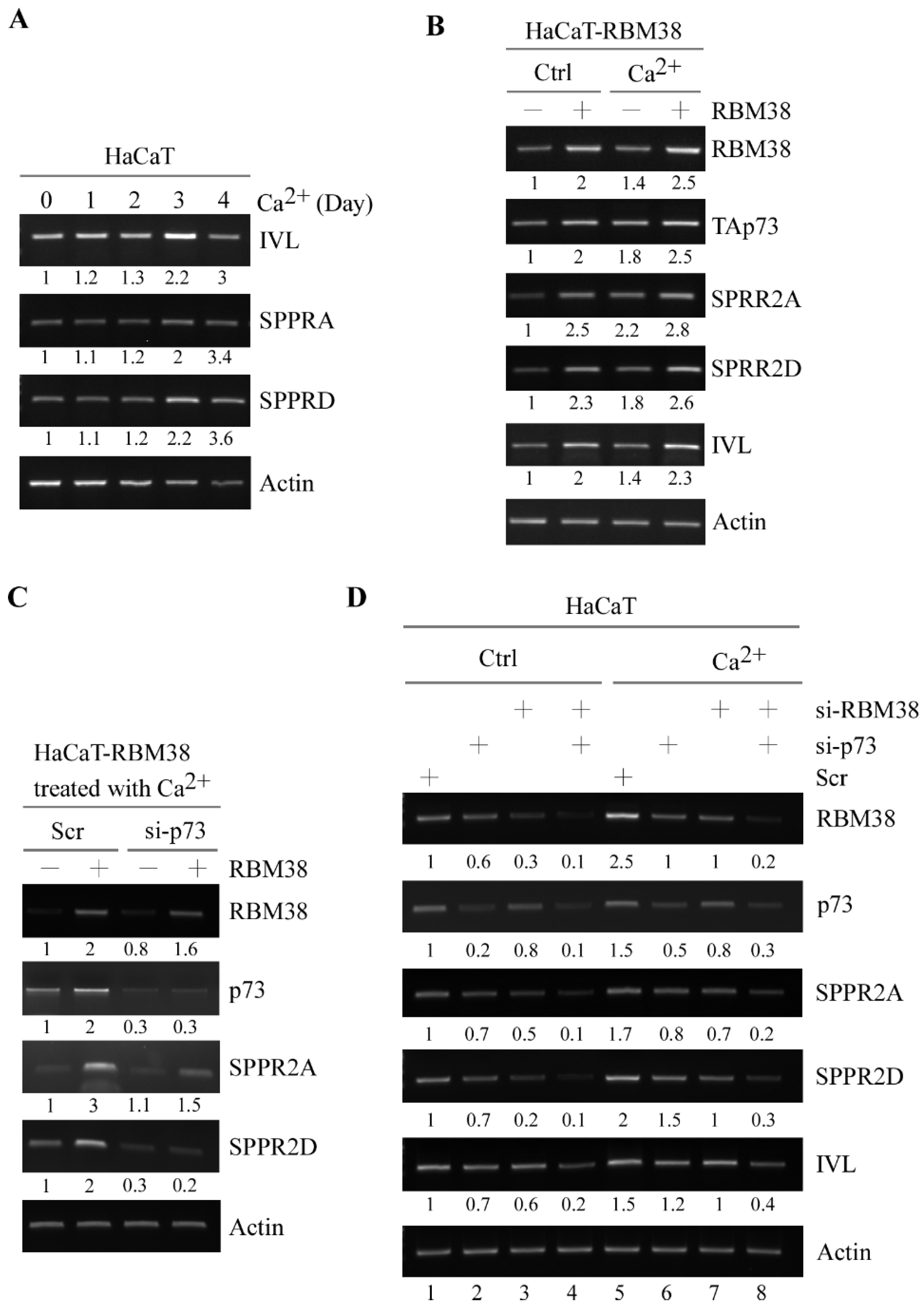
3.3. SPRR2A and SPRR2D Are Induced by p73 and RBM38 for Keratinocyte Differentiation
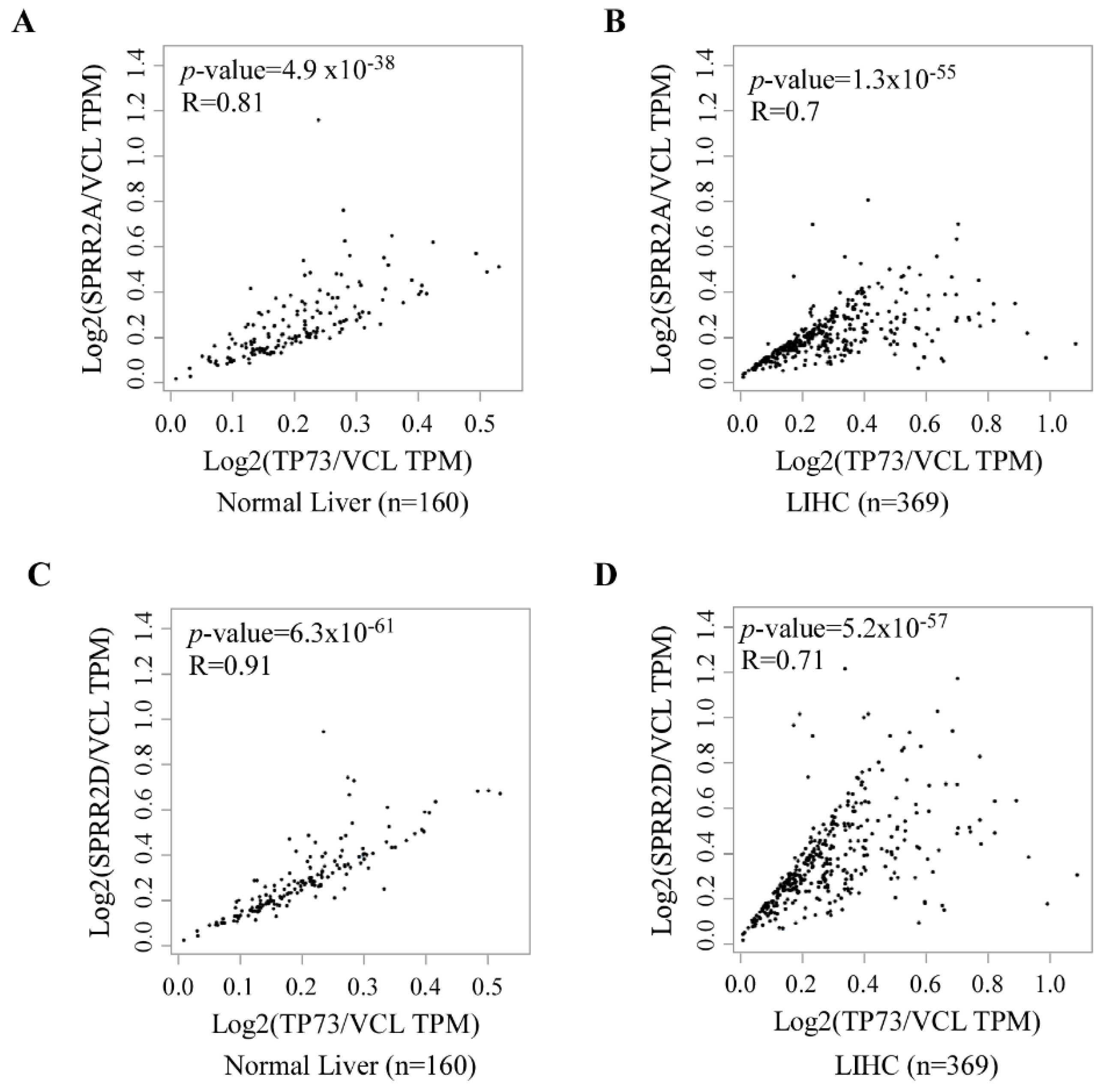
3.4. p73 and SPRR2A/2D Are Coordinately Expressed in Normal and Neoplastic Tissues
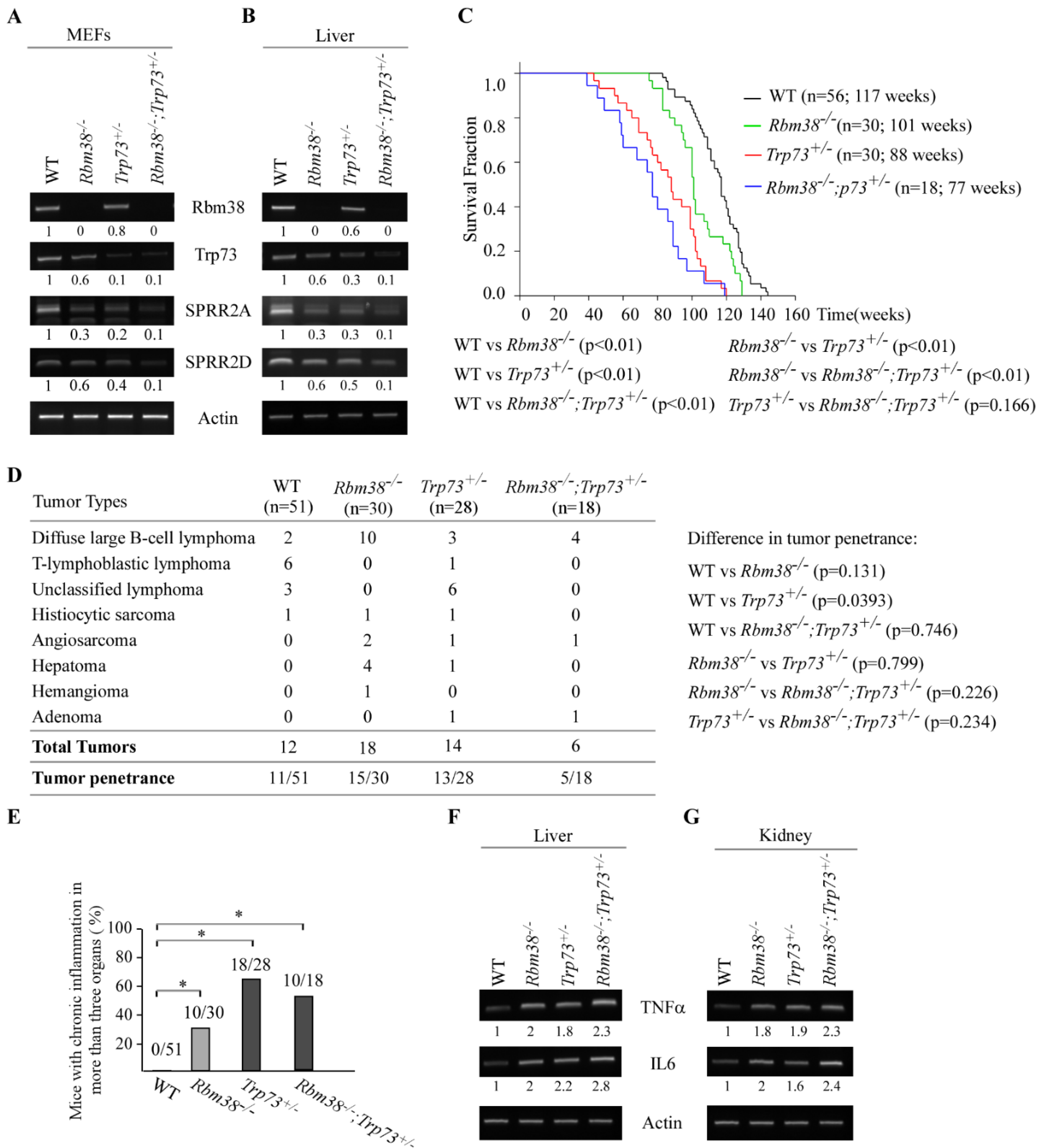
Loss of Rbm38 Cooperates with Trp73 Deficiency to Modulate Chronic Inflammation Potentially Via SPRR2A/2D
4. Discussion
5. Conclusion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Harms, K.; Nozell, S.; Chen, X. The common and distinct target genes of the p53 family transcription factors. Cell. Mol. Life Sci. 2004, 61, 822–842. [Google Scholar] [CrossRef]
- Kaghad, M.; Bonnet, H.; Yang, A.; Creancier, L.; Biscan, J.-C.; Valent, A.; Minty, A.; Chalon, P.; Lelias, J.-M.; Dumont, X.; et al. Monoallelically Expressed Gene Related to p53 at 1p36, a Region Frequently Deleted in Neuroblastoma and Other Human Cancers. Cell 1997, 90, 809–819. [Google Scholar] [CrossRef] [Green Version]
- Melino, G.; De Laurenzi, V.; Vousden, K.H. p73: Friend or foe in tumorigenesis. Nat. Rev. Cancer 2002, 2, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Walker, N.; Bronson, R.; Kaghad, M.; Oosterwegel, M.; Bonnin, J.; Vagner, C.; Bonnet, H.; Dikkes, P.; Sharpe, A.; et al. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nat. Cell Biol. 2000, 404, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Deyoung, M.P.; Ellisen, L.W. p63 and p73 in human cancer: Defining the network. Oncogene 2007, 26, 5169–5183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moll, U.M.; Slade, N. p63 and p73: Roles in development and tumor formation. Mol. Cancer Res. 2004, 2, 371–386. [Google Scholar] [PubMed]
- Vikhreva, P.; Melino, G.; Amelio, I. p73 Alternative Splicing: Exploring a Biological Role for the C-Terminal Isoforms. J. Mol. Biol. 2018, 430, 1829–1838. [Google Scholar] [CrossRef]
