Magnesium Effect in K/Co-Mg-Mn-Al Mixed Oxide Catalyst for Direct NO Decomposition

 , , ,
, , ,  and
and 
Abstract
:1. Introduction
2. Results
2.1. Characterization of Catalysts
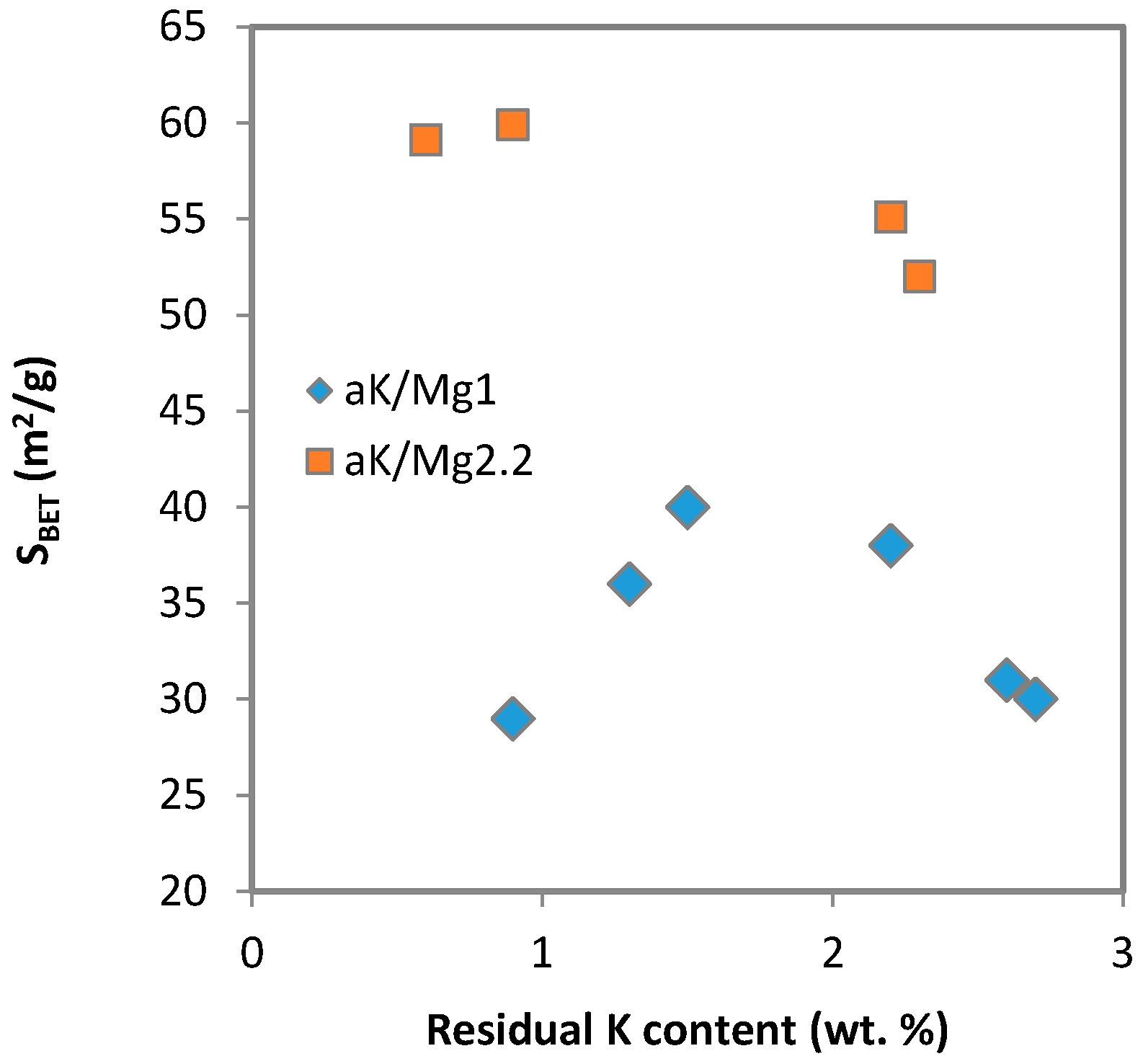
2.1.1. Chemical Composition and Specific Surface Area
2.1.2. Phase Composition
2.1.3. TPR-H2
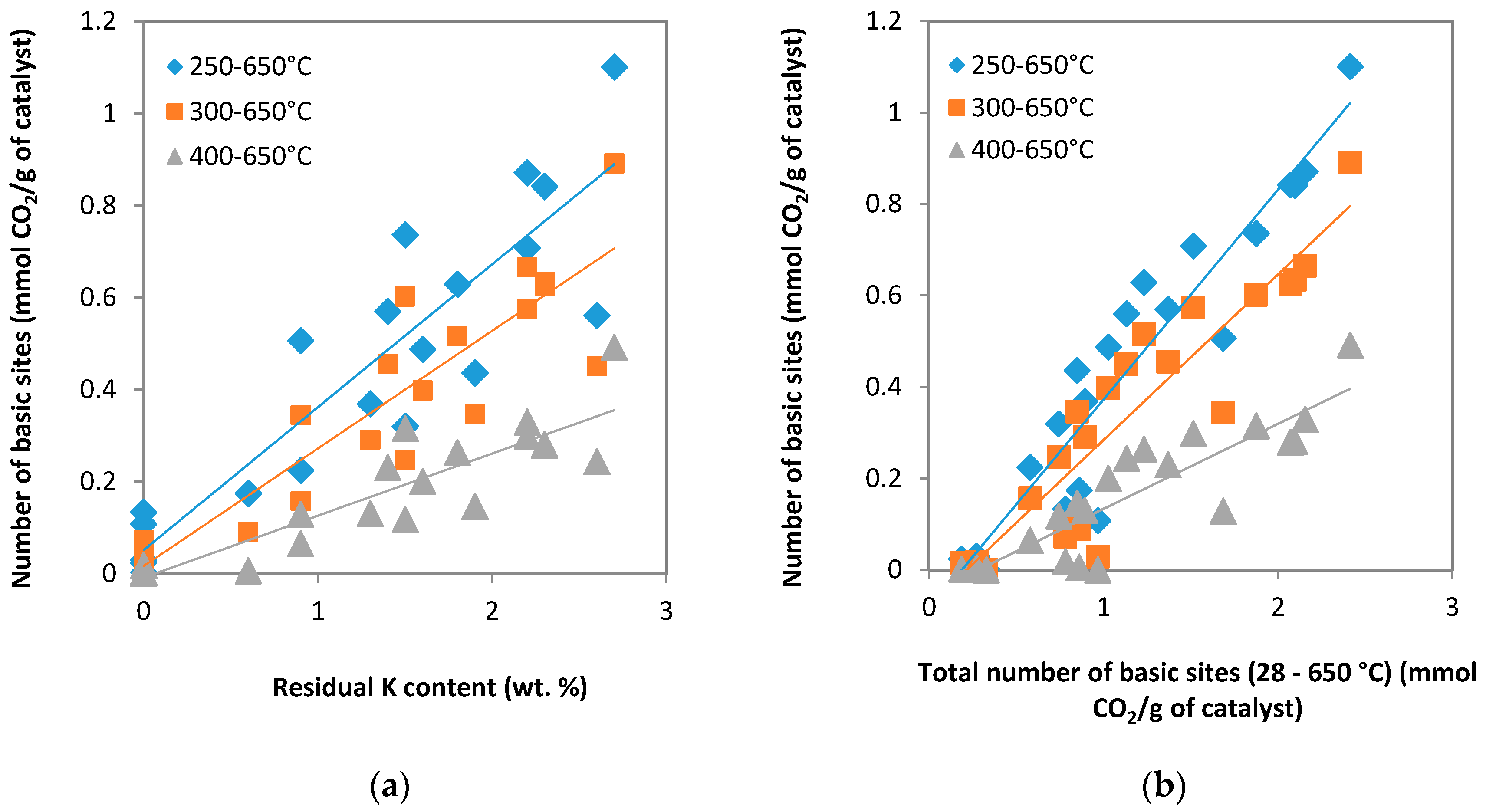
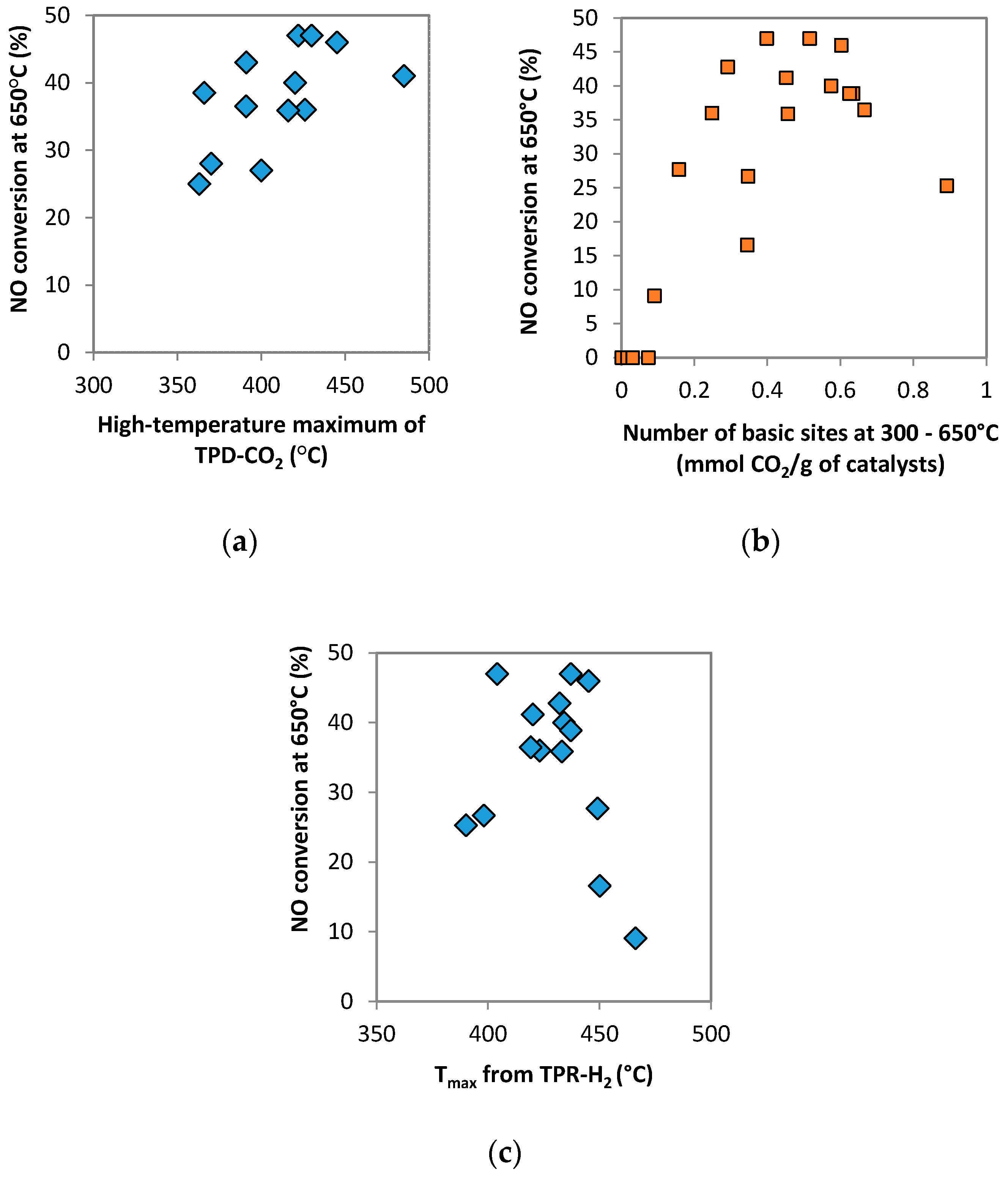
2.1.4. TPD-CO2
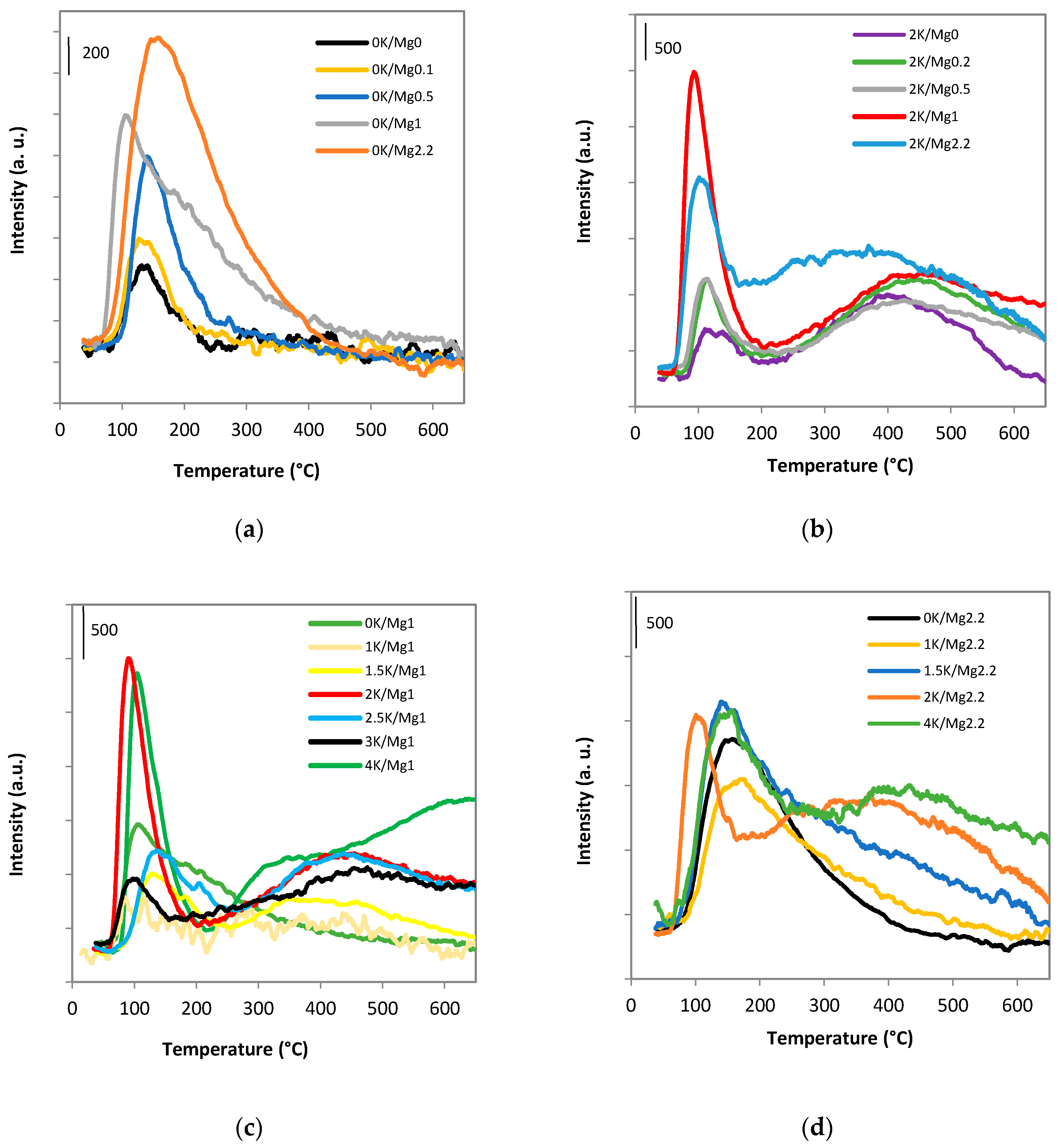
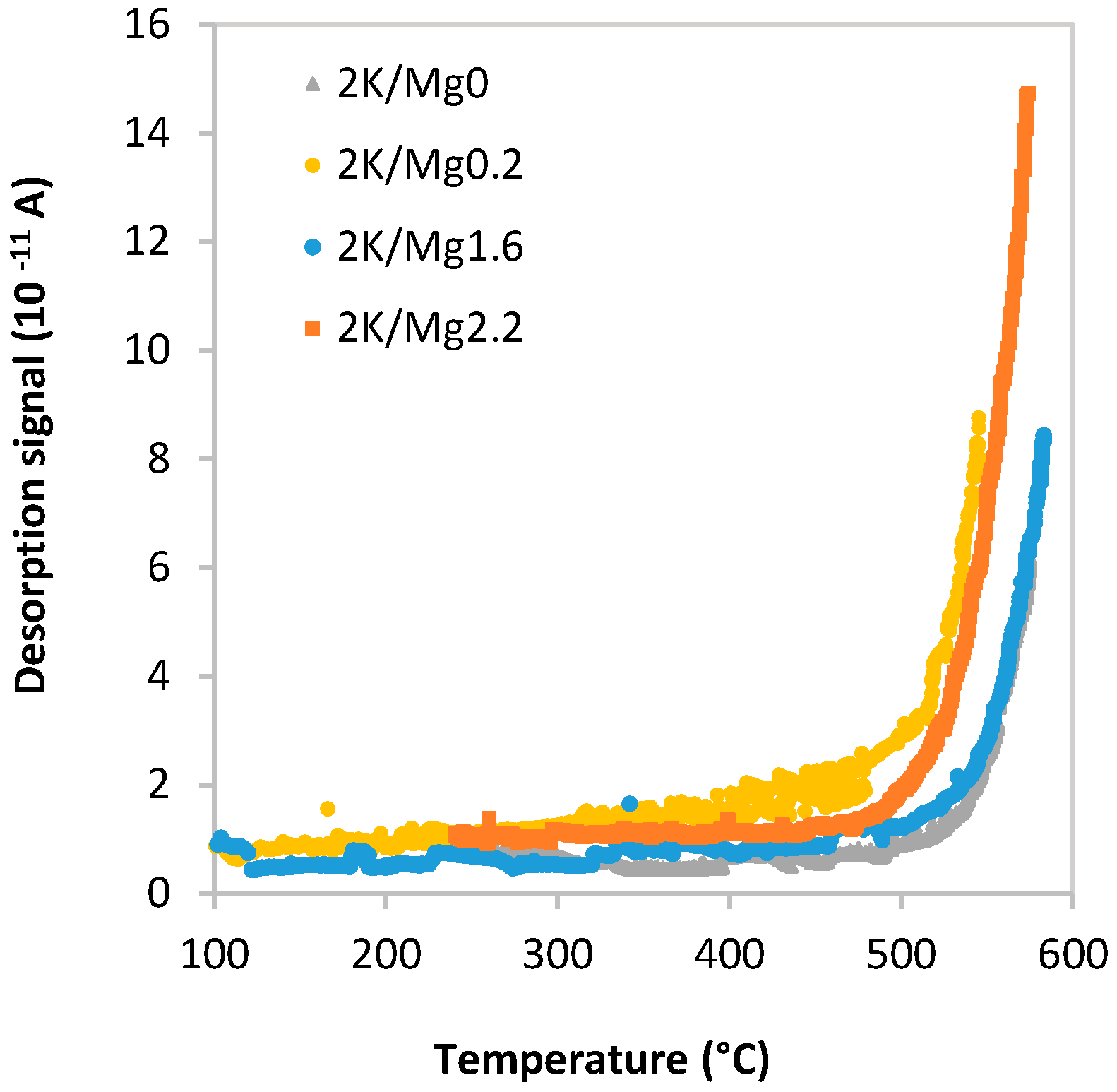
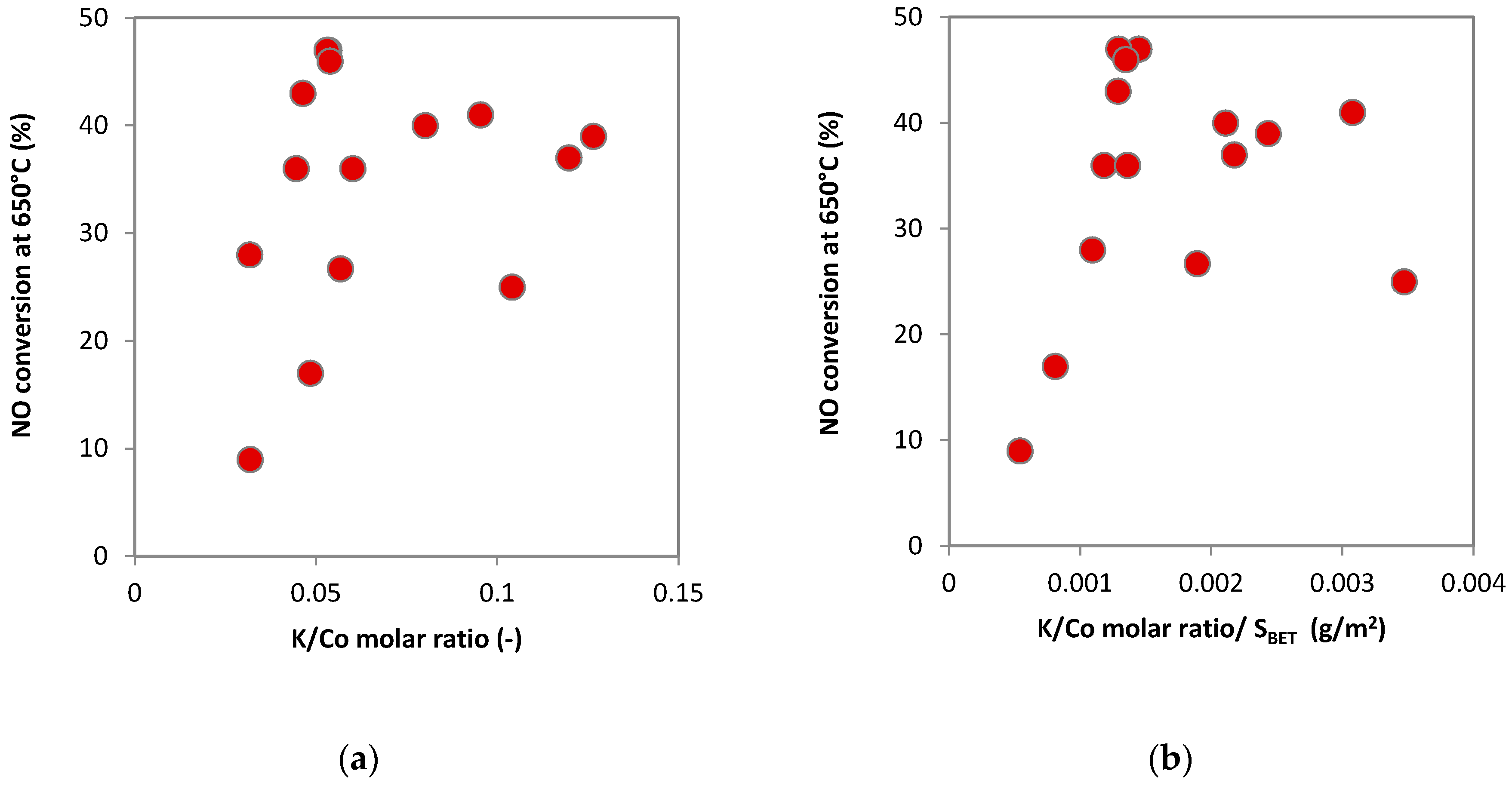
2.1.5. Species-Resolved Thermal Alkali Desorption
2.2. NO Decomposition
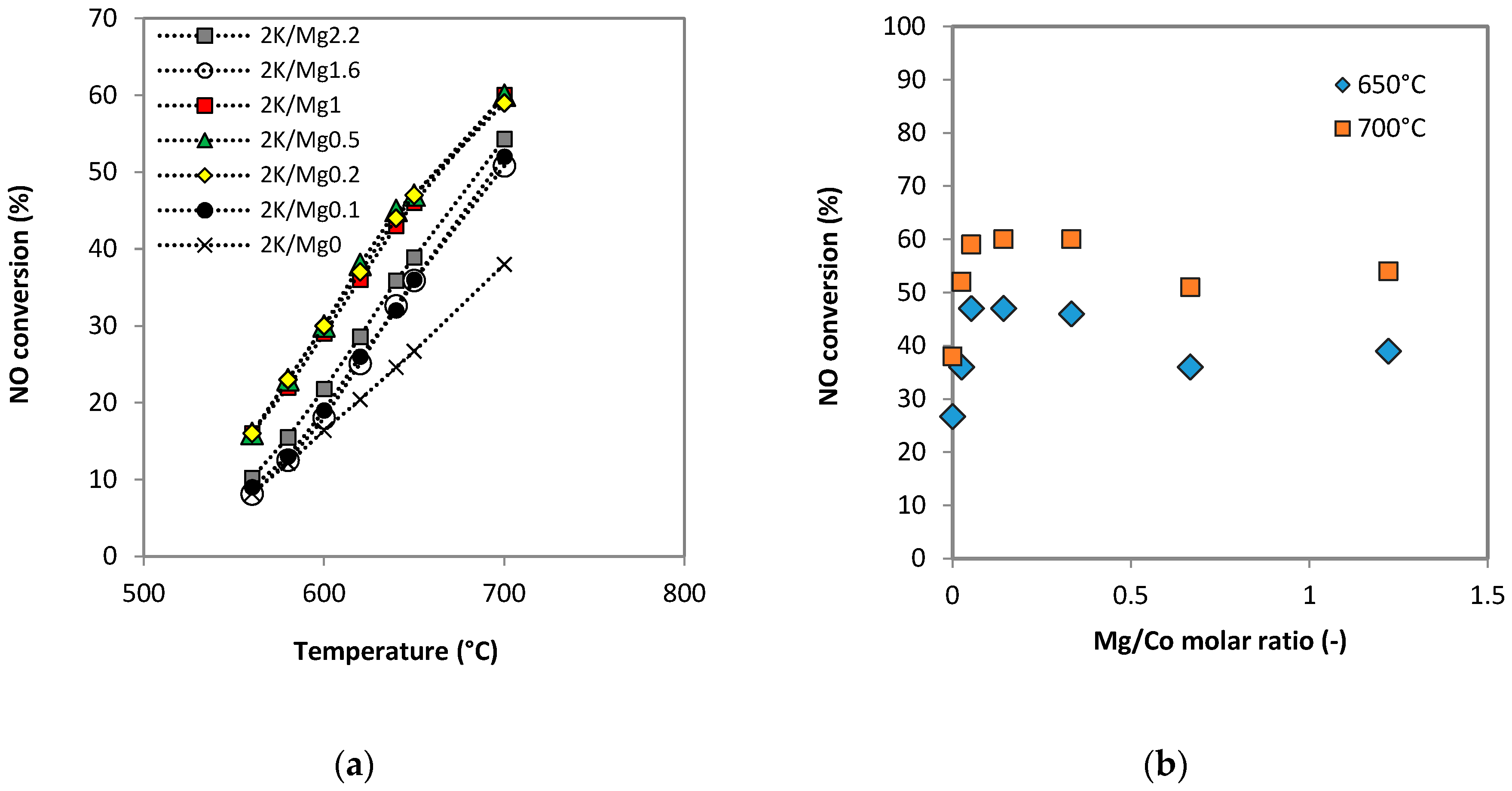
2.2.1. Catalytic Activity in Inert Conditions
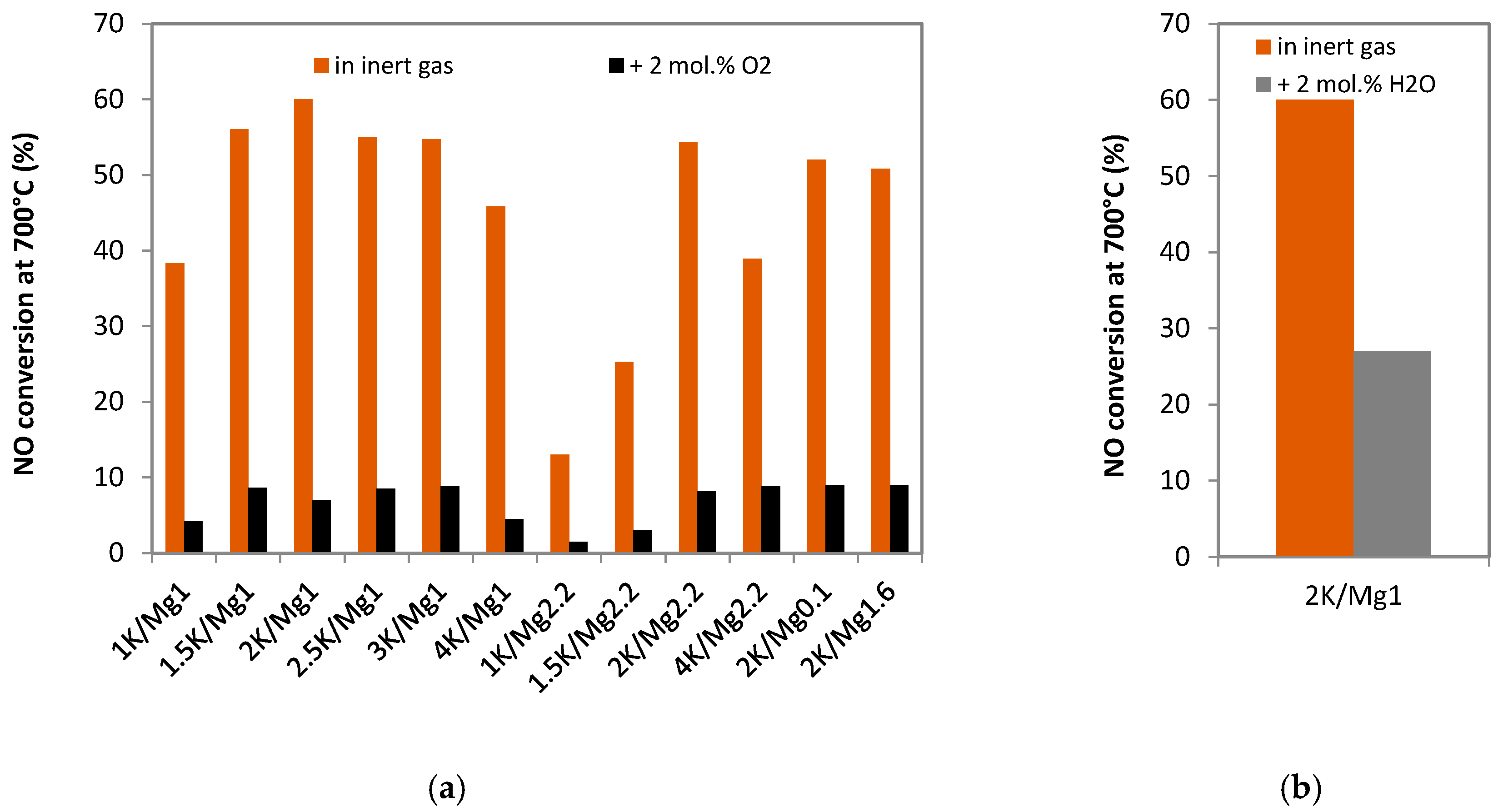
2.2.2. Catalytic Activity in the Presence of Oxygen and Water Vapor
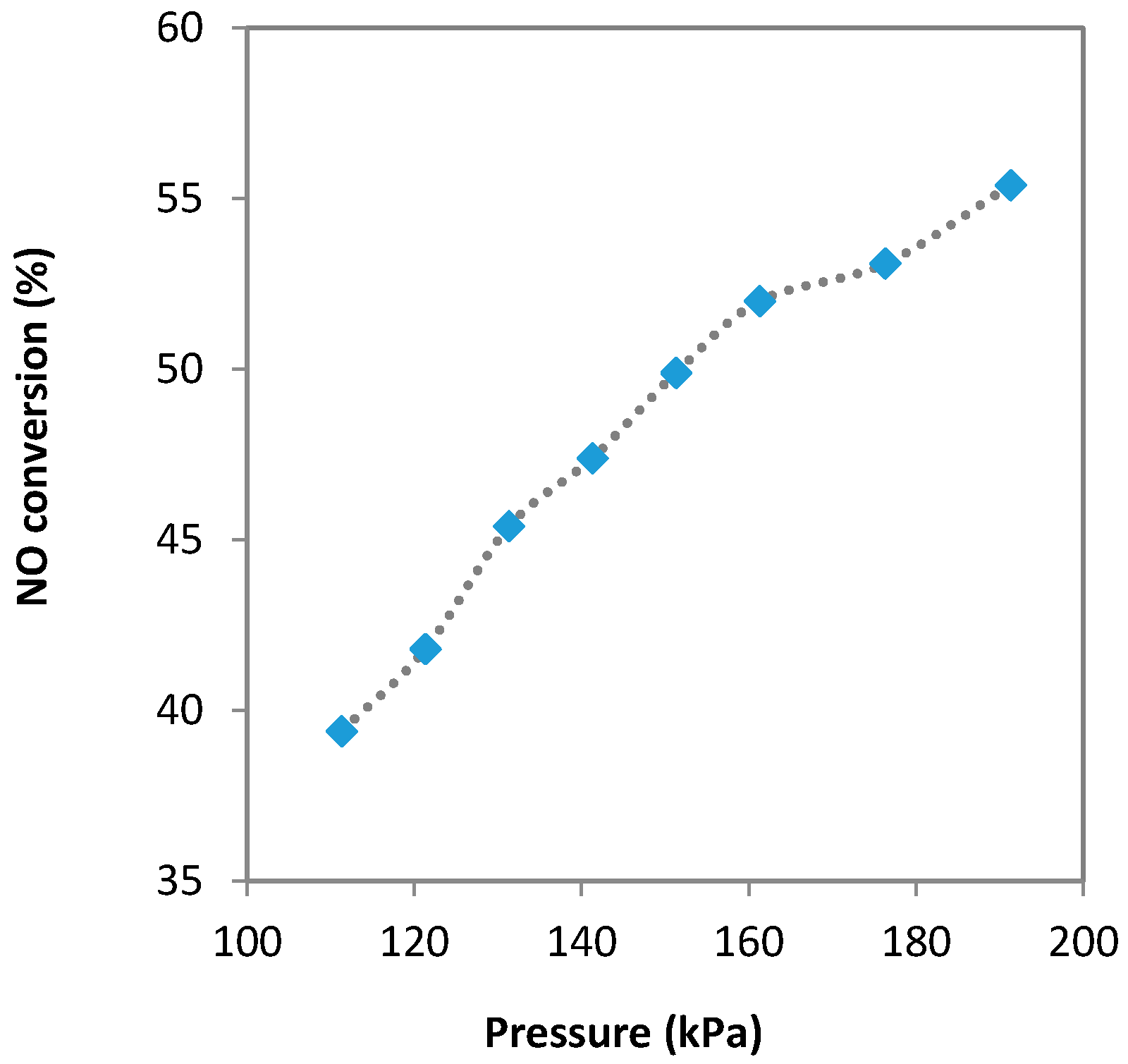
2.2.3. The Effect of Pressure
3. Discussions
4. Materials and Methods
4.1. Catalyst Preparation
4.2. Catalyst Characterization
4.3. Catalytic Measurement
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Haneda, M.; Hamada, H. Recent progress in catalytic NO decomposition. Comptes Rendus Chim. 2016, 19, 1254–1265. [Google Scholar] [CrossRef]
- Winter, E.R.S. The catalytic decomposition of nitric oxide by metallic oxides. J. Catal. 1971, 22, 158–170. [Google Scholar] [CrossRef]
- Haneda, M.; Kintaichi, Y.; Bion, N.; Hamada, H. Alkali metal-doped cobalt oxide catalysts for NO decomposition. Appl. Catal. B: Environ. 2003, 46, 473–482. [Google Scholar] [CrossRef]
- Haneda, M.; Nakamura, I.; Fujitani, T.; Hamada, H. Catalytic Active Site for NO Decomposition Elucidated by Surface Science and Real Catalyst. Catal. Surv. Asia 2005, 9, 207–215. [Google Scholar] [CrossRef]
- Park, P.W.; Kil, J.K.; Kung, H.H.; Kung, M.C. NO decomposition over sodium-promoted cobalt oxide. Catal. Today 1998, 42, 51–60. [Google Scholar] [CrossRef]
- Konsolakis, M.; Yentekakis, I.V. The reduction of NO by propene over Ba-promoted Pt/gamma-Al2O3 catalysts. J. Catal. 2001, 198, 142–150. [Google Scholar] [CrossRef] [Green Version]
