
Homologous Recombination Repair in Biliary Tract Cancers: A Prime Target for PARP Inhibition?

 ,
,
Abstract
:Simple Summary
Abstract

1. Introduction
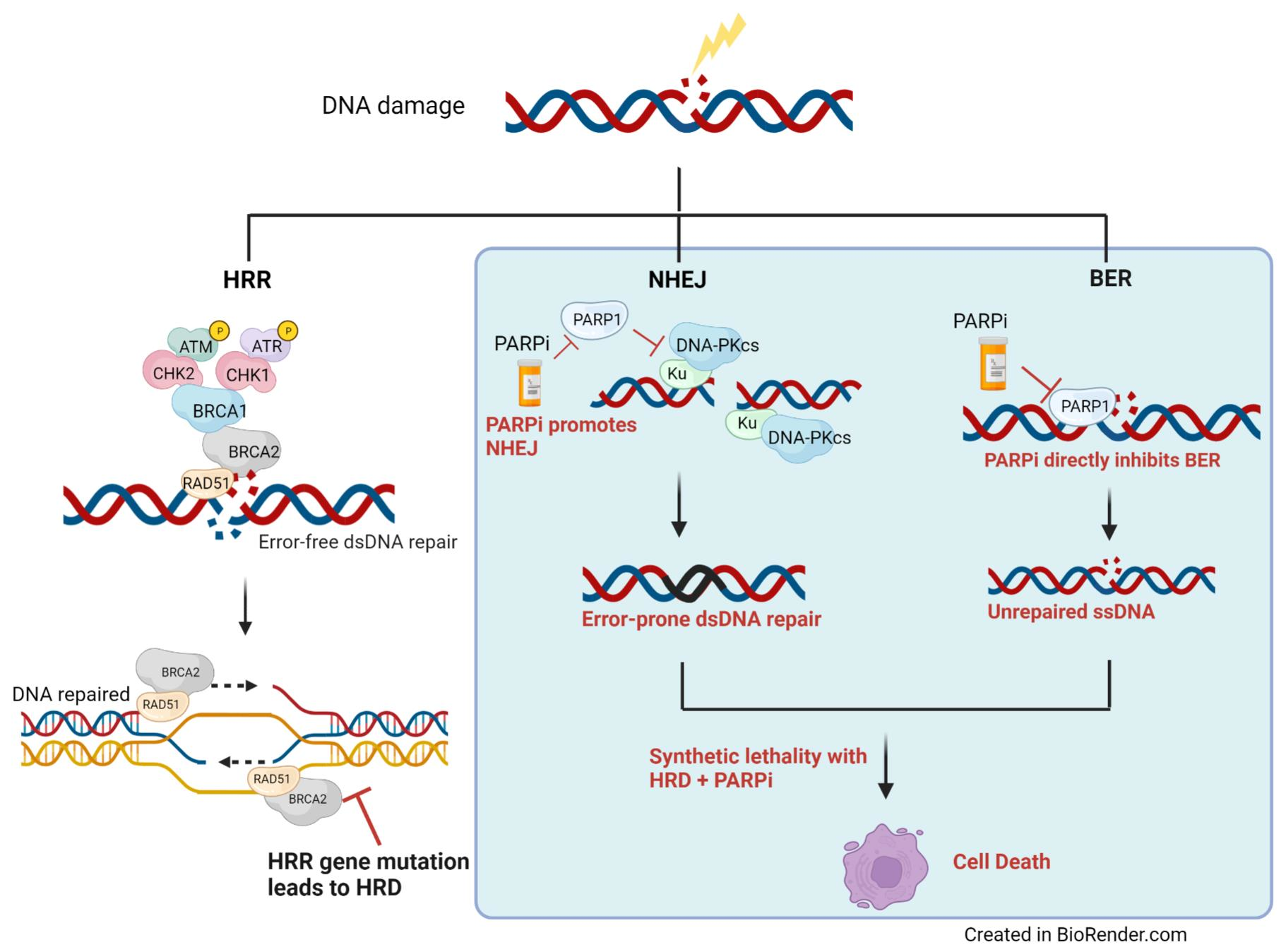
2. The DNA Damage Repair Pathway
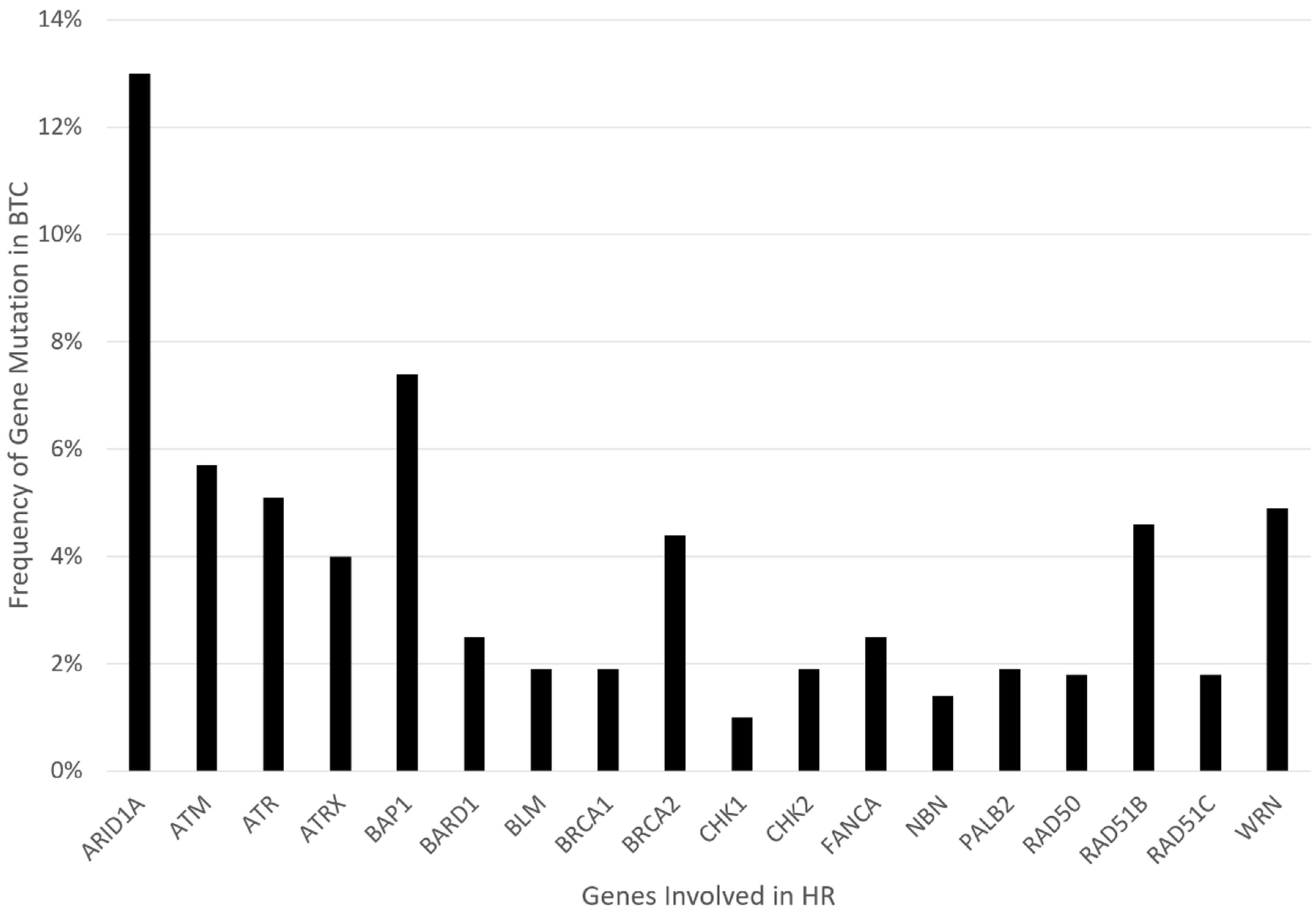
3. Biomarkers of HRD and Predictors of PARPi Response in BTC
4. ARID1A
5. ATM and ATR
6. IDH1
7. RAD52
8. RAD51 and RAD51C
9. Genomic Loss of Heterozygosity
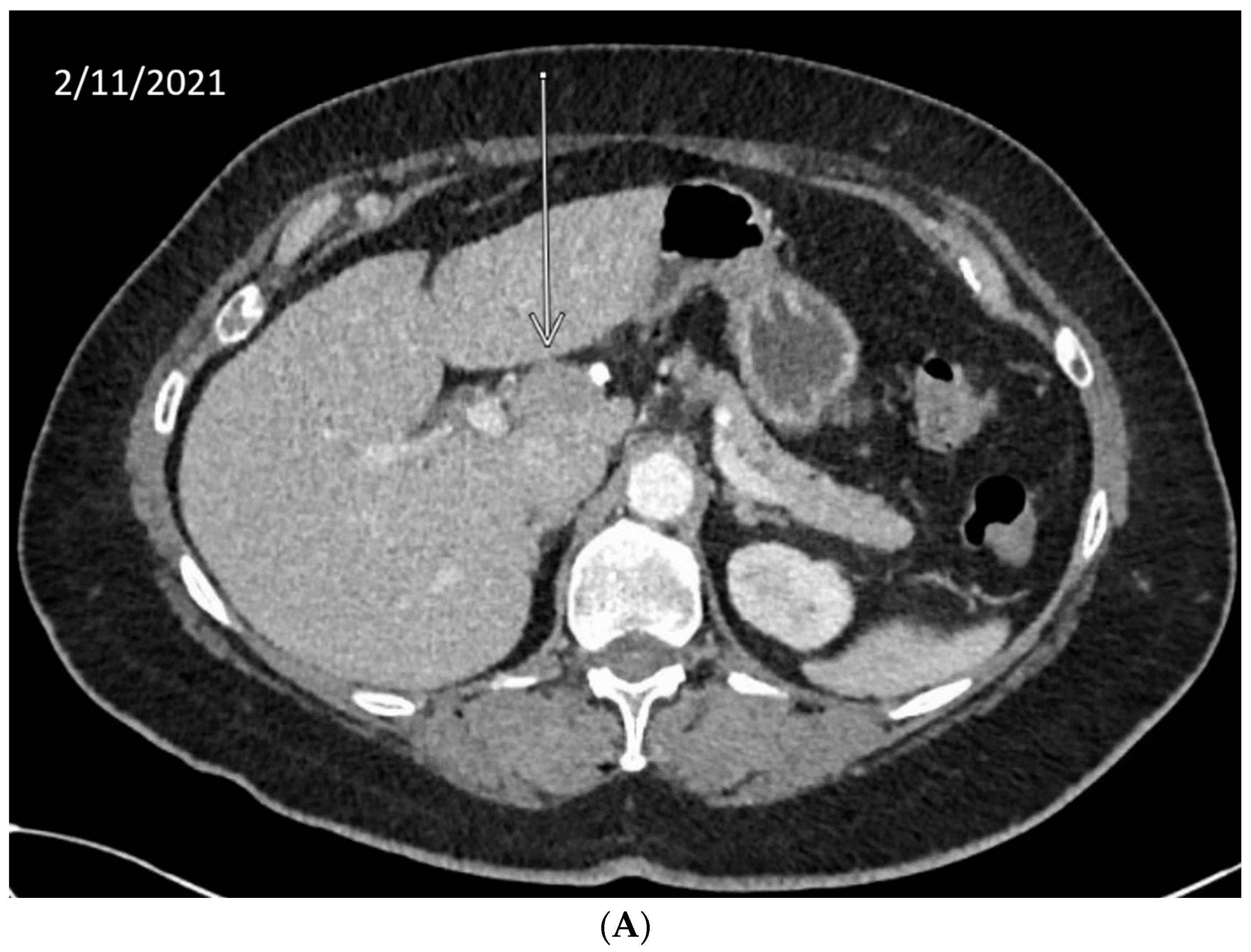
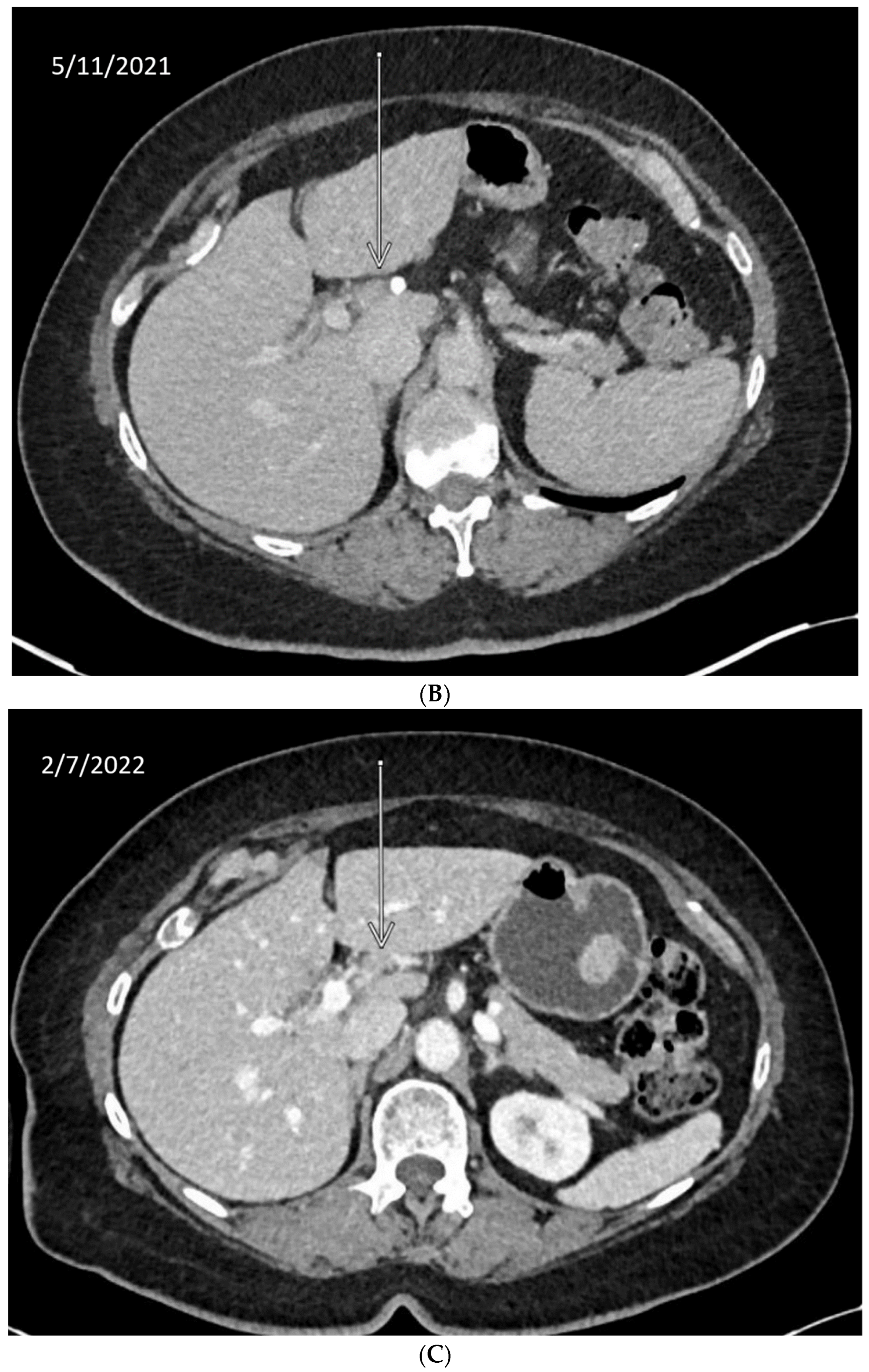
10. Anecdotal Case of RAD51 Mutation in CCA
11. Augmenting PARPi Efficacy
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Malaguarnera, G.; Giordano, M.; Paladina, I.; Rando, A.; Uccello, M.; Basile, F.; Biondi, A.; Carnazzo, S.; Alessandria, I.; Mazzarino, C. Markers of bile duct tumors. World J. Gastrointest. Oncol. 2011, 3, 49–59. [Google Scholar] [CrossRef]
- Rizvi, S.; Khan, S.A.; Hallemeier, C.L.; Kelley, R.K.; Gores, G.J. Cholangiocarcinoma—Evolving concepts and therapeutic strategies. Nat. Rev. Clin. Oncol. 2018, 15, 95–111. [Google Scholar] [CrossRef] [Green Version]
- Everhart, J.E.; Ruhl, C.E. Burden of Digestive Diseases in the United States Part III: Liver, Biliary Tract, and Pancreas. Gastroenterology 2009, 136, 1134–1144. [Google Scholar] [CrossRef]
- Forner, A.; Vidili, G.; Rengo, M.; Bujanda, L.; Ponz-Sarvisé, M.; Lamarca, A. Clinical presentation, diagnosis and staging of cholangiocarcinoma. Liver Int. 2019, 39, 98–107. [Google Scholar] [CrossRef] [Green Version]
- Koerkamp, B.G.; Wiggers, J.K.; Gönen, M.; Doussot, A.; Allen, P.J.; Besselink, M.G.H.; Blumgart, L.H.; Busch, O.R.C.; D’Angelica, M.I.; DeMatteo, R.P.; et al. Survival after resection of perihilar cholangiocarcinoma—Development and external validation of a prognostic nomogram. Ann. Oncol. 2016, 27, 753. [Google Scholar] [CrossRef] [Green Version]
- DeOliveira, M.L.; Cunningham, S.C.; Cameron, J.L.; Kamangar, F.; Winter, J.M.; Lillemoe, K.D.; Choti, M.A.; Yeo, C.J.; Schulick, R.D. Cholangiocarcinoma. Ann. Surg. 2007, 245, 755–762. [Google Scholar] [CrossRef]