- Costanzo, A.; Pediconi, N.; Narcisi, A.; Guerrieri, F.; Belloni, L.; Fausti, F.; Botti, E.; Levrero, M. TP63andTP73in cancer, an unresolved “family” puzzle of complexity, redundancy and hierarchy. FEBS Lett. 2014, 588, 2590–2599. [Google Scholar] [CrossRef] [Green Version]
- Nemajerova, A.; Moll, U.M. Tissue-specific roles of p73 in development and homeostasis. J. Cell Sci. 2019, 132, jcs233338. [Google Scholar] [CrossRef] [Green Version]
- Sabapathy, K. p73: A Positive or Negative Regulator of Angiogenesis, or Both? Mol. Cell. Biol. 2015, 36, 848–854. [Google Scholar] [CrossRef] [Green Version]
- Napoli, M.; Flores, E.R. Unifying the p73 knockout phenotypes: TAp73 orchestrates multiciliogenesis. Genes Dev. 2016, 30, 1253–1254. [Google Scholar] [CrossRef] [Green Version]
- Marshall, C.B.; Mays, D.J.; Beeler, J.S.; Rosenbluth, J.M.; Boyd, K.L.; Guasch, G.L.S.; Shaver, T.M.; Tang, L.J.; Liu, Q.; Shyr, Y.; et al. p73 Is Required for Multiciliogenesis and Regulates the Foxj1-Associated Gene Network. Cell Rep. 2016, 14, 2289–2300. [Google Scholar] [CrossRef] [Green Version]
- Tissir, F.; Ravni, A.; Achouri, Y.; Riethmacher, D.; Meyer, G.; Goffinet, A.M. DeltaNp73 regulates neuronal survival in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 16871–16876. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, M.T.; Rufini, A.; Wetzel, M.K.; Tsuchihara, K.; Inoue, S.; Tomasini, R.; Itie-Youten, A.; Wakeham, A.; Arsenian-Henriksson, M.; Melino, G.; et al. Isoform-specific p73 knockout mice reveal a novel role for Np73 in the DNA damage response pathway. Genes Dev. 2010, 24, 549–560. [Google Scholar] [CrossRef] [Green Version]
- Tomasini, R.; Tsuchihara, K.; Wilhelm, M.; Fujitani, M.; Rufini, A.; Cheung, C.C.; Khan, F.; Itie-Youten, A.; Wakeham, A.; Tsao, M.-S.; et al. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev. 2008, 22, 2677–2691. [Google Scholar] [CrossRef] [Green Version]