- Konsolakis, M.; Yentekakis, I.V. Strong promotional effects of Li, K, Rb and Cs on the Pt-catalysed reduction of NO by propene. Appl. Catal. B: Environ. 2001, 29, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Iwamoto, S.; Takahashi, R.; Inoue, M. Direct decomposition of nitric oxide over Ba catalysts supported on CeO2-based mixed oxides. Appl. Catal. B: Environ. 2007, 70, 146–150. [Google Scholar] [CrossRef]
- Nakamura, I.; Haneda, M.; Hamada, H.; Fujitani, T. Direct decomposition of nitrogen monoxide over a K-deposited Co (0001) surface: Comparison to K-doped cobalt oxide catalysts. J. Electron. Spectrosc. Relat. Phenom. 2006, 150, 150–154. [Google Scholar] [CrossRef]
- Borowiecki, T.; Denis, A.; Rawski, M.; Gołębiowski, A.; Stołecki, K.; Dmytrzyk, J.; Kotarba, A. Studies of potassium-promoted nickel catalysts for methane steam reforming: Effect of surface potassium location. Appl. Surf. Sci. 2014, 300, 191–200. [Google Scholar] [CrossRef]
- Karásková, K.; Obalová, L.; Kovanda, F. N2O catalytic decomposition and temperature programmed desorption tests on alkali metals promoted Co-Mn-Al mixed oxide. Catal. Today 2011, 176, 208–211. [Google Scholar] [CrossRef]
- Obalová, L.; Karásková, K.; Jirátová, K.; Kovanda, F. Effect of potassium in calcined Co-Mn-Al layered double hydroxide on the catalytic decomposition of N2O. Appl. Catal. B: Environ. 2009, 90, 132–140. [Google Scholar] [CrossRef]
- Obalová, L.; Karásková, K.; Wach, A.; Kustrowski, P.; Mamulová-Kutláková, K.; Michalik, S.; Jirátová, K. Alkali metals as promoters in Co-Mn-Al mixed oxide for N2O decomposition. Appl. Catal. A-Gen. 2013, 462, 227–235. [Google Scholar] [CrossRef]
- Pacultová, K.; Draštíková, V.; Chromčáková, Ž.; Bílková, T.; Kutláková, K.M.; Kotarba, A.; Obalová, L. On the stability of alkali metal promoters in Co mixed oxides during direct NO catalytic decomposition. Mol. Catal. 2017, 428, 33–40. [Google Scholar] [CrossRef]
- Pacultová, K.; Bílková, T.; Klegova, A.; Karásková, K.; Fridrichová, D.; Jirátová, K.; Kiška, T.; Balabánová, J.; Koštejn, M.; Kotarba, A.; et al. Co-Mn-Al Mixed Oxides Promoted by K for Direct NO Decomposition: Effect of Preparation Parameters. Catalysts 2019, 9, 593. [Google Scholar] [CrossRef] [Green Version]