- Lubienski, A. Hepatocellular Carcinoma: Interventional Bridging to Liver Transplantation. Transplantation 2005, 80, S113–S119. [Google Scholar] [CrossRef]
- Endo, I.; Gonen, M.; Yopp, A.C.; Dalal, K.M.; Zhou, Q.; Klimstra, D.; D’Angelica, M.; DeMatteo, R.P.; Fong, Y.; Schwartz, L.; et al. Intrahepatic Cholangiocarcinoma. Ann. Surg. 2008, 248, 84–96. [Google Scholar] [CrossRef]
- Spolverato, G.; Kim, Y.; Alexandrescu, S.; Marques, H.P.; Lamelas, J.; Aldrighetti, L.; Gamblin, T.C.; Maithel, S.K.; Pulitano, C.; Bauer, T.W.; et al. Management and Outcomes of Patients with Recurrent Intrahepatic Cholangiocarcinoma Following Previous Curative-Intent Surgical Resection. Ann. Surg. Oncol. 2015, 23, 235–243. [Google Scholar] [CrossRef]
- Jarnagin, W.R.; Ruo, L.; Little, S.A.; Klimstra, D.; D’Angelica, M.; DeMatteo, R.P.; Wagman, R.; Blumgart, L.H.; Fong, Y. Patterns of initial disease recurrence after resection of gallbladder carcinoma and hilar cholangiocarcinoma: Implications for adjuvant therapeutic strategies. Cancer 2003, 98, 1689–1700. [Google Scholar] [CrossRef]
- Hasegawa, S.; Ikai, I.; Fujii, H.; Hatano, E.; Shimahara, Y. Surgical Resection of Hilar Cholangiocarcinoma: Analysis of Survival and Postoperative Complications. World J. Surg. 2007, 31, 1258–1265. [Google Scholar] [CrossRef]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Pereira, S.P.; et al. Cisplatin plus Gemcitabine versus Gemcitabine for Biliary Tract Cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef] [Green Version]
- Lamarca, A.; Palmer, D.H.; Wasan, H.S.; Ross, P.J.; Ma, Y.T.; Arora, A.; Falk, S.; Gillmore, R.; Wadsley, J.; Patel, K.; et al. Second-line FOLFOX chemotherapy versus active symptom control for advanced biliary tract cancer (ABC-06): A Phase 3, Open-;Label, Randomised, Controlled Trial. Lancet Oncol. 2021, 22, 690–701. [Google Scholar] [CrossRef]
- Park, I.; Lee, J.-L.; Ryu, M.-H.; Kim, T.-W.; Lee, S.S.; Park, D.H.; Lee, S.S.; Seo, D.W.; Kim, M.-H. Prognostic factors and predictive model in patients with advanced biliary tract adenocarcinoma receiving first-line palliative chemotherapy. Cancer 2009, 115, 4148–4155. [Google Scholar] [CrossRef]