- Inoue, S.; Tomasini, R.; Rufini, A.; Elia, A.J.; Agostini, M.; Amelio, I.; Cescon, D.; Dinsdale, D.; Zhou, L.; Harris, I.S.; et al. TAp73 is required for spermatogenesis and the maintenance of male fertility. Proc. Natl. Acad. Sci. USA 2014, 111, 1843–1848. [Google Scholar] [CrossRef] [Green Version]
- Shu, L.; Yan, W.; Chen, X. RNPC1, an RNA-binding protein and a target of the p53 family, is required for maintaining the stability of the basal and stress-induced p21 transcript. Genes Dev. 2006, 20, 2961–2972. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Cho, S.-J.; Shu, L.; Yan, W.; Guerrero, T.; Kent, M.; Skorupski, K.; Chen, H.; Chen, X. Translational repression of p53 by RNPC1, a p53 target overexpressed in lymphomas. Genes Dev. 2011, 25, 1528–1543. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Cho, S.J.; Chen, X. RNPC1, an RNA-binding protein and a target of the p53 family, regulates p63 expression through mRNA stability. Proc. Natl. Acad. Sci. USA 2010, 107, 9614–9619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, W.; Zhang, J.; Zhang, Y.; Jung, Y.-S.; Chen, X. p73 Expression Is Regulated by RNPC1, a Target of the p53 Family, via mRNA Stability. Mol. Cell. Biol. 2012, 32, 2336–2348. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Kong, X.; Zhang, Y.; Sun, W.; Wang, J.; Chen, M.; Chen, X. FDXRregulatesTP73tumor suppressor viaIRP2to modulate aging and tumor suppression. J. Pathol. 2020, 251, 284–296. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, E.; Ren, C.; Yan, W.; Zhang, M.; Chen, M.; Cardiff, R.D.; Imai, D.M.; Wisner, E.; Chen, X. Mice deficient in Rbm38, a target of the p53 family, are susceptible to accelerated aging and spontaneous tumors. Proc. Natl. Acad. Sci. USA 2014, 111, 18637–18642. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Sun, W.; Kong, X.; Zhang, Y.; Yang, H.J.; Ren, C.; Jiang, Y.; Chen, M.; Chen, X. Mutant p53 antagonizes p63/p73-mediated tumor suppression via Notch1. Proc. Natl. Acad. Sci. USA 2019, 116, 24259–24267. [Google Scholar] [CrossRef]