- Jirátová, K.; Pacultová, K.; Balabánová, J.; Karásková, K.; Klegova, A.; Bílková, T.; Jandová, V.; Koštejn, M.; Martaus, A.; Kotarba, A.; et al. Precipitated K-Promoted Co-Mn-Al Mixed Oxides for Direct NO Decomposition: Preparation and Properties. Catalysts 2019, 9, 592. [Google Scholar] [CrossRef] [Green Version]
- Kotarba, A.; Rożek, W.; Serafin, I.; Sojka, Z. Reverse effect of doping on stability of principal components of styrene catalyst: KFeO2 and K2Fe22O34. J. Catal. 2007, 247, 238–244. [Google Scholar] [CrossRef]
- An, H.; McGinn, P.J. Catalytic behavior of potassium containing compounds for diesel soot combustion. Appl. Catal. B: Environ. 2006, 62, 46–56. [Google Scholar] [CrossRef]
- Bieniasz, W.; Trębala, M.; Sojka, Z.; Kotarba, A. Irreversible deactivation of styrene catalyst due to potassium loss—Development of antidote via mechanism pinning. Catal. Today 2010, 154, 224–228. [Google Scholar] [CrossRef]
- Trębala, M.; Bieniasz, W.; Holmlid, L.; Molenda, M.; Kotarba, A. Potassium stabilization in β-K2Fe22O34 by Cr and Ce doping studied by field reversal method. Solid State Ion. 2011, 192, 664–667. [Google Scholar] [CrossRef]
- Kotarba, A.; Bieniasz, W.; Kuśtrowski, P.; Stadnicka, K.; Sojka, Z. Composite ferrite catalyst for ethylbenzene dehydrogenation: Enhancement of potassium stability and catalytic performance by phase selective doping. Appl. Catal. A: Gen. 2011, 407, 100–105. [Google Scholar] [CrossRef]
- Stelmachowski, P.; Maniak, G.; Kaczmarczyk, J.; Zasada, F.; Piskorz, W.; Kotarba, A.; Sojka, Z. Mg and Al substituted cobalt spinels as catalysts for low temperature deN2O—Evidence for octahedral cobalt active sites. Appl. Catal. A: Gen. 2014, 146, 105–111. [Google Scholar] [CrossRef]
- Basag, S.; Kovanda, F.; Piwowarska, Z.; Kowalczyk, A.; Pamin, K.; Chmielarz, L. Hydrotalcite-derived Co-containing mixed metal oxide catalysts for methanol incineration Role of cobalt content, Mg/Al ratio and calcination temperature. J. Therm. Anal. Calorim. 2017, 129, 1301–1311. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.X.; Yu, J.J.; Liu, C.C.; Hao, Z.P. N2O catalytic decomposition over mixed oxides derived from Co-Mg/Al hydrotalcite-like compounds. Acta Phys.-Chim. Sin. 2007, 23, 162–168. [Google Scholar]