- Jusakul, A.; Cutcutache, I.; Yong, C.H.; Lim, J.Q.; Ni Huang, M.; Padmanabhan, N.; Nellore, V.; Kongpetch, S.; Ng, A.W.T.; Ng, L.M.; et al. Whole-Genome and Epigenomic Landscapes of Etiologically Distinct Subtypes of Cholangiocarcinoma. Cancer Discov. 2017, 7, 1116–1135. [Google Scholar] [CrossRef] [Green Version]
- Farshidfar, F.; Zheng, S.; Gingras, M.-C.; Newton, Y.; Shih, J.; Robertson, A.G.; Hinoue, T.; Hoadley, K.A.; Gibb, E.A.; Roszik, J.; et al. Integrative Genomic Analysis of Cholangiocarcinoma Identifies Distinct IDH-Mutant Molecular Profiles. Cell Rep. 2017, 19, 2878–2880. [Google Scholar] [CrossRef]
- Fiste, O.; Ntanasis-Stathopoulos, I.; Gavriatopoulou, M.; Liontos, M.; Koutsoukos, K.; Dimopoulos, M.; Zagouri, F. The Emerging Role of Immunotherapy in Intrahepatic Cholangiocarcinoma. Vaccines 2021, 9, 422. [Google Scholar] [CrossRef]
- Rizzo, A.; Ricci, A.D.; Brandi, G. Pemigatinib: Hot topics behind the first approval of a targeted therapy in cholangiocarcinoma. Cancer Treat. Res. Commun. 2021, 27, 100337. [Google Scholar] [CrossRef]
- Zhu, A.X.; Macarulla, T.; Javle, M.M.; Kelley, R.K.; Lubner, S.J.; Adeva, J.; Cleary, J.M.; Catenacci, D.V.T.; Borad, M.J.; Bridgewater, J.A.; et al. Final Overall Survival Efficacy Results of Ivosidenib for Patients with Advanced Cholangiocarcinoma With IDH1 Mutation. JAMA Oncol. 2021, 7, 1669. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Sahai, V.; Hollebecque, A.; Vaccaro, G.; Melisi, D.; Al-Rajabi, R.; Paulson, A.S.; Borad, M.J.; Gallinson, D.; Murphy, A.G.; et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: A Multicentre, Open-Label, Phase 2 Study. Lancet Oncol. 2020, 21, 671–684. [Google Scholar] [CrossRef]
- Heeke, A.L.; Pishvaian, M.J.; Lynce, F.; Xiu, J.; Brody, J.R.; Chen, W.-J.; Baker, T.M.; Marshall, J.L.; Isaacs, C. Prevalence of Homologous Recombination–Related Gene Mutations Across Multiple Cancer Types. JCO Precis. Oncol. 2018, 2018, 1–13. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- COSMIC: The Catalogue of Somatic Mutations in Cancer. Available online: Cancer.sanger.ac.uk (accessed on 25 March 2022).
