
Photoelectrochemical Water Oxidation by Cobalt Cytochrome C Integrated-ATO Photoanode

 ,
,  and
and 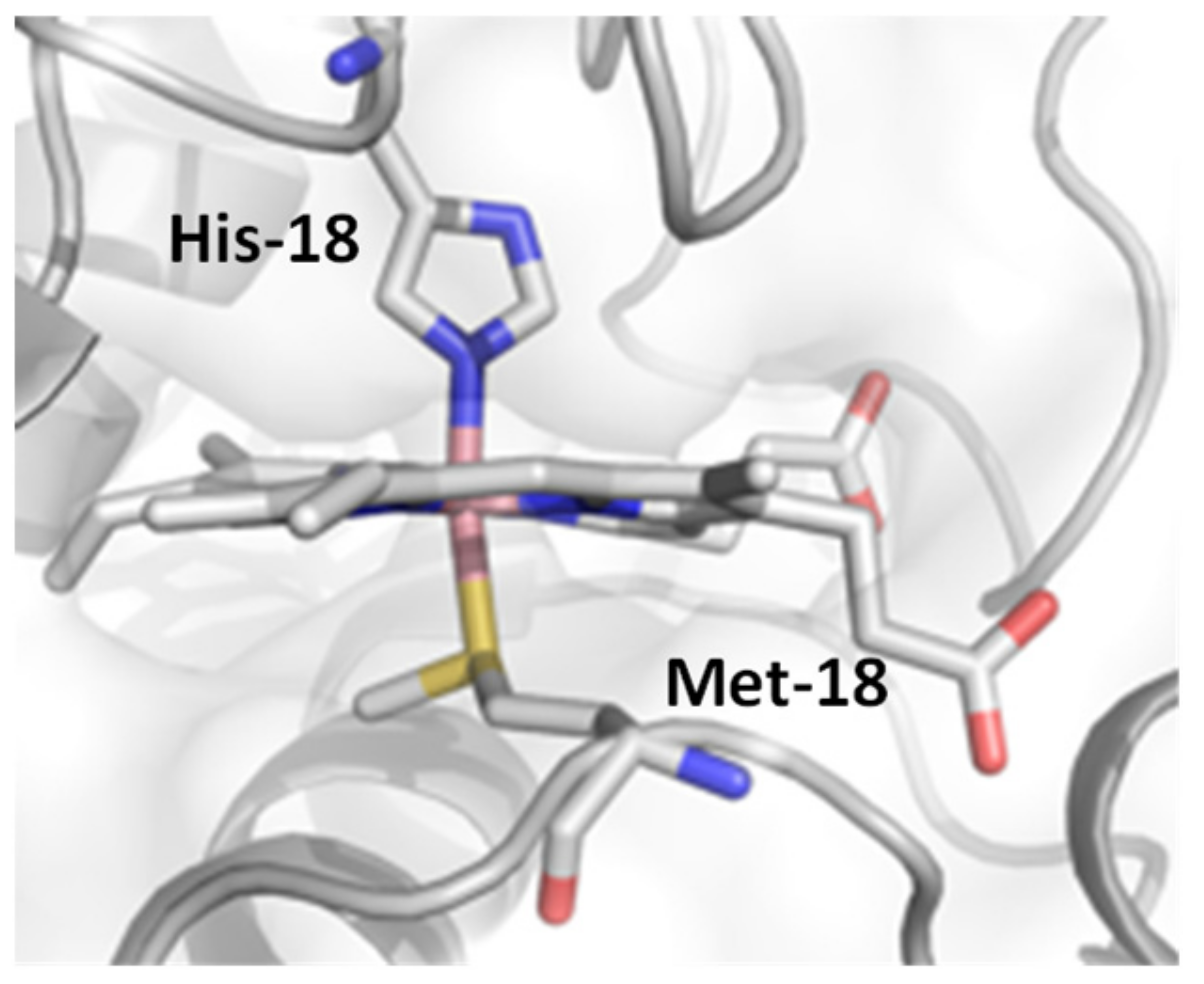{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
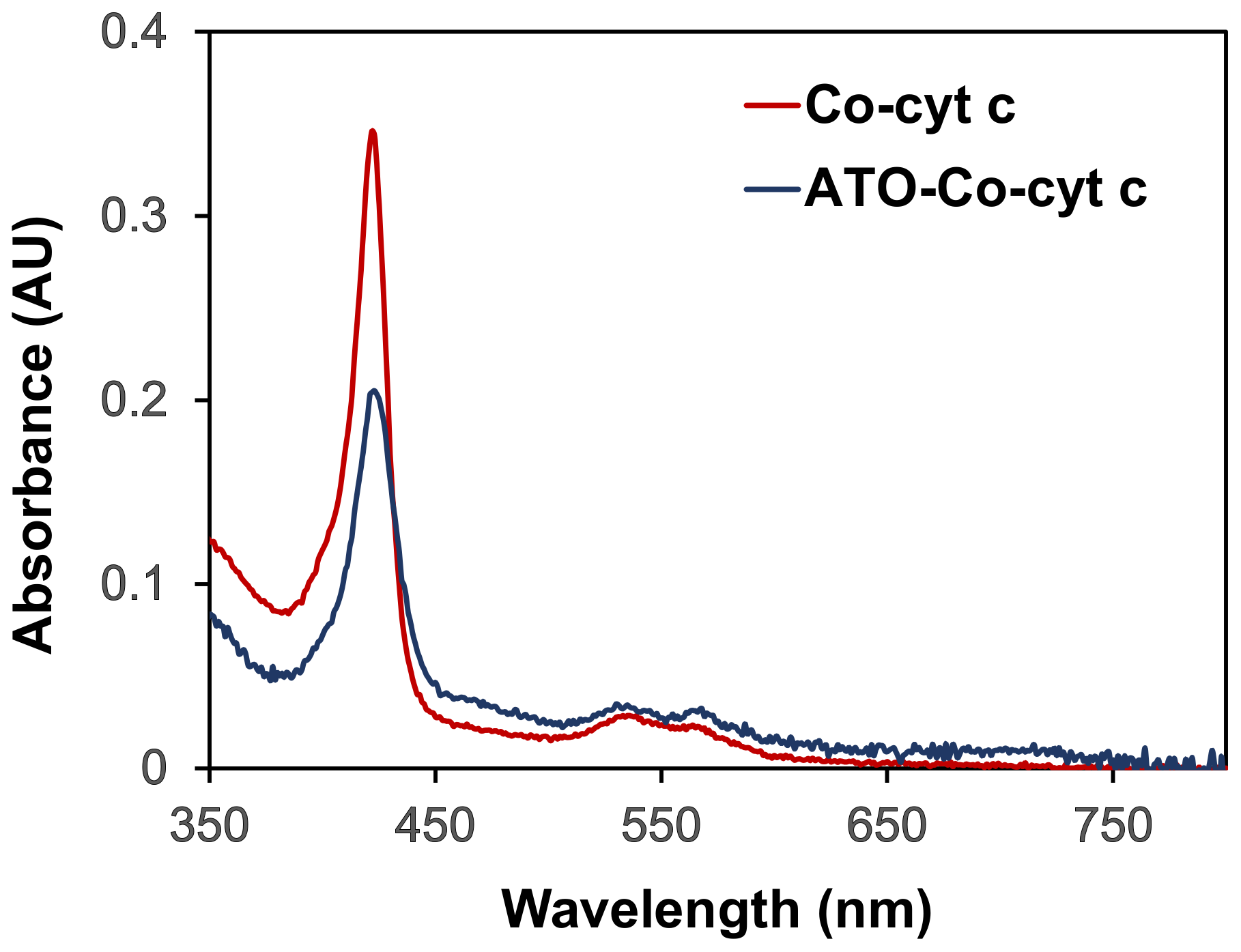
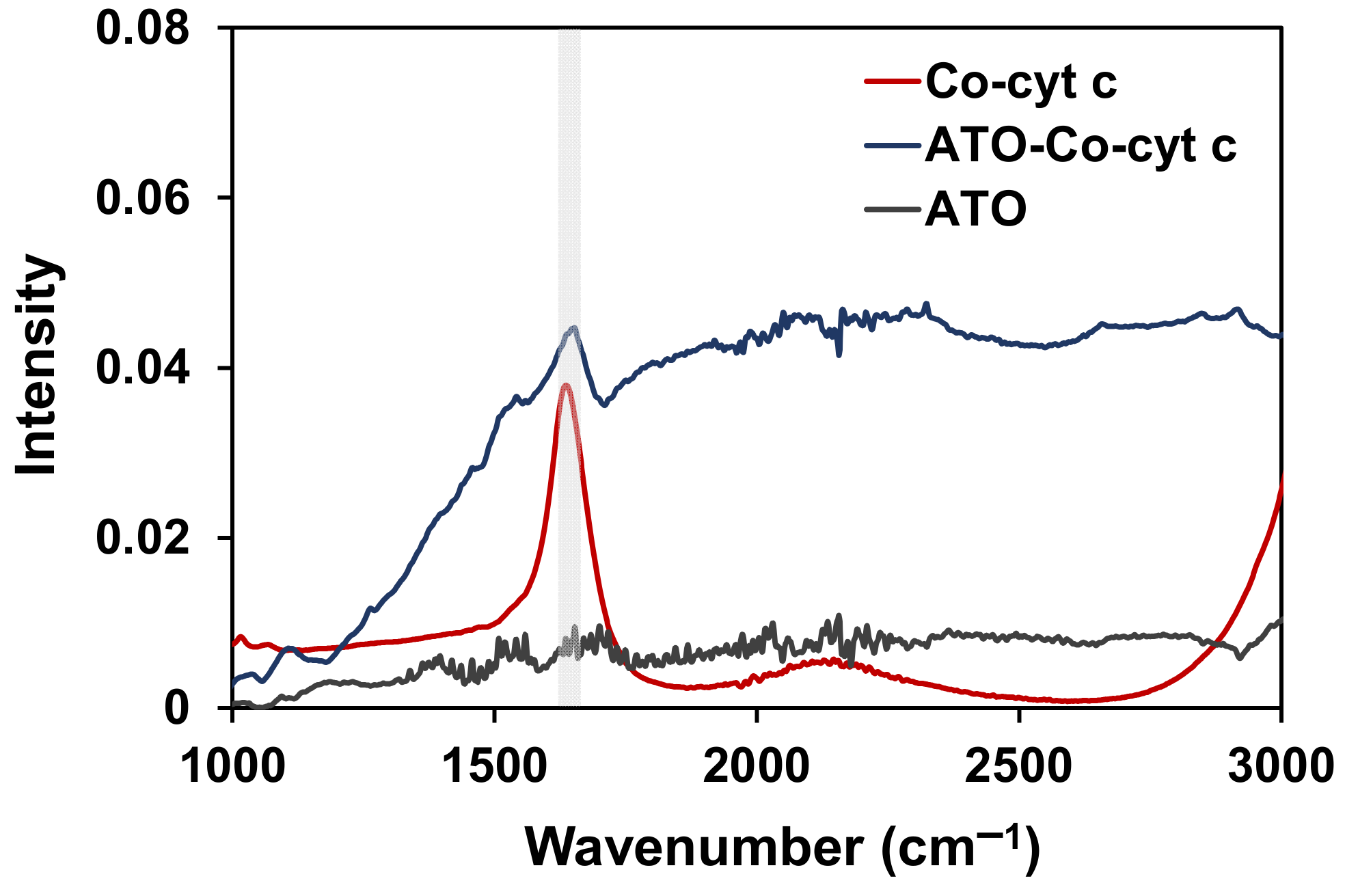
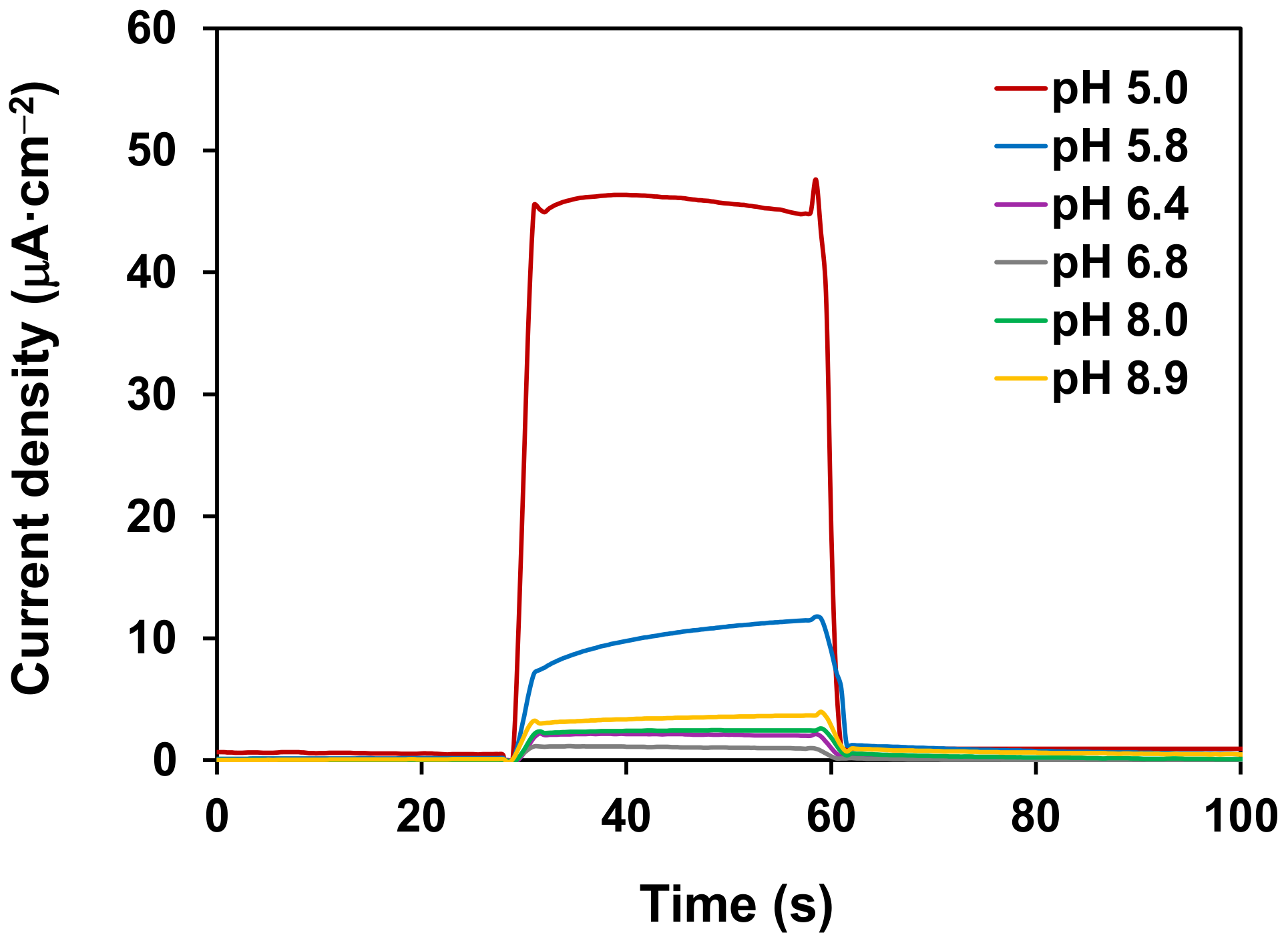
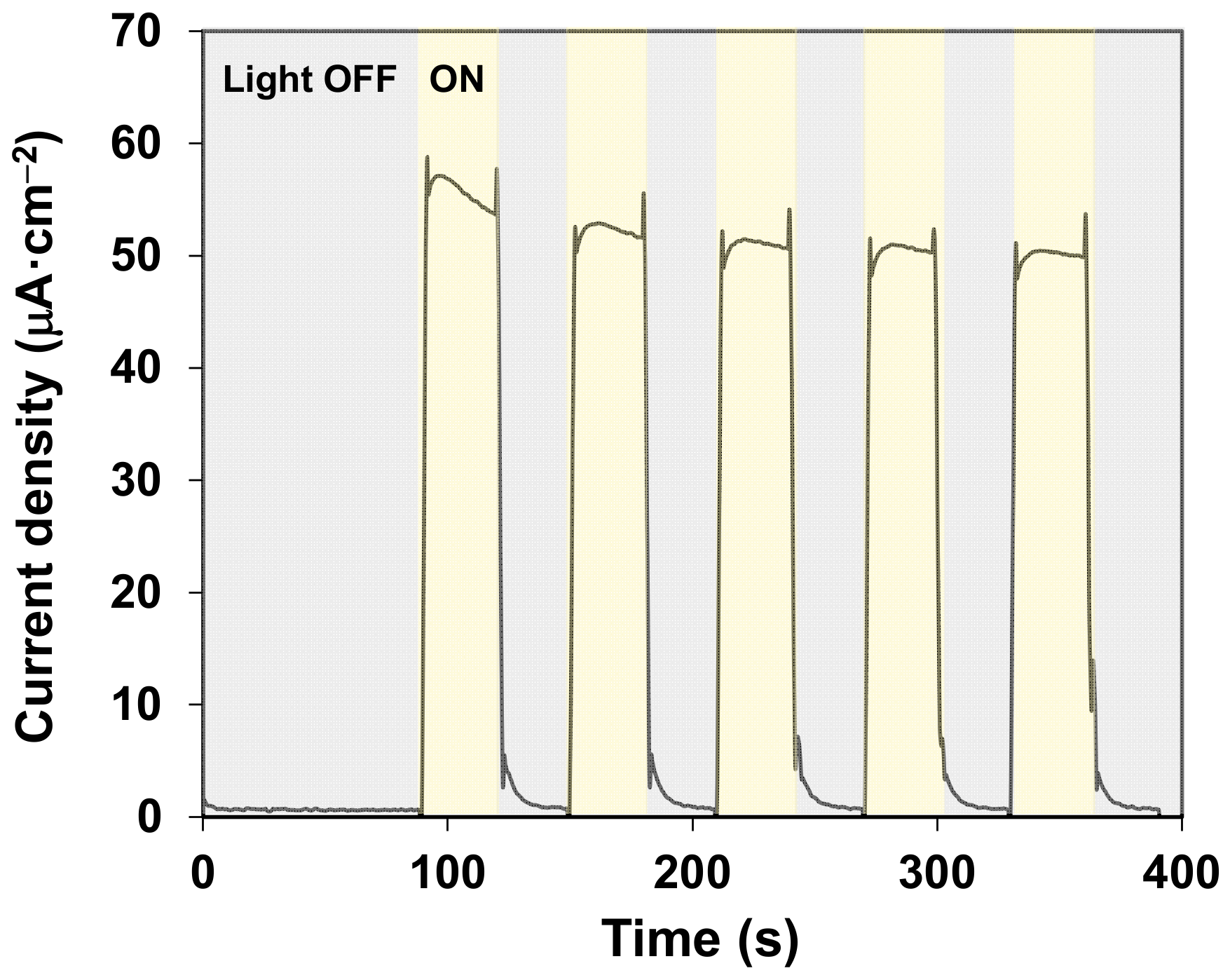
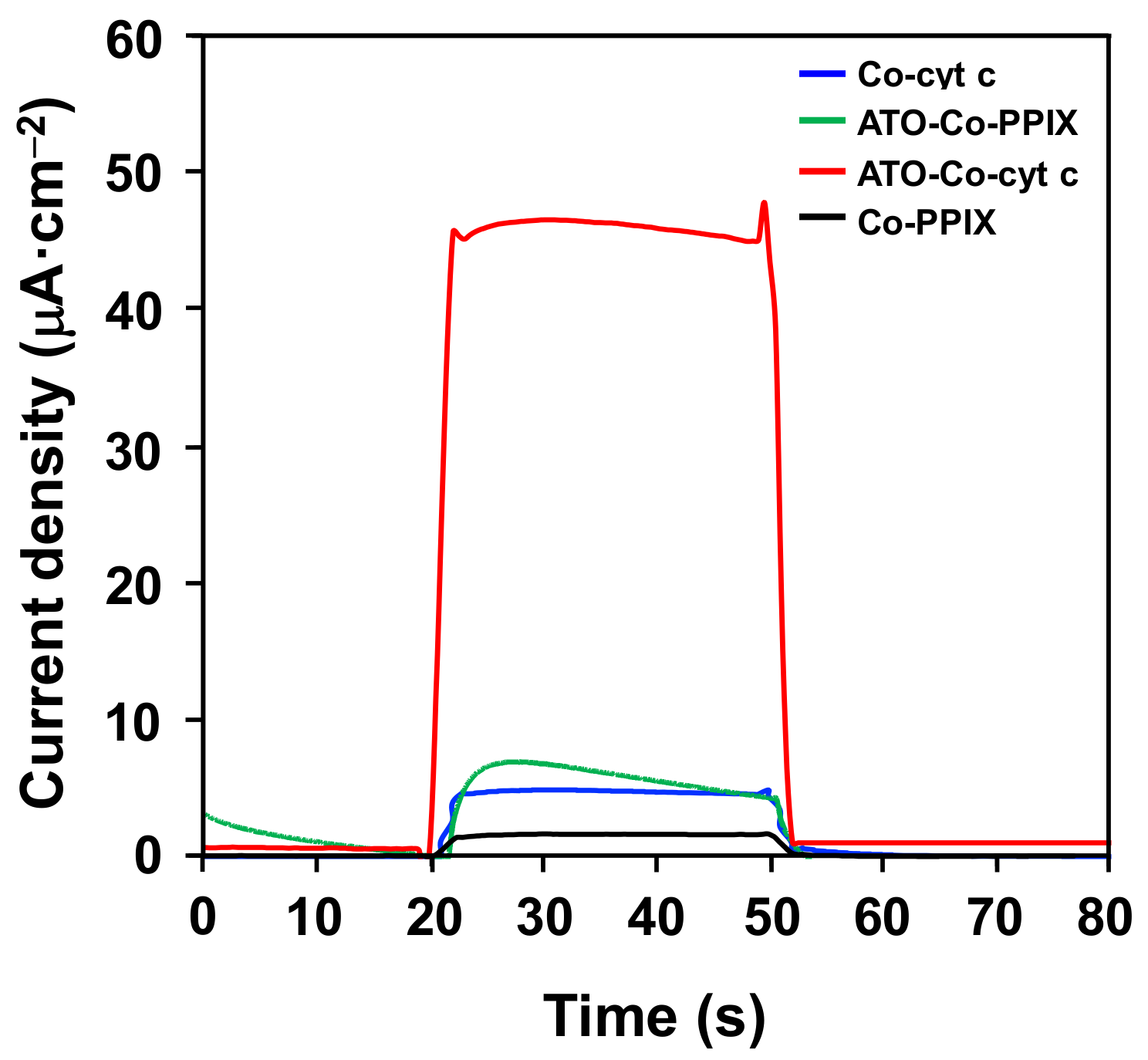
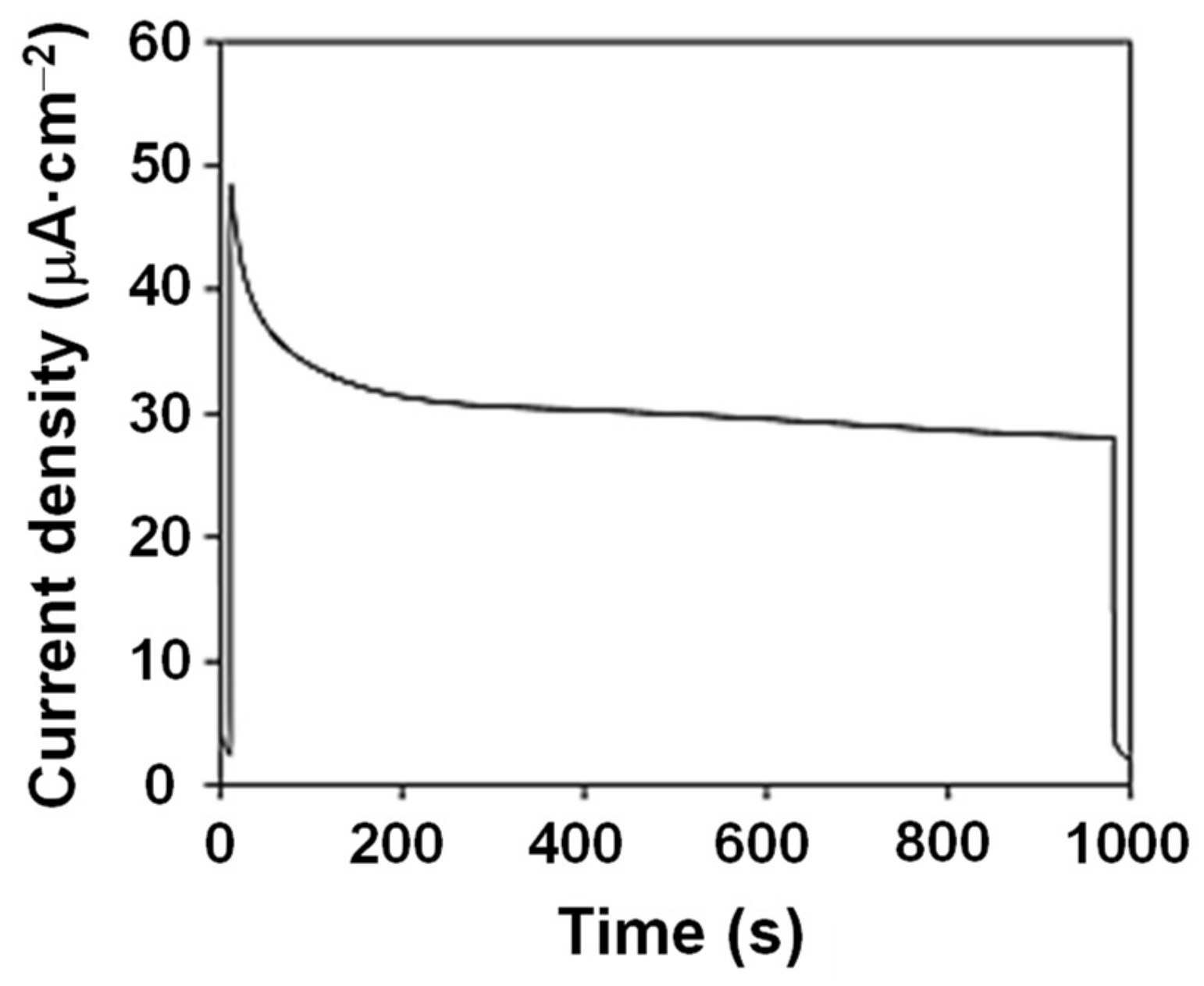
2. Results and Discussion
3. Materials and Methods
3.1. Preparation of ATO Film on FTO Glass
3.2. Metal Substitution Procedure
3.3. Cobalt Insertion
3.4. Incorporation of Co-cyt C into ATO Films
3.5. Photocurrent Measurements and Cyclic Voltammetry
3.6. ICP-MS
3.7. Photoelectrochemical Water Splitting
3.8. FTIR