- Palomares, E.; Uzcátegui, A.; Franch, C.; Corma, A. Multifunctional catalyst for maximizing NOx oxidation/storage/reduction: The role of the different active sites. Appl. Catal. B: Environ. 2013, 142, 795–800. [Google Scholar] [CrossRef] [Green Version]
- Mokhtar, M.; Basahel, S.N.; Al-Angary, Y.O. Nanosized spinel oxide catalysts for CO-oxidation prepared via CoMnMgAl quaternary hydrotalcite route. J. Alloys Compd. 2010, 493, 376–384. [Google Scholar] [CrossRef]
- Castaño, M.H.; Molina, R.; Moreno, S. Mn–Co–Al–Mg mixed oxides by auto-combustion method and their use as catalysts in the total oxidation of toluene. J. Mol. Catal. A: Chem. 2013, 370, 167–174. [Google Scholar] [CrossRef]
- Choya, A.; de Rivas, B.; González-Velasco, J.R.; Gutiérrez-Ortiz, J.I.; Lopéz-Fonseca, R. On the beneficial effect of MgO promoter on the performance of Co3O4/Al2O3 catalysts for combustion of dilute methane. Appl. Catal. A-Gen. 2019, 582, 117099. [Google Scholar] [CrossRef]
- Ulla, M.A.; Spretz, R.; Lombardo, E.; Daniell, W.; Knozinger, H. Catalytic combustion of methane on Co/MgO: Characterisation of active cobalt sites. Appl. Catal. B: Environ. 2001, 29, 217–229. [Google Scholar] [CrossRef]
- Ji, S.F.; Xiao, T.C.; Wang, H.T.; Flahaut, E.; Coleman, K.S.; Green, M.L.H. Catalytic combustion of methane over cobalt-magnesium oxide solid solution catalysts. Catal. Lett. 2001, 75, 65–71. [Google Scholar] [CrossRef]
- Haneda, M.; Kintaichi, Y.; Hamada, H. Reaction mechanism of NO decomposition over alkali metal-doped cobalt oxide catalysts. Appl. Catal. B: Environ. 2005, 55, 169–175. [Google Scholar] [CrossRef]
- Bai, Z.; Chen, B.; Zhao, Q.; Shi, C.; Crocker, M. Positive effects of K+ in hybrid CoMn-K and Pd/Ba/Al2O3 catalysts for NOx storage and reduction. Appl. Catal. B 2019, 249, 333–345. [Google Scholar] [CrossRef]
- Li, Q.; Meng, M.; Tsubaki, N.; Li, X.; Li, Z.; Xie, Y.; Hu, T.; Zhang, J. Performance of K-promoted hydrotalcite-derived CoMgAlO catalysts used for soot combustion, NOx storage and simultaneous soot–NOx removal. Appl. Catal. B 2009, 91, 406–415. [Google Scholar] [CrossRef]
- Le-Phuc, N. Removal of NOx in the presence of oxygen over Mn/BaO/Al2O3 catalysts. Mater. Sci. Nanotechnol. 2017, 2, 37–40. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, Y.; Xin, Y.; Li, Q.; Zhang, Z.; Jiang, Z.; Ma, Y.; Zhou, H.; Zhang, J. Enhanced NOx conversion by coupling NOx storage-reduction with CO adsorption-oxidation over the combined Pd−K/MgAlO and Pd/MgAlO catalysts. Catal. Today 2015, 258, 416–423. [Google Scholar] [CrossRef]