- Yang, W.; Sun, Y. Promising Molecular Targets for the Targeted Therapy of Biliary Tract Cancers: An Overview. OncoTargets Ther. 2021, 14, 1341–1366. [Google Scholar] [CrossRef]
- Ricci, A.D.; Rizzo, A.; Bonucci, C.; Tober, N.; Palloni, A.; Mollica, V.; Maggio, I.; Deserti, M.; Tavolari, S.; Brandi, G. PARP Inhibitors in Biliary Tract Cancer: A New Kid on the Block? Medicines 2020, 7, 54. [Google Scholar] [CrossRef]
- Minten, E.V.; Yu, D.S. DNA Repair: Translation to the Clinic. Clin. Oncol. 2019, 31, 303–310. [Google Scholar] [CrossRef]
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef]
- Goldstein, M.; Kastan, M.B. The DNA Damage Response: Implications for Tumor Responses to Radiation and Chemotherapy. Annu. Rev. Med. 2015, 66, 129–143. [Google Scholar] [CrossRef] [Green Version]
- Caja, F.; Vodickova, L.; Kral, J.; Vymetalkova, V.; Naccarati, A.; Vodicka, P. DNA Mismatch Repair Gene Variants in Sporadic Solid Cancers. Int. J. Mol. Sci. 2020, 21, 5561. [Google Scholar] [CrossRef]
- Pearl, L.H.; Schierz, A.C.; Ward, S.E.; Al-Lazikani, B.; Pearl, F.M. Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer 2015, 15, 166–180. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Peng, Y.; Wei, L.; Zhang, W.; Yang, L.; Lan, L.; Kapoor, P.; Ju, Z.; Mo, Q.; Shih, I.-M.; et al. ARID1A Deficiency Impairs the DNA Damage Checkpoint and Sensitizes Cells to PARP Inhibitors. Cancer Discov. 2015, 5, 752–767. [Google Scholar] [CrossRef] [Green Version]
- Mullen, J.; Kato, S.; Sicklick, J.K.; Kurzrock, R. Targeting ARID1A mutations in cancer. Cancer Treat. Rev. 2021, 100, 102287. [Google Scholar] [CrossRef]
- Park, Y.; Chui, M.H.; Rahmanto, Y.S.; Yu, Z.-C.; Shamanna, R.A.; Bellani, M.A.; Gaillard, S.; Ayhan, A.; Viswanathan, A.; Seidman, M.M.; et al. Loss of ARID1A in Tumor Cells Renders Selective Vulnerability to Combined Ionizing Radiation and PARP Inhibitor Therapy. Clin. Cancer Res. 2019, 25, 5584–5594. [Google Scholar] [CrossRef]
- Hu, H.-M.; Zhao, X.; Kaushik, S.; Robillard, L.; Barthelet, A.; Lin, K.K.; Shah, K.N.; Simmons, A.D.; Raponi, M.; Harding, T.C.; et al. A Quantitative Chemotherapy Genetic Interaction Map Reveals Factors Associated with PARP Inhibitor Resistance. Cell Rep. 2018, 23, 918–929. [Google Scholar] [CrossRef] [Green Version]
- Toh, M.; Ngeow, J. Homologous Recombination Deficiency: Cancer Predispositions and Treatment Implications. The Oncologist 2021, 26, e1526–e1537. [Google Scholar] [CrossRef]
- Neeb, A.; Herranz, N.; Arce-Gallego, S.; Miranda, S.; Buroni, L.; Yuan, W.; Athie, Y.W.A.; Casals, T.; Carmichael, J.; Rodrigues, D.N.; et al. Advanced Prostate Cancer with ATM Loss: PARP and ATR Inhibitors. Eur. Urol. 2021, 79, 200–211. [Google Scholar] [CrossRef]
- Gout, J.; Perkhofer, L.; Morawe, M.; Arnold, F.; Ihle, M.; Biber, S.; Lange, S.; Roger, E.; Kraus, J.M.; Stifter, K.; et al. Synergistic targeting and resistance to PARP inhibition in DNA damage repair-deficient pancreatic cancer. Gut 2021, 70, 743–760. [Google Scholar] [CrossRef]
- Wang, C.; Jette, N.; Moussienko, D.; Bebb, D.G.; Lees-Miller, S.P. ATM-Deficient Colorectal Cancer Cells Are Sensitive to the PARP Inhibitor Olaparib. Transl. Oncol. 2017, 10, 190–196. [Google Scholar] [CrossRef]
- Bang, Y.-J.; Xu, R.-H.; Chin, K.; Lee, K.-W.; Park, S.H.; Rha, S.Y.; Shen, L.; Qin, S.; Xu, N.; Im, S.-A.; et al. Olaparib in combination with paclitaxel in patients with advanced gastric cancer who have progressed following first-line therapy (GOLD): A Double-Blind, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2017, 18, 1637–1651. [Google Scholar] [CrossRef]
- Menolfi, D.; Zha, S. ATM, ATR and DNA-PKcs kinases—The lessons from the mouse models: Inhibition ≠ deletion. Cell Biosci. 2020, 10, 81. [Google Scholar] [CrossRef]
- Kim, H.; Xu, H.; George, E.; Hallberg, D.; Kumar, S.; Jagannathan, V.; Medvedev, S.; Kinose, Y.; Devins, K.; Verma, P.; et al. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat. Commun. 2020, 11, 3726. [Google Scholar] [CrossRef]
- Juhász, S.; Elbakry, A.; Mathes, A.; Löbrich, M. ATRX Promotes DNA Repair Synthesis and Sister Chromatid Exchange during Homologous Recombination. Mol. Cell 2018, 71, 11–24.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garbarino, J.; Eckroate, J.; Sundaram, R.K.; Jensen, R.B.; Bindra, R.S. Loss of ATRX confers DNA repair defects and PARP inhibitor sensitivity. Transl. Oncol. 2021, 14, 101147. [Google Scholar] [CrossRef] [PubMed]
- Bononi, A.; Giorgi, C.; Patergnani, S.; Larson, D.; Verbruggen, K.; Tanji, M.; Pellegrini, L.; Signorato, V.; Olivetto, F.; Pastorino, S.; et al. BAP1 regulates IP3R3-mediated Ca2+ flux to mitochondria suppressing cell transformation. Nature 2017, 546, 549–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ismail, I.H.; Davidson, R.; Gagné, J.-P.; Xu, Z.Z.; Poirier, G.G.; Hendzel, M.J. Germline Mutations in BAP1 Impair Its Function in DNA Double-Strand Break Repair. Cancer Res. 2014, 74, 4282–4294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabbatino, F.; Liguori, L.; Malapelle, U.; Schiavi, F.; Tortora, V.; Conti, V.; Filippelli, A.; Tortora, G.; Ferrone, C.R.; Pepe, S. Case Report: BAP1 Mutation and RAD21 Amplification as Predictive Biomarkers to PARP Inhibitor in Metastatic Intrahepatic Cholangiocarcinoma. Front. Oncol. 2020, 10, 567289. [Google Scholar] [CrossRef]
- A Fennell, D.; King, A.; Mohammed, S.; Branson, A.; Brookes, C.; Darlison, L.; Dawson, A.G.; Gaba, A.; Hutka, M.; Morgan, B.; et al. Rucaparib in patients with BAP1-deficient or BRCA1-deficient mesothelioma (MiST1): An Open-Label, Single-Arm, Phase 2a Clinical Trial. Lancet Respir. Med. 2021, 9, 593–600. [Google Scholar] [CrossRef]
- Fabbro, M.; Rodriguez, J.A.; Baer, R.; Henderson, B.R. BARD1 Induces BRCA1 Intranuclear Foci Formation by Increasing RING-dependent BRCA1 Nuclear Import and Inhibiting BRCA1 Nuclear Export. J. Biol. Chem. 2002, 277, 21315–21324. [Google Scholar] [CrossRef] [Green Version]
- Özden, O.; Bishehsari, F.; Bauer, J.; Park, S.-H.; Jana, A.; Baik, S.H.; Sporn, J.C.; Staudacher, J.; Yazici, C.; Krett, N.; et al. Expression of an Oncogenic BARD1 Splice Variant Impairs Homologous Recombination and Predicts Response to PARP-1 Inhibitor Therapy in Colon Cancer. Sci. Rep. 2016, 6, 26273. [Google Scholar] [CrossRef]
- Kong, E.A.Y.; Xu, C.; Sun, X.; Sun, H.; Zhao, X.; He, N.; Ji, K.; Wang, Q.; Du, L.; Wang, J.; et al. BLM helicase inhibition synergizes with PARP inhibition to improve the radiosensitivity of olaparib resistant non-small cell lung cancer cells by inhibiting homologous recombination repair. Cancer Biol. Med. 2021, 18, 1–32. [Google Scholar] [CrossRef]