- Scoumanne, A.; Cho, S.J.; Zhang, J.; Chen, X. The cyclin-dependent kinase inhibitor p21 is regulated by RNA-binding protein PCBP4 via mRNA stability. Nucleic Acids Res. 2010, 39, 213–224. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Xu, E.; Ren, C.; Yang, H.J.; Zhang, Y.; Sun, W.; Kong, X.; Zhang, W.; Chen, M.; Huang, E.C.; et al. Genetic Ablation of Rbm38 Promotes Lymphomagenesis in the Context of Mutant p53 by Downregulating PTEN. Cancer Res. 2018, 78, 1511–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dohn, M.; Zhang, S.; Chen, X. p63α and ΔNp63α can induce cell cycle arrest and apoptosis and differentially regulate p53 target genes. Oncogene 2001, 20, 3193–3205. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Yan, W.; Chen, X. p53 is required for nerve growth factor-mediated differentiation of PC12 cells via regulation of TrkA levels. Cell Death Differ. 2006, 13, 2118–2128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carregaro, F.; Stefanini, A.C.B.; Henrique, T.; Tajara, E.H. Study of small proline-rich proteins (SPRRs) in health and disease: A review of the literature. Arch. Dermatol. Res. 2013, 305, 857–866. [Google Scholar] [CrossRef]
- Rochman, M.; Travers, J.; Miracle, C.E.; Bedard, M.C.; Wen, T.; Azouz, N.; Caldwell, J.M.; Kc, K.; Sherrill, J.D.; Davis, B.; et al. Profound loss of esophageal tissue differentiation in patients with eosinophilic esophagitis. J. Allergy Clin. Immunol. 2017, 140, 738–749.e3. [Google Scholar] [CrossRef] [Green Version]
- Hohl, D.; de Viragh, P.A.; Arniguet-Barras, F.O.; Gibbs, S.; Backendorf, C.; Huber, M. The Small Proline-Rich Proteins Constitute a Multigene Family of Differentially Regulated Cornified Cell Envelope Precursor Proteins. J. Investig. Dermatol. 1995, 104, 902–909. [Google Scholar] [CrossRef] [Green Version]