- Grzybek, G.; Wójcik, S.; Legutko, P.; Gryboś, J.; Indyka, P.; Leszczyński, B.; Kotarba, A.; Sojka, Z. Thermal stability and repartition of potassium promoter between the support and active phase in the K-Co2.6Zn0.4O4|α-Al2O3 catalyst for N2O decomposition: Crucial role of activation temperature on catalytic performance. Appl. Catal. B: Environ. 2017, 205, 597–604. [Google Scholar] [CrossRef]
- Ladgaonkar, B.P.; Vaingankar, A.S. X-ray diffraction investigation of cation distribution in CdχCu1-χFe2O4 ferrite system. Mater. Chem. Phys. 1998, 56, 280–283. [Google Scholar] [CrossRef]
- Raghuvanshi, S.; Mazaleyrat, F.; Kane, S.N. Mg1-xZnxFe2O4 nanoparticles: Interplay between cation distribution and magnetic properties. AIP Adv. 2018, 8, 047804. [Google Scholar] [CrossRef] [Green Version]
- Sukandhiya, S. Effect of Mn2+ ions on Structural and Magnetic properties of Co-precipitated Ni-Cr nano ferrite for Potential applications as MRI Contrast agent. Int. J. Res. Appl. Sci. Eng. Technol. 2018, 6, 3713–3721. [Google Scholar] [CrossRef]
- Wu, M.; Zhan, W.; Guo, Y.; Guo, Y.; Wang, Y.; Wang, L.; Lu, G. An effective Mn-Co mixed oxide catalyst for the solvent-free selective oxidation of cyclohexane with molecular oxygen. Appl. Catal. A: Gen. 2016, 523, 97–106. [Google Scholar] [CrossRef]
- Trivedi, S.; Prasad, R. Reactive calcination route for synthesis of active Mn–Co3O4 spinel catalysts for abatement of CO–CH4 emissions from CNG vehicles. J. Environ. Chem. Eng. 2016, 4, 1017–1028. [Google Scholar] [CrossRef]
- Larimi, A.S.; Kazemeini, M.; Khorasheh, F. Highly selective doped Pt-MgO nano-sheets for renewable hydrogen production from APR of glycerol. Int. J. Hydrog. Energy 2016, 41, 17390–17398. [Google Scholar] [CrossRef]
- Jung, D.H.; Umirov, N.; Kim, T.; Bakenov, Z.; Kim, J.S.; Kim, S.S. Thermal and Structural Stabilities of LixCoO2 Cathode for Li Secondary Battery Studied by a Temperature Programmed Reduction. Eurasian Chem.-Technol. J. 2019, 21, 3–12. [Google Scholar] [CrossRef]