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Walter, M.G.; Warren, E.L.; McKone, J.R.; Boettcher, S.W.; Mi, Q.; Santori, E.A.; Lewis, N.S. Solar Water Splitting Cells. Chem. Rev. 2010, 110, 6446–6473. [Google Scholar] [CrossRef] [PubMed]
- Reece, S.Y.; Hamel, J.A.; Sung, K.; Jarvi, T.D.; Esswein, A.J.; Pijpers, J.J.H.; Nocera, D.G. Wireless Solar Water Splitting Using Silicon-Based Semiconductors and Earth-Abundant Catalysts. Science 2011, 334, 645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokol, K.P.; Robinson, W.E.; Warnan, J.; Kornienko, N.; Nowaczyk, M.M.; Ruff, A.; Zhang, J.Z.; Reisner, E. Bias-free photoelectrochemical water splitting with photosystem II on a dye-sensitized photoanode wired to hydrogenase. Nat. Energy 2018, 3, 944–951. [Google Scholar] [CrossRef]
- Dalle, K.E.; Warnan, J.; Leung, J.J.; Reuillard, B.; Karmel, I.S.; Reisner, E. Electro- and Solar-Driven Fuel Synthesis with First Row Transition Metal Complexes. Chem. Rev. 2019, 119, 2752–2875. [Google Scholar] [CrossRef]