- Wang, C.-X.; Zhang, Z.-L.; Yin, Q.-K.; Tu, J.-L.; Wang, J.-E.; Xu, Y.-H.; Rao, Y.; Ou, T.-M.; Huang, S.-L.; Li, D.; et al. Design, Synthesis, and Evaluation of New Quinazolinone Derivatives that Inhibit Bloom Syndrome Protein (BLM) Helicase, Trigger DNA Damage at the Telomere Region, and Synergize with PARP Inhibitors. J. Med. Chem. 2020, 63, 9752–9772. [Google Scholar] [CrossRef]
- Smith, J.; Mun Tho, L.; Xu, N.A.; Gillespie, D. Chapter 3—The ATM–Chk2 and ATR–Chk1 pathways in DNA damage signaling and cancer. In Advances in Cancer Research; Vande Woude, G.F., Klein, G., Eds.; Academic Press: Cambridge, MA, USA, 2010; pp. 73–112. [Google Scholar]
- Yin, Y.; Shen, Q.; Zhang, P.; Tao, R.; Chang, W.; Li, R.; Xie, G.; Liu, W.; Zhang, L.; Kapoor, P.; et al. Chk1 inhibition potentiates the therapeutic efficacy of PARP inhibitor BMN673 in gastric cancer. Am. J. Cancer Res. 2017, 7, 473–483. [Google Scholar] [PubMed]
- Walden, H.; Deans, A.J. The Fanconi Anemia DNA Repair Pathway: Structural and Functional Insights into a Complex Disorder. Annu. Rev. Biophys. 2014, 43, 257–278. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Campbell, D.; Patnaik, A.; Shapiro, J.D.; Sautois, B.; Vogelzang, N.J.; Voog, E.G.; Bryce, A.H.; McDermott, R.; Ricci, F.; et al. Non-BRCA DNA Damage Repair Gene Alterations and Response to the PARP Inhibitor Rucaparib in Metastatic Castration-Resistant Prostate Cancer: Analysis From the Phase II TRITON2 Study. Clin. Cancer Res. 2020, 26, 2487–2496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramus, S.J.; Song, H.; Dicks, E.; Tyrer, J.P.; Rosenthal, A.N.; Intermaggio, M.P.; Fraser, L.; Gentry-Maharaj, A.; Hayward, J.; Philpott, S.; et al. Germline Mutations in the BRIP1, BARD1, PALB2, and NBN Genes in Women with Ovarian Cancer. JNCI J. Natl. Cancer Inst. 2015, 107, djv214. [Google Scholar] [CrossRef]
- Lajud, S.A.; Nagda, D.A.; Yamashita, T.; Zheng, J.; Tanaka, N.; Abuzeid, W.M.; Civantos, A.; Bezpalko, O.; O’Malley, B.W.; Li, D. Dual Disruption of DNA Repair and Telomere Maintenance for the Treatment of Head and Neck Cancer. Clin. Cancer Res. 2014, 20, 6465–6478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tung, N.M.; Robson, M.E.; Ventz, S.; Santa-Maria, C.A.; Nanda, R.; Marcom, P.K.; Shah, P.D.; Ballinger, T.J.; Yang, E.S.; Vinayak, S.; et al. TBCRC 048: Phase II Study of Olaparib for Metastatic Breast Cancer and Mutations in Homologous Recombination-Related Genes. J. Clin. Oncol. 2020, 38, 4274–4282. [Google Scholar] [CrossRef]
- Alblihy, A.; Alabdullah, M.L.; Toss, M.S.; Algethami, M.; Mongan, N.P.; Rakha, E.A.; Madhusudan, S. RAD50 deficiency is a predictor of platinum sensitivity in sporadic epithelial ovarian cancers. Mol. Biomed. 2020, 1, 19. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, G.; Xue, F.; Edwards, R.; Sood, A.K.; Zhang, W.; Yang, D. Copy number deletion of RAD50 as predictive marker of BRCAness and PARP inhibitor response in BRCA wild type ovarian cancer. Gynecol. Oncol. 2016, 141, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef]
- Vispe, S.; Cazaux, C.; Lesca, C.; Defais, M. Overexpression of Rad51 protein stimulates homologous recombination and increases resistance of mammalian cells to ionizing radiation. Nucleic Acids Res. 1998, 26, 2859–2864. [Google Scholar] [CrossRef] [Green Version]
- Baumann, P.; West, S.C. Role of the human RAD51 protein in homologous recombination and double-stranded-break repair. Trends Biochem. Sci. 1998, 23, 247–251. [Google Scholar] [CrossRef]
- Cruz, C.; Castroviejo-Bermejo, M.; Gutiérrez-Enríquez, S.; Llop-Guevara, A.; Ibrahim, Y.; Oliver, A.G.; Bonache, S.; Morancho, B.; Bruna, A.; Rueda, O.; et al. RAD51 foci as a functional biomarker of homologous recombination repair and PARP inhibitor resistance in germline BRCA-mutated breast cancer. Ann. Oncol. 2018, 29, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Waks, A.; Cohen, O.; Kochupurakkal, B.; Kim, D.; Dunn, C.; Buendia, J.B.; Wander, S.; Helvie, K.; Lloyd, M.; Marini, L.; et al. Reversion and non-reversion mechanisms of resistance to PARP inhibitor or platinum chemotherapy in BRCA1/2-mutant metastatic breast cancer. Ann. Oncol. 2020, 31, 590–598. [Google Scholar] [CrossRef]
- Gottifredi, V.; Wiesmüller, L. Current Understanding of RAD52 Functions: Fundamental and Therapeutic Insights. Cancers 2020, 12, 705. [Google Scholar] [CrossRef] [Green Version]
- Davis, A.P. The Rad52–Rad59 complex interacts with Rad51 and replication protein A. DNA Repair 2003, 2, 1127–1134. [Google Scholar] [CrossRef]
- Toma, M.; Sullivan-Reed, K.; Śliwiński, T.; Skorski, T. RAD52 as a Potential Target for Synthetic Lethality-Based Anticancer Therapies. Cancers 2019, 11, 1561. [Google Scholar] [CrossRef] [Green Version]
- Sullivan-Reed, K.; Bolton-Gillespie, E.; Dasgupta, Y.; Langer, S.; Siciliano, M.; Nieborowska-Skorska, M.; Hanamshet, K.; Belyaeva, E.A.; Bernhardy, A.J.; Lee, J.; et al. Simultaneous Targeting of PARP1 and RAD52 Triggers Dual Synthetic Lethality in BRCA-Deficient Tumor Cells. Cell Rep. 2018, 23, 3127–3136. [Google Scholar] [CrossRef]
- Shamanna, R.A.; Lu, H.; de Freitas, J.K.; Tian, J.; Croteau, D.L.; Bohr, V.A. WRN regulates pathway choice between classical and alternative non-homologous end joining. Nat. Commun. 2016, 7, 13785. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Sinha, D.; Bhattacharya, S.; Srinivasan, K.; Abdisalaam, S.; Asaithamby, A. Werner Syndrome Protein and DNA Replication. Int. J. Mol. Sci. 2018, 19, 3442. [Google Scholar] [CrossRef] [Green Version]
- Arai, A.; Chano, T.; Futami, K.; Furuichi, Y.; Ikebuchi, K.; Inui, T.; Tameno, H.; Ochi, Y.; Shimada, T.; Hisa, Y.; et al. RECQL1 and WRN Proteins Are Potential Therapeutic Targets in Head and Neck Squamous Cell Carcinoma. Cancer Res. 2011, 71, 4598–4607. [Google Scholar] [CrossRef] [Green Version]
- Sachdev, E.; Tabatabai, R.; Roy, V.; Rimel, B.J.; Mita, M.M. PARP Inhibition in Cancer: An Update on Clinical Development. Target. Oncol. 2019, 14, 657–679. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-S.; Camacho, C.V.; Kraus, W.L. Alternate therapeutic pathways for PARP inhibitors and potential mechanisms of resistance. Exp. Mol. Med. 2021, 53, 42–51. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, A.D. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 2018, 71, 172–176. [Google Scholar] [CrossRef]
- Caron, M.-C.; Sharma, A.K.; O’Sullivan, J.; Myler, L.R.; Ferreira, M.T.; Rodrigue, A.; Coulombe, Y.; Ethier, C.; Gagné, J.-P.; Langelier, M.-F.; et al. Poly(ADP-ribose) polymerase-1 antagonizes DNA resection at double-strand breaks. Nat. Commun. 2019, 10, 2954. [Google Scholar] [CrossRef] [Green Version]