- Pacultová, K.; Karásková, K.; Kovanda, F.; Jirátová, K.; Šrámek, J.; Kustrowski, P.; Kotarba, A.; Chromčáková, Ž.; Kočí, K.; Obalová, L. K-doped Co-Mn-Al mixed oxide catalyst for N2O abatement from nitric acid plant waste gases. Pilot plant studies. Ind. Eng. Chem. Res. 2016, 55, 7076–7084. [Google Scholar] [CrossRef]
- Cheng, H.; Huang, Y.; Wang, A.; Li, L.; Wang, X.; Zhang, T. N2O decomposition over K-promoted Co-Al catalysts prepared from hydrotalcite-like precursors. Appl. Catal. B: Environ. 2009, 89, 391–397. [Google Scholar] [CrossRef]
- Imanaka, N.; Masui, T. Advances in direct NOx decomposition catalysts. Appl. Catal. A: Gen. 2012, 431, 1–8. [Google Scholar] [CrossRef]
- Smoláková, L.; Frolich, K.; Troppová, I.; Kutálek, P.; Kroft, E.; Čapek, L. Determination of basic sites in Mg-Al mixed oxides by combination of TPD-CO2 and CO2 adsorption calorimetry. J. Therm. Anal. Calorim. 2017, 127, 1921–1929. [Google Scholar] [CrossRef]
- Hong, W.-J.; Iwamoto, S.; Inoue, M. Direct NO decomposition over a Ce–Mn mixed oxide modified with alkali and alkaline earth species and CO2-TPD behavior of the catalysts. Catal. Today 2011, 164, 489–494. [Google Scholar] [CrossRef]
- Kotarba, A.; Hagstrom, M.; Engvall, K.; Pettersson, J.B.C. High pressure desorption of K+ from iron ammonia catalyst migration of the promoter towards Fe active planes. Catal. Lett. 2004, 95, 93–97. [Google Scholar] [CrossRef]
- Iwakuni, H.; Shinmyou, Y.; Yano, H.; Matsumoto, H.; Ishihara, T. Direct decomposition of NO into N2 and O2 on BaMnO3-based perovskite oxides. Appl. Catal. B: Environ. 2007, 74, 299–306. [Google Scholar] [CrossRef]
- Goto, K.; Ishihara, T. Direct decomposition of NO into N2 and O2 over Ba3Y3.4Sc0.6O9. Appl. Catal. A: Gen. 2011, 409, 66–73. [Google Scholar] [CrossRef]
- Tofan, C.; Klvana, D.; Kirchnerova, J. Decomposition of nitric oxide over perovskite oxide catalysts: Effect of CO2, H2O and CH4. Appl. Catal. B 2002, 36, 311–323. [Google Scholar] [CrossRef]
- Obalová, L.; Maniak, G.; Karásková, K.; Kovanda, F.; Kotarba, A. Electronic nature of potassium promotion effect in Co–Mn–Al mixed oxide on the catalytic decomposition of N2O. Catal. Commun. 2011, 12, 1055–1058. [Google Scholar] [CrossRef] [Green Version]