- Ye, S.; Ding, C.; Liu, M.; Wang, A.; Huang, Q.; Li, C. Water Oxidation Catalysts for Artificial Photosynthesis. Adv. Mater. 2019, 31, 1902069. [Google Scholar] [CrossRef]
- Hisatomi, T.; Kubota, J.; Domen, K. Recent advances in semiconductors for photocatalytic and photoelectrochemical water splitting. Chem. Soc. Rev. 2014, 43, 7520–7535. [Google Scholar] [CrossRef]
- Zhang, B.; Sun, L. Artificial photosynthesis: Opportunities and challenges of molecular catalysts. Chem. Soc. Rev. 2019, 48, 2216–2264. [Google Scholar] [CrossRef] [Green Version]
- Pannwitz, A.; Klein, D.M.; Rodríguez-Jiménez, S.; Casadevall, C.; Song, H.; Reisner, E.; Hammarström, L.; Bonnet, S. Roadmap towards solar fuel synthesis at the water interface of liposome membranes. Chem. Soc. Rev. 2021, 50, 4833–4855. [Google Scholar] [CrossRef]
- Ferreira, K.N.; Iverson, T.M.; Maghlaoui, K.; Barber, J.; Iwata, S. Architecture of the photosynthetic oxygen-evolving center. Science 2004, 303, 1831–1838. [Google Scholar] [CrossRef] [Green Version]
- Lubitz, W.; Chrysina, M.; Cox, N. Water oxidation in photosystem II. Photosynth Res. 2019, 142, 105–125. [Google Scholar] [CrossRef] [Green Version]
- Abdi, F.F.; van de Krol, R. Nature and Light Dependence of Bulk Recombination in Co-Pi-Catalyzed BiVO4 Photoanodes. J. Phys. Chem. C 2012, 116, 9398–9404. [Google Scholar] [CrossRef]
- Zhong, D.K.; Choi, S.; Gamelin, D.R. Near-complete suppression of surface recombination in solar photoelectrolysis by "Co-Pi" catalyst-modified W:BiVO4. J. Am. Chem. Soc. 2011, 133, 18370–18377. [Google Scholar] [CrossRef]
- Blakemore, J.D.; Crabtree, R.H.; Brudvig, G.W. Molecular Catalysts for Water Oxidation. Chem. Rev. 2015, 115, 12974–13005. [Google Scholar] [CrossRef]