- Penson, R.T.; Valencia, R.V.; Cibula, D.; Colombo, N.; Leath, C.A., III; Bidziński, M.; Kim, J.-W.; Nam, J.H.; Madry, R.; Hernández, C.; et al. Olaparib Versus Nonplatinum Chemotherapy in Patients With Platinum-Sensitive Relapsed Ovarian Cancer and a Germline BRCA1/2 Mutation (SOLO3): A Randomized Phase III Trial. J. Clin. Oncol. 2020, 38, 1164–1174. [Google Scholar] [CrossRef] [PubMed]
- Spizzo, G.; Puccini, A.; Xiu, J.; Goldberg, R.M.; Grothey, A.; Shields, A.F.; Arora, S.P.; Khushman, M.M.; Salem, M.E.; Battaglin, F.; et al. Frequency of BRCA mutation in biliary tract cancer and its correlation with tumor mutational burden (TMB) and microsatellite instability (MSI). J. Clin. Oncol. 2019, 37, 4085. [Google Scholar] [CrossRef]
- Zhao, S.; Xu, Y.; Wu, W.; Wang, P.; Wang, Y.; Jiang, H.; Zhu, J. ARID1A Variations in Cholangiocarcinoma: Clinical Significances and Molecular Mechanisms. Front. Oncol. 2021, 11, 2360. [Google Scholar] [CrossRef]
- Sun, X.; Wang, S.; Wei, Y.; Luo, X.; Jia, Y.; Li, L.; Gopal, P.; Zhu, M.; Nassour, I.; Chuang, J.-C.; et al. Arid1a Has Context-Dependent Oncogenic and Tumor Suppressor Functions in Liver Cancer. Cancer Cell 2017, 32, 574–589.e6. [Google Scholar] [CrossRef] [Green Version]
- Trizzino, M.; Barbieri, E.; Petracovici, A.; Wu, S.; Welsh, S.A.; Owens, T.A.; Licciulli, S.; Zhang, R.; Gardini, A. The Tumor Suppressor ARID1A Controls Global Transcription via Pausing of RNA Polymerase II. Cell Rep. 2018, 23, 3933–3945. [Google Scholar] [CrossRef]
- Reske, J.J.; Wilson, M.R.; Holladay, J.; Siwicki, R.A.; Skalski, H.; Harkins, S.; Adams, M.; Risinger, J.I.; Hostetter, G.; Lin, K.; et al. Co-existing TP53 and ARID1A mutations promote aggressive endometrial tumorigenesis. PLoS Genet. 2021, 17, e1009986. [Google Scholar] [CrossRef]
- Chaisaingmongkol, J.; Budhu, A.; Dang, H.; Rabibhadana, S.; Pupacdi, B.; Kwon, S.M.; Forgues, M.; Pomyen, Y.; Bhudhisawasdi, V.; Lertprasertsuke, N.; et al. Common Molecular Subtypes Among Asian Hepatocellular Carcinoma and Cholangiocarcinoma. Cancer Cell 2017, 32, 57–70.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathur, R. ARID1A loss in cancer: Towards a mechanistic understanding. Pharmacol. Ther. 2018, 190, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.; Zhao, T.; Tse, K.-H.; Chow, H.-M.; Cui, Y.; Jiang, L.; Du, S.; Loy, M.M.T.; Herrup, K. ATM and ATR play complementary roles in the behavior of excitatory and inhibitory vesicle populations. Proc. Natl. Acad. Sci. USA 2018, 115, E292–E301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bang, Y.-J.; Im, S.-A.; Lee, K.-W.; Cho, J.Y.; Song, E.-K.; Lee, K.H.; Kim, Y.H.; Park, J.O.; Chun, H.G.; Zang, D.Y.; et al. Randomized, Double-Blind Phase II Trial with Prospective Classification by ATM Protein Level to Evaluate the Efficacy and Tolerability of Olaparib Plus Paclitaxel in Patients with Recurrent or Metastatic Gastric Cancer. J. Clin. Oncol. 2015, 33, 3858–3865. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Ye, D.; Guan, K.-L.; Xiong, Y. IDH1 and IDH2 Mutations in Tumorigenesis: Mechanistic Insights and Clinical Perspectives. Clin. Cancer Res. 2012, 18, 5562–5571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulkowski, P.L.; Corso, C.D.; Robinson, N.D.; Scanlon, S.E.; Purshouse, K.R.; Bai, H.; Liu, Y.; Sundaram, R.K.; Hegan, D.C.; Fons, N.R.; et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci. Transl. Med. 2017, 9, eaal2463. [Google Scholar] [CrossRef] [Green Version]
- Inoue, S.; Li, W.Y.; Tseng, A.; Beerman, I.; Elia, A.J.; Bendall, S.C.; Lemonnier, F.; Kron, K.J.; Cescon, D.W.; Hao, Z.; et al. Mutant IDH1 Downregulates ATM and Alters DNA Repair and Sensitivity to DNA Damage Independent of TET. Cancer Cell 2016, 30, 337–348. [Google Scholar] [CrossRef] [Green Version]
- Eder, J.P.; Doroshow, D.B.; Do, K.T.; Keedy, V.L.; Sklar, J.S.; Glazer, P.; Bindra, R.; Shapiro, G.I. Clinical Efficacy of Olaparib in IDH1/IDH2-Mutant Mesenchymal Sarcomas. JCO Precis. Oncol. 2021, 5, 466–472. [Google Scholar] [CrossRef]
- Zan, H.; Tat, C.; Qiu, Z.; Taylor, J.R.; Guerrero, J.A.; Shen, T.; Casali, P. Rad52 competes with Ku70/Ku86 for binding to S-region DSB ends to modulate antibody class-switch DNA recombination. Nat. Commun. 2017, 8, 14244. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.E.; Meghani, K.; Brault, M.-E.; Leclerc, L.; He, Y.; Day, T.A.; Elias, K.M.; Drapkin, R.; Weinstock, D.M.; Dao, F.; et al. Platinum and PARP Inhibitor Resistance Due to Overexpression of MicroRNA-622 in BRCA1-Mutant Ovarian Cancer. Cell Rep. 2016, 14, 429–439. [Google Scholar] [CrossRef] [Green Version]
- Murfuni, I.; Rass, U. Chapter 8—Targeting homologous recombination repair in cancer. In DNA Repair in Cancer Therapy, 2nd ed.; Kelley, M.R., Fishel, M.L., Eds.; Academic Press: Cambridge, MA, USA, 2016; pp. 225–275. [Google Scholar]
- Graeser, M.; McCarthy, A.; Lord, C.; Savage, K.; Hills, M.; Salter, J.; Orr, N.; Parton, M.; Smith, I.E.; Reis-Filho, J.S.; et al. A Marker of Homologous Recombination Predicts Pathologic Complete Response to Neoadjuvant Chemotherapy in Primary Breast Cancer. Clin. Cancer Res. 2010, 16, 6159–6168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tennstedt, P.; Fresow, R.; Simon, R.; Marx, A.; Terracciano, L.; Petersen, C.; Sauter, G.; Dikomey, E.; Borgmann, K. RAD51 overexpression is a negative prognostic marker for colorectal adenocarcinoma. Int. J. Cancer 2012, 132, 2118–2126. [Google Scholar] [CrossRef] [PubMed]
- Wiegmans, A.P.; Al-Ejeh, F.; Chee, N.; Yap, P.-Y.; Gorski, J.J.; Da Silva, L.; Bolderson, E.; Chenevix-Trench, G.; Anderson, R.; Simpson, P.T.; et al. Rad51 supports triple negative breast cancer metastasis. Oncotarget 2014, 5, 3261–3272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.-L.; Liu, S.-S.; Xiong, Z.-F.; Wang, F.; Li, X.-Y.; Deng, H. Clinical significance of RAD51C and its contribution to ovarian carcinogenesis. Int. J. Clin. Exp. Pathol. 2020, 13, 14–20. [Google Scholar] [PubMed]
- Suwaki, N.; Klare, K.; Tarsounas, M. RAD51 paralogs: Roles in DNA damage signalling, recombinational repair and tumorigenesis. Semin. Cell Dev. Biol. 2011, 22, 898–905. [Google Scholar] [CrossRef] [PubMed]
- Rein, H.L.; A Bernstein, K.; A Baldock, R. RAD51 paralog function in replicative DNA damage and tolerance. Curr. Opin. Genet. Dev. 2021, 71, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Song, H.; Leslie, G.; Engel, C.; Hahnen, E.; Auber, B.; Horváth, J.; Kast, K.; Niederacher, D.; Turnbull, C.; et al. Ovarian and Breast Cancer Risks Associated With Pathogenic Variants in RAD51C and RAD51D. JNCI J. Natl. Cancer Inst. 2020, 112, 1242–1250. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; McInerny, S.; Zethoven, M.; Cheasley, D.; Lim, B.W.X.; Rowley, S.M.; Devereux, L.; Grewal, N.; Ahmadloo, S.; Byrne, D.; et al. Combined Tumor Sequencing and Case-Control Analyses of RAD51C in Breast Cancer. J. Natl. Cancer Inst. 2019, 111, 1332–1338. [Google Scholar] [CrossRef]