- Klyushina, A.; Pacultová, K.; Karásková, K.; Jirátová, K.; Ritz, M.; Fridrichová, D.; Volodarskaja, A.; Obalová, L. Effect of preparation method on catalytic properties of Co-Mn-Al mixed oxides for N2O decomposition. J. Mol. Catal. A: Chem. 2016, 425, 237–247. [Google Scholar] [CrossRef]

















{kind=link}
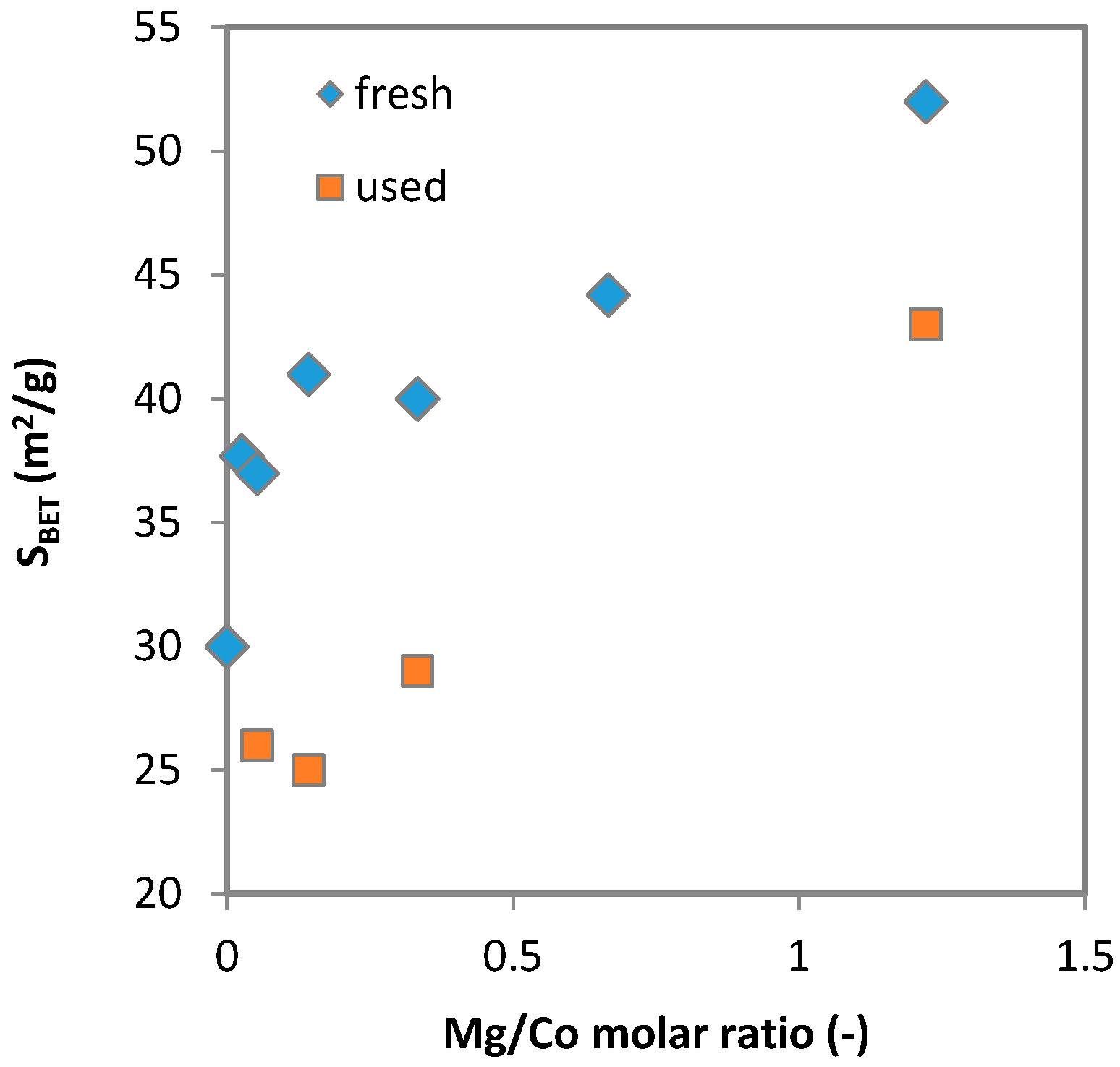{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Metal Content (wt. %) | Real Molar Ratio | Theoretical Molar Ratio | SBET (m2 g−1) 1 | |||||
|---|---|---|---|---|---|---|---|---|---|
| K | Co | Mg | Mn | Al | Co:Mg:Mn:Al | Co:Mg:Mn:Al | Fresh | Used | |
| 0K/Mg0 | 0.0 | 52.1 | – | 11.7 | – | 4:0:1:- | 4:0:1:1 | 36 | – |
| 0K/Mg0.1 | – | – | – | – | – | – | 3.9:0.1:1:1 | 39 | – |
| 0K/Mg0.2 | – | – | – | – | – | – | 3.8:0.2:1:1 | 41 | – |
| 0K/Mg0.5 | – | – | – | – | – | – | 3.5:0.5:1:1 | 41 | – |
| 0K/Mg1 | 0.0 | 42.4 | 5.3 | 12.5 | 6.0 | 3:0.9:0.9:1 | 3:1:1:1 | 44 | 38 |
| 0K/Mg1.6 | – | – | – | – | – | – | 2.4:1.6:1:1 | 50 | – |
| 0K/Mg2.2 | – | – | – | – | – | – | 1.8:2.2:1:1 | 63 | – |
| 2K/Mg0 | 1.9 | 50.5 | 0.0 | 12.0 | 6.0 | 4:0:1:1 | 4:0:1:1 | 30 | – |
| 2K/Mg0.1 | 1.5 | 50.8 | 0.5 | 11.4 | 5.5 | 3.9:0.1:0.9:0.9 | 3.9:0.1:1:1 | 38 | – |
| 2K/Mg0.2 | 1.8 | 50.7 | 1.1 | 11.9 | 6.5 | 3.8:0.2:1:1.1 | 3.8:0.2:1:1 | 37 | 26 |
| 2K/Mg0.5 | 1.6 | 45.5 | 2.5 | 11.7 | 6.3 | 3.5:0.5:1:1.1 | 3.5:0.5:1:1 | 41 | 25 |
| 2K/Mg1 | 1.5 | 42.0 | 5.3 | 12.5 | 6.7 | 3:0.9:1:1 | 3:1:1:1 | 40 | 29 |
| 2K/Mg1.6 | 1.4 | 35.1 | 8.4 | 13.1 | 6.4 | 2.4:1.4:1:1 | 2.4:1.6:1:1 | 44 | – |
| 2K/Mg2.2 | 2.3 | 27.4 | 11.9 | 13.0 | 6.5 | 1.8:1.9:0.9:0.9 | 1.8:2.2:1:1 | 52 | 43 |
| 1K/Mg1 | 0.9 | 42.8 | 5.2 | 12.2 | 6.3 | 3:0.9:0:9:1 | 3:1:1:1 | 29 | 28 |
| 1.5K/Mg1 | 1.3 | 42.3 | 5.2 | 12.4 | 6.2 | 3:0.9:0:9:1 | 3:1:1:1 | 36 | 31 |
| 2.5K/Mg1 | 2.2 | 41.4 | 5.2 | 12.1 | 6.0 | 3:0.9:0:9:0.9 | 3:1:1:1 | 38 | 34 |
| 3K/Mg1 | 2.6 | 41.1 | 5.1 | 11.8 | 6.0 | 3:0.9:0:9:0.9 | 3:1:1:1 | 31 | 28 |
| 4K/Mg1 | 2.7 | 39.1 | 5.1 | 11.7 | 6.1 | 3:0.9:1:1 | 3:1:1:1 | 30 | 27 |
| 1K/Mg2.2 | 0.6 | 28.4 | 12.7 | 13.7 | 7.2 | 1.8:2:0.9:1 | 1.8:2.2:1:1 | 59 | – |
| 1.5K/Mg2.2 | 0.9 | 28.0 | 12.5 | 13.6 | 7.3 | 1.8:1.9:0.9:1 | 1.8:2.2:1:1 | 60 | – |