- Cabral, A.; Voskamp, P.; Cleton-Jansen, A.-M.; South, A.; Nizetic, D.; Backendorf, C. Structural Organization and Regulation of the Small Proline-rich Family of Cornified Envelope Precursors Suggest a Role in Adaptive Barrier Function. J. Biol. Chem. 2001, 276, 19231–19237. [Google Scholar] [CrossRef] [Green Version]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef] [Green Version]
- Breitkreutz, D.; Stark, H.-J.; Plein, P.; Baur, M.; Fusenig, N.E. Differential modulation of epidermal keratinization in immortalized (HaCaT) and tumorigenic human skin keratinocytes (HaCaT-ras) by retinoic acid and extracellular Ca2+. Differentiation 1993, 54, 201–217. [Google Scholar] [CrossRef]
- Eckert, R.L.; Welter, J.F. Transcription factor regulation of epidermal keratinocyte gene expression. Mol. Biol. Rep. 1996, 23, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.J.; Zhang, J.; Yan, W.; Cho, S.-J.; Lucchesi, C.; Chen, M.; Huang, E.C.; Scoumanne, A.; Zhang, W.; Chen, X. Ninjurin 1 has two opposing functions in tumorigenesis in a p53-dependent manner. Proc. Natl. Acad. Sci. USA 2017, 114, 11500–11505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Qian, Y.; Zhang, J.; Yanhong, Z.; Jung, Y.-S.; Chen, M.; Huang, E.; Lloyd, K.; Duan, Y.; Wang, J.; et al. Ferredoxin reductase is critical for p53-dependent tumor suppression via iron regulatory protein 2. Genes Dev. 2017, 31, 1243–1256. [Google Scholar] [CrossRef] [Green Version]
- Steinert, P.M.; Marekov, L.N. The Proteins Elafin, Filaggrin, Keratin Intermediate Filaments, Loricrin, and Small Proline-rich Proteins 1 and 2 Are Isodipeptide Cross-linked Components of the Human Epidermal Cornified Cell Envelope. J. Biol. Chem. 1995, 270, 17702–17711. [Google Scholar] [CrossRef] [Green Version]
- Segre, J.A. Epidermal barrier formation and recovery in skin disorders. J. Clin. Investig. 2006, 116, 1150–1158. [Google Scholar] [CrossRef] [PubMed]