- Boussios, S.; Karathanasi, A.; Cooke, D.; Neille, C.; Sadauskaite, A.; Moschetta, M.; Zakynthinakis-Kyriakou, N.; Pavlidis, N. PARP Inhibitors in Ovarian Cancer: The Route to “Ithaca”. Diagnostics 2019, 9, 55. [Google Scholar] [CrossRef] [Green Version]
- Min, A.; Im, S.-A.; Yoon, Y.-K.; Song, S.-H.; Nam, H.-J.; Hur, H.-S.; Kim, H.-P.; Lee, K.-H.; Han, S.-W.; Oh, D.-Y.; et al. RAD51C-Deficient Cancer Cells Are Highly Sensitive to the PARP Inhibitor Olaparib. Mol. Cancer Ther. 2013, 12, 865–877. [Google Scholar] [CrossRef] [Green Version]
- Kondrashova, O.; Nguyen, M.; Shield-Artin, K.; Tinker, A.V.; Teng, N.N.H.; Harrell, M.I.; Kuiper, M.J.; Ho, G.Y.; Barker, H.; Jasin, M.; et al. Secondary Somatic Mutations Restoring RAD51C and RAD51D Associated with Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2017, 7, 984–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokol, E.S.; Pavlick, D.; Khiabanian, H.; Frampton, G.M.; Ross, J.S.; Gregg, J.P.; Lara, P.N.; Oesterreich, S.; Agarwal, N.; Necchi, A.; et al. Pan-Cancer Analysis of BRCA1 and BRCA2 Genomic Alterations and Their Association with Genomic Instability as Measured by Genome-Wide Loss of Heterozygosity. JCO Precis. Oncol. 2020, 4, 442–465. [Google Scholar] [CrossRef] [PubMed]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An International, Multicentre, Open-Label, Phase 2 Trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Westphalen, C.B.; Fine, A.D.; André, F.; Ganesan, S.; Heinemann, V.; Rouleau, E.; Turnbull, C.; Palacios, L.G.; Lopez, J.-A.; Sokol, E.S.; et al. Pan-cancer Analysis of Homologous Recombination Repair–associated Gene Alterations and Genome-wide Loss-of-Heterozygosity Score. Clin. Cancer Res. 2021, 28, 1412–1421. [Google Scholar] [CrossRef] [PubMed]
- Noordermeer, S.M.; van Attikum, H. PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol. 2019, 29, 820–834. [Google Scholar] [CrossRef] [Green Version]
- Norquist, B.; Wurz, K.A.; Pennil, C.C.; Garcia, R.; Gross, J.; Sakai, W.; Karlan, B.Y.; Taniguchi, T.; Swisher, E.M. Secondary Somatic Mutations Restoring BRCA1/2 Predict Chemotherapy Resistance in Hereditary Ovarian Carcinomas. J. Clin. Oncol. 2011, 29, 3008–3015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klotz, D.M.; Wimberger, P. Overcoming PARP inhibitor resistance in ovarian cancer: What Are the Most Promising Strategies? Arch. Gynecol. Obstet. 2020, 302, 1087–1102. [Google Scholar] [CrossRef]
- Ibrahim, Y.H.; García-García, C.; Serra, V.; He, L.; Torres-Lockhart, K.; Prat, A.; Anton, P.; Cozar, P.; Guzmán, M.; Grueso, J.; et al. PI3K Inhibition Impairs BRCA1/2 Expression and Sensitizes BRCA-Proficient Triple-Negative Breast Cancer to PARP Inhibition. Cancer Discov. 2012, 2, 1036–1047. [Google Scholar] [CrossRef] [Green Version]
- A Konstantinopoulos, P.; Barry, W.T.; Birrer, M.; Westin, S.N.; A Cadoo, K.; I Shapiro, G.; Mayer, E.L.; E O’Cearbhaill, R.; Coleman, R.L.; Kochupurakkal, B.; et al. Olaparib and α-specific PI3K inhibitor alpelisib for patients with epithelial ovarian cancer: A Dose-Escalation and Dose-Expansion Phase 1b Trial. Lancet Oncol. 2019, 20, 570–580. [Google Scholar] [CrossRef]
- Mehta, R.; Wood, A.C.; Yu, J.; Kim, R. Investigational PARP inhibitors for the treatment of biliary tract cancer: Spotlight on Preclinical and Clinical Studies. Expert Opin. Investig. Drugs 2021, 30, 451–461. [Google Scholar] [CrossRef]
- Higuchi, T.; Flies, D.B.; Marjon, N.A.; Mantia-Smaldone, G.; Ronner, L.; Gimotty, P.A.; Adams, S.F. CTLA-4 Blockade Synergizes Therapeutically with PARP Inhibition in BRCA1-Deficient Ovarian Cancer. Cancer Immunol. Res. 2015, 3, 1257–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.K.; Hsu, J.M.; Hsu, J.L.; Yu, W.H.; Du, Y.; Lee, H.H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, C.; Armstrong, S.A.; Agarwal, S.; Wang, H.; Noel, M.S.; Weinberg, B.A.; Marshall, J.; He, A.R. Phase II study of combination pembrolizumab and olaparib in patients with advanced cholangiocarcinoma: Interim results. J. Clin. Oncol. 2022, 40, 452. [Google Scholar] [CrossRef]
- Chen, E.X.; O’Kane, G.M.; Mason, W.P.; Knox, J.J.; Razak, A.R.A.; Zadeh, G. Phase II basket trial of olaparib and durvalumab in patients (pts) with isocitrate dehydrogenase (IDH) mutated solid tumors. J. Clin. Oncol. 2020, 38, TPS3167. [Google Scholar] [CrossRef]
- Mak, J.P.; Ma, H.T.; Poon, R.Y. Synergism between ATM and PARP1 Inhibition Involves DNA Damage and Abrogating the G2 DNA Damage Checkpoint. Mol. Cancer Ther. 2019, 19, 123–134. [Google Scholar] [CrossRef] [Green Version]
- Smith, H.L.; Prendergast, L.; Curtin, N.J. Exploring the Synergy between PARP and CHK1 Inhibition in Matched BRCA2 Mutant and Corrected Cells. Cancers 2020, 12, 878. [Google Scholar] [CrossRef] [Green Version]
- Ricci, A.D.; Rizzo, A.; Brandi, G. The DNA damage repair (DDR) pathway in biliary tract cancer (BTC): A New Pandora’s Box? ESMO Open 2020, 5, e001042. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Role/Mechanism in HR | Frequency in BTC [22,23] | Highest Level of Evidence Isolating Gene Mutation to PARPi Sensitivity |
|---|---|---|---|
| ARID1A | Mostly exerts a role in NHEJ mechanisms. It is a member of the SWI/SNF complex, which is involved in chromatin remodeling and essential for DNA repair [31,32]. | 13% | Cell lines: Loss of ARID1A shows sensitivity to PARPi [33]. Clinical: Retrospective data suggest that ARID1A loss may result in PARPi resistance in breast and ovarian cancer [34]. |
| ATM | Functions upstream in the HR pathway. It helps phosphorylate and activate downstream HR effectors, such as BAP1, CHK2, and WRN [35]. | 5.7% | Animal model: ATM loss in prostate cancer leads to sensitivity to PARPi and ATR inhibition [36,37]. Cell lines: ATM loss in colorectal cell lines demonstrate sensitivity to Olaparib [38]. Clinical: Phase 3 trial of olaparib plus paclitaxel in advanced gastric cancer did not show improved mOS in the olaparib arm for patients deficient in ATM (via immunostaining) [39]. ATM inhibitors are being tested in early phase trials. |
| ATR | Regulates cell cycle checkpoint and mitotic entry. It phosphorylates and activates downstream CHK1 and WRN [40]. | 5.1% | Xenograft: PARPi with ATR inhibitor helps overcomes platinum resistance in ovarian cancer models [41]. |
| ATRX | Involved in chromatin remodeling and is part of the SWI/SNF family. Operates downstream of RAD51. It is necessary for DNA repair synthesis and formation of sister chromatid exchanges at DSBs [42]. | 4% | Cell line: ATRX knockout cells were susceptible to PARPi (and ATR inhibition) [43]. |