| 4K/Mg2.2 | 2.2 | 27.7 | 12.2 | 13.1 | 7.0 | 1.8:1.9:0.9:1 | 1.8:2.2:1:1 | 55 | – |
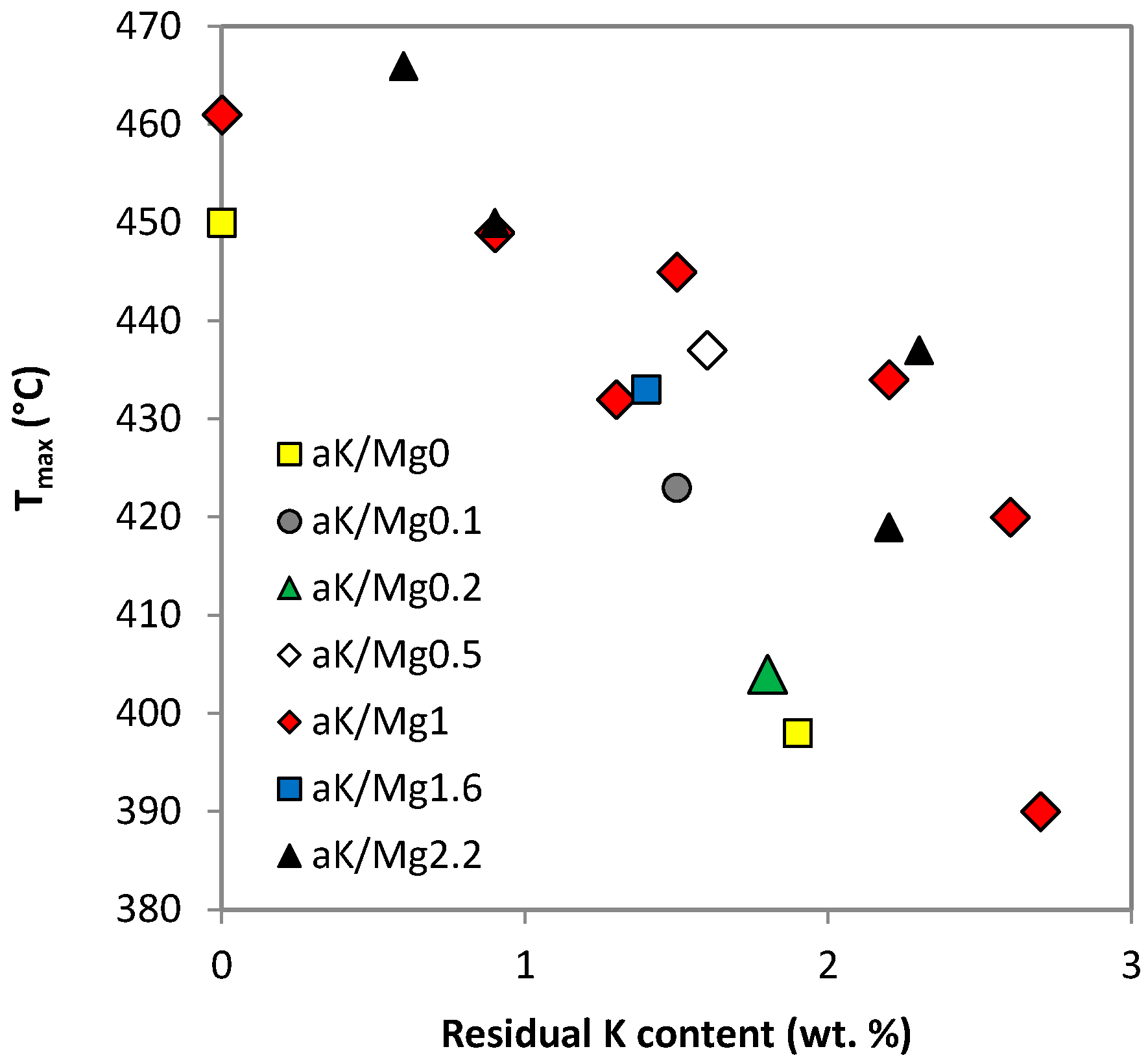
| Sample | TPR-H2 (mmol g−1) | Tmax (°C) 1 | TPD-CO2 (mmol g−1) | Tmax (°C) 2 |
|---|---|---|---|---|
| 40–600 °C 1 | 28–650 °C | |||
| 0K/Mg0 | 5.8 | 206, 450 | 0.2 | – |
| 0K/Mg0.1 | 6.9 | 435 | 0.3 | – |
| 0K/Mg0.2 | – | – | – | – |
| 0K/Mg0.5 | 5.5 | 454 | 0.3 | – |
| 0K/Mg1 | 5.1 | 461 | 0.8 | – |
| 0K/Mg1.6 | – | – | – | – |
| 0K/Mg2.2 | 3.5 | 452 | 1.0 | – |
| 2K/Mg0 | 6.1 | 317, 398 | 0.9 | 404 |
| 2K/Mg0.1 | 6.0 | 164, 423 | 0.7 | 426 |
| 2K/Mg0.2 | 6.0 | 143, 404 | 1.2 | 430 |
| 2K/Mg0.5 | 5.1 | 437 | 1.0 | 422 |
| 2K/Mg1 | 5.1 | 445 | 1.9 | 445 |
| 2K/Mg1.6 | 4.6 | 433 | 1.4 | 416 |
| 2K/Mg2.2 | 4.1 | 437 | 2.1 | 366 |
| 1K/Mg1 | 5.2 | 145, 449 | 0.6 | 370 |
| 1.5K/Mg1 | 5.3 | 142, 432 | 0.9 | 391 |
| 2.5K/Mg1 | 5.4 | 153, 434 | 1.5 | 420 |
| 3K/Mg1 | 5.5 | 420 | 1.1 | 485 |
| 4K/Mg1 | 6.3 | 169, 303, 390 | 2.4 | 363, >600 |
| 1K/Mg2.2 | 4.0 | 251, 466 | 0.9 | – |
| 1.5K/Mg2.2 | 4.1 | 213, 450 | 1.7 | – |
| 4K/Mg2.2 | 4.2 | 212, 419 | 2.2 | 391 |
| Sample | Activation Energy of Potassium Desorption (eV) |
|---|---|
| 2K/Mg0 | 2.1 |
| 2K/Mg0.2 | 1.4 |
| 2K/Mg1.6 | 1.8 |
| 2K/Mg2.2 | 1.8 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karásková, K.; Pacultová, K.; Klegova, A.; Fridrichová, D.; Valášková, M.; Jirátová, K.; Stelmachowski, P.; Kotarba, A.; Obalová, L. Magnesium Effect in K/Co-Mg-Mn-Al Mixed Oxide Catalyst for Direct NO Decomposition. Catalysts 2020, 10, 931. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10080931
Karásková K, Pacultová K, Klegova A, Fridrichová D, Valášková M, Jirátová K, Stelmachowski P, Kotarba A, Obalová L. Magnesium Effect in K/Co-Mg-Mn-Al Mixed Oxide Catalyst for Direct NO Decomposition. Catalysts. 2020; 10(8):931. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10080931
Chicago/Turabian StyleKarásková, Kateřina, Kateřina Pacultová, Anna Klegova, Dagmar Fridrichová, Marta Valášková, Květuše Jirátová, Paweł Stelmachowski, Andrzej Kotarba, and Lucie Obalová. 2020. "Magnesium Effect in K/Co-Mg-Mn-Al Mixed Oxide Catalyst for Direct NO Decomposition" Catalysts 10, no. 8: 931. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10080931