- Lechuga, S.; Ivanov, A.I. Disruption of the epithelial barrier during intestinal inflammation: Quest for new molecules and mechanisms. Biochim. Biophys. Acta (BBA) Bioenerg. 2017, 1864, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Schleimer, R.P.; Berdnikovs, S. Etiology of epithelial barrier dysfunction in patients with type 2 inflammatory diseases. J. Allergy Clin. Immunol. 2017, 139, 1752–1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishida-Yamamoto, A.; Iizuka, H.; Manabe, M.; O’Guin, W.M.; Hohl, D.; Kartasova, T.; Kuroki, T.; Roop, D.R.; Eady, R.A.J. Altered distribution of keratinization markers in epidermolytic hyperkeratosis. Arch. Dermatol. Res. 1995, 287, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Beeler, J.S.; Marshall, C.B.; Gonzalez-Ericsson, P.I.; Shaver, T.M.; Guasch, G.L.S.; Lea, S.T.; Johnson, K.N.; Jin, H.; Venters, B.J.; Sanders, M.E.; et al. p73 regulates epidermal wound healing and induced keratinocyte programming. PLOS ONE 2019, 14, e0218458. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations All authors have read and agreed to the published version of the manuscript. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kong, X.; Wang, D.; Sun, W.; Chen, M.; Chen, J.; Shi, J.; Zhang, J.; Chen, X. Small Proline-Rich Protein 2A and 2D Are Regulated by the RBM38-p73 Axis and Associated with p73-Dependent Suppression of Chronic Inflammation. Cancers 2021, 13, 2829. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13112829
Kong X, Wang D, Sun W, Chen M, Chen J, Shi J, Zhang J, Chen X. Small Proline-Rich Protein 2A and 2D Are Regulated by the RBM38-p73 Axis and Associated with p73-Dependent Suppression of Chronic Inflammation. Cancers. 2021; 13(11):2829. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13112829
Chicago/Turabian StyleKong, Xiangmudong, Dan Wang, Wenqiang Sun, Mingyi Chen, Jinhui Chen, Jisen Shi, Jin Zhang, and Xinbin Chen. 2021. "Small Proline-Rich Protein 2A and 2D Are Regulated by the RBM38-p73 Axis and Associated with p73-Dependent Suppression of Chronic Inflammation" Cancers 13, no. 11: 2829. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13112829