- Macchioni, A. The Middle-Earth between Homogeneous and Heterogeneous Catalysis in Water Oxidation with Iridium. Eur. J. Inorg. Chem. 2019, 2019, 7–17. [Google Scholar] [CrossRef]
- Lloret-Fillol, J.; Costas, M. Chapter One—Water oxidation at base metal molecular catalysts. In Advances in Organometallic Chemistry; Pérez, P.J., Ed.; Academic Press/Elsevier: Cambridge, MA, USA, 2019; Volume 71, pp. 1–52. [Google Scholar]
- Casadevall, C.; Bucci, A.; Costas, M.; Lloret-Fillol, J. Chapter Four—Water oxidation catalysis with well-defined molecular iron complexes. In Advances in Inorganic Chemistry; Van Eldik, R., Hubbard, C.D., Eds.; Academic Press/Elsevier: Cambridge, MA, USA, 2019; Volume 74, pp. 151–196. [Google Scholar]
- Sayama, K.; Nomura, A.; Arai, T.; Sugita, T.; Abe, R.; Yanagida, M.; Oi, T.; Iwasaki, Y.; Abe, Y.; Sugihara, H. Photoelectrochemical decomposition of water into H2 and O2 on porous BiVO4 thin-film electrodes under visible light and significant effect of Ag ion treatment. J. Phys. Chem. B 2006, 110, 11352–11360. [Google Scholar] [CrossRef]
- Carey, A.-M.; Zhang, H.; Mieritz, D.; Volosin, A.; Gardiner, A.T.; Cogdell, R.J.; Yan, H.; Seo, D.-K.; Lin, S.; Woodbury, N.W. Photocurrent Generation by Photosynthetic Purple Bacterial Reaction Centers Interfaced with a Porous Antimony-Doped Tin Oxide (ATO) Electrode. ACS Appl. Mater. Interfaces 2016, 8, 25104–25110. [Google Scholar] [CrossRef]
- Jo, W.; Jang, J.-W.; Kong, K.-J.; Kang, H.J.; Kim, J.; Jun, H.; Parmar, K.; Lee, J.S. Phosphate Doping into Monoclinic BiVO4 for Enhanced Photoelectrochemical Water Oxidation Activity. Angew. Chem. Int. Ed. Engl. 2012, 124, 3201–3205. [Google Scholar] [CrossRef]