| BAP1 | Involved in chromatin modulation and transcriptional regulation. It localizes in the endoplasmic reticulum, where it binds, deubiquitylates, and stabilizes IP3R3, modulating calcium release from the endoplasmic reticulum and apoptosis [44]. In HR, it regulates and recruits key downstream effectors, including p53, BRCA1, and RAD51. It is phosphorylated by ATM [45]. | 7.4% | Case report: Patient with refractory metastatic CCA with novel BAP1 mutation (splice site c.581-17_585del22) had a good, prolonged response to olaparib (>11 months) [46]. Clinical: Rucaparib in patients with BAP1-deficient (by immunostaining) or BRCA1-deficient recurrent mesothelioma showed early signs of efficacy (disease control rate at 12 weeks was 58%) [47]. |
| BARD1 | BRCA1-associated RING Domain Protein 1 upon genotoxic stress, BARD1 serves as a BRCA1 nuclear chaperone that promotes the formation and retention of BRCA1 foci, and these foci are colocalized with DNA repair effectors such as BRCA2 and RAD51 [48]. | 2.5% | Cell line: Colon cancer cells with BARD1 loss of function are more aggressive but sensitive to PARPi [49]. |
| BLM | Unwinds dsDNA and regulates RAD51 foci formation. It is part of the BTR complex [35]. | 1.9% | Cell line: in NSCLC cells, BLM inhibitor sensitized sells to PARPi-medicated radiosensitization [50]. Another study demonstrated the synergy of BLM helicase inhibitor with PARPi in colon cancer cells [51]. |
| BRCA1/2 | BRCA1 promotes HR over NHEJ by directly interacting with PALB2 and recruiting BRCA2/RAD51 to DSBs. | 1.9% for BRAC1, 4.4% for BRAC2 | Clinical: Very limited data in CCA; however, there is robust data in phase III trials with PARPi for other cancers of the prostate, breast, ovarian, and pancreas. |
| CHK1/2 | CHK1 is mostly phosphorylated by ATR; CHK2 by ATM. Regulates cell cycle checkpoint and DNA fork stabilization [52]. | 1.0% for CHK1, 1.9% for CHK2 | Cell line: CHK1 knockout gastric cancer cell line was suspectable to PARPi. Synergy was shown between PARPi and CHK1 inhibitor [53]. |
| FANC | A group of proteins forming the Fanconi Anemia core complex, which participates in HR by attracting HR effectors to the DSB site. FANCD1 gene is otherwise known as BRCA2. [54] FANCA is the most commonly altered FANC gene. | 2.5%, | Clinical: Very limited data in CCA; however, the data for use of PARPi in FANCD1/BRCA2 mutation in other cancers is robust. In the TRITON2 study (rucaparib in prostate cancer), of 4 patients with a FANCA alteration, one patient with a monoallelic truncating alteration had a complete response [55]. |
| NBN or NBS1 | Recognizes and localizes to DSB sites. It recruits ATM and ATR. It is part of the MRN complex (MRE11-RAD50-NBS1) [56]. | 1.4% | Cell line: Dual disruption of MRN complex and PARP inhibition showed synergy in BRCA-proficient head and neck cancer cells [57]. |
| PALB2 | Localizes with BRCA2 and recruits RAD51 to the DBS site [35]. | 1.9% | Clinical: Phase II trial for metastatic breast cancer with HR mutations showed an ORR of 82% for germline PALB2 mutations when treated with olaparib [58]. A phase II study of maintenance rucaparib in advanced pancreatic cancer showed an ORR of 50% in germline PALB2 mutation. |
| RAD50 | A critical part of the MRN complex (MRE11-RAD50-NBS1). Recognizes, localizes, and recruits HR effectors to DSB sites [56]. | 1.8% | Cell line: RAD50 depletion using siRNA in cancer cells showed increased platinum sensitivity [59]. Knockout of RAD50 in ovarian cancer cell lines yielded better responses to olaparib and rucaparib [60]. Clinical: A retrospective study of BRCA wild-type ovarian cancer showed somatic copy number deletion of RAD50 (by mRNA testing) led to a higher genome-wide mutation rate and increased sensitivity to olaparib and rucaparib [60]. |
| RAD51 and paralogs | Physical interaction between BRCA2 and RAD51 is essential for error-free DSB repair [61]. BRCA2 is required for the localization of RAD51 to sites of DNA damage, where RAD51 forms the nucleoprotein filament required for recombination. The foci of the RAD51 protein are apparent in the nucleus after certain forms of DNA damage, and these likely represent sites of repair by HR. BRCA2-deficient cells do not form RAD51 foci in response to DNA damage. [62,63]. | 4.6% for RAD51B, 1.8% for RAD51C | Cell line: Silencing of RAD51 expression increases sensitivity to PARPi [31]. Clinical: In breast cancer patients, immunostaining of RAD51 nuclear foci showed that increased expression of RAD51 nuclear foci correlated with PARPi resistance in BRCA mutated tumors [64]. In breast cancer specimens, the presence of RAD51 foci by immunostaining predicted resistance to DNA-damaging therapy [65]. |
| RAD52 | Binds single-stranded DNA and plays a key part in single-strand annealing and HRR of DSBs. In mammals, RAD52 is diminished in HRR compared to other proteins, including BRCA1/2 but may compensate for BRCA1/2 deficiencies. In checkpoint-deficient cells, it facilitates break-induced replication (BIR) [66,67,68].One of its most prominent functions is in the formation of a Rad51–Rad52–Rad59 complex [67]. | Cell line: Dual suppression of RAD52 and PARP1 via inhibitors demonstrate a synergistic effect in BRCA1/2-deficient cells in vitro and in vivo [69]. | |
| WRN | Recruited to the sites of collapsed replication forks and is phosphorylated at multiple Ser/Thr sites by ATM, ATR, and CDK1 kinases. WRN binding to perturbed replication forks not only stabilizes RAD51 and the replication fork but also prevents excessive nuclease activities of MRE11 and/or EXO1 [70,71] | 4.9% | Cell line: Combining siRNA-mediated silencing of WRN in head and neck squamous cell carcinoma augmented sensitivity to cisplatin [72]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, C.; Kulasekaran, M.; Roy, T.; Decker, B.; Alexander, S.; Margolis, M.; Jha, R.C.; Kupfer, G.M.; He, A.R. Homologous Recombination Repair in Biliary Tract Cancers: A Prime Target for PARP Inhibition? Cancers 2022, 14, 2561. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14102561
Yin C, Kulasekaran M, Roy T, Decker B, Alexander S, Margolis M, Jha RC, Kupfer GM, He AR. Homologous Recombination Repair in Biliary Tract Cancers: A Prime Target for PARP Inhibition? Cancers. 2022; 14(10):2561. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14102561
Chicago/Turabian StyleYin, Chao, Monika Kulasekaran, Tina Roy, Brennan Decker, Sonja Alexander, Mathew Margolis, Reena C. Jha, Gary M. Kupfer, and Aiwu R. He. 2022. "Homologous Recombination Repair in Biliary Tract Cancers: A Prime Target for PARP Inhibition?" Cancers 14, no. 10: 2561. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14102561