- Nakazono, T.; Parent, A.R.; Sakai, K. Cobalt porphyrins as homogeneous catalysts for water oxidation. Chem. Commun. 2013, 49, 6325–6327. [Google Scholar] [CrossRef]
- Wang, D.; Groves, J.T. Efficient water oxidation catalyzed by homogeneous cationic cobalt porphyrins with critical roles for the buffer base. Proc. Natl. Acad. Sci. USA 2013, 110, 15579–15584. [Google Scholar] [CrossRef] [Green Version]
- Dogutan, D.K.; McGuire, R.; Nocera, D.G. Electocatalytic Water Oxidation by Cobalt(III) Hangman β-Octafluoro Corroles. J. Am. Chem. Soc. 2011, 133, 9178–9180. [Google Scholar] [CrossRef]
- Han, A.; Jia, H.; Ma, H.; Ye, S.; Wu, H.; Lei, H.; Han, Y.; Cao, R.; Du, P. Cobalt porphyrin electrode films for electrocatalytic water oxidation. Phys. Chem. Chem. Phys. 2014, 16, 11224–11232. [Google Scholar] [CrossRef]
- Liu, B.; Li, J.; Wu, H.-L.; Liu, W.-Q.; Jiang, X.; Li, Z.-J.; Chen, B.; Tung, C.-H.; Wu, L.-Z. Improved Photoelectrocatalytic Performance for Water Oxidation by Earth-Abundant Cobalt Molecular Porphyrin Complex-Integrated BiVO4 Photoanode. Acs Appl. Mater. Interfaces 2016, 8, 18577–18583. [Google Scholar] [CrossRef]
- Sun, Z.; Li, J.; Zheng, H.; Liu, X.; Ye, S.; Du, P. Pyrolyzed cobalt porphyrin-modified carbon nanomaterial as an active catalyst for electrocatalytic water oxidation. Int. J. Hydrog. Energy 2015, 40, 6538–6545. [Google Scholar] [CrossRef]
- Daniel, Q.; Ambre, R.B.; Zhang, B.; Philippe, B.; Chen, H.; Li, F.; Fan, K.; Ahmadi, S.; Rensmo, H.; Sun, L. Re-Investigation of Cobalt Porphyrin for Electrochemical Water Oxidation on FTO Surface: Formation of CoOx as Active Species. ACS Catal. 2017, 7, 1143–1149. [Google Scholar] [CrossRef]
- Lloret-Fillol, J.; Costas, M. Water oxidation at base metal molecular catalysts. Adv. Organomet. Chem. 2019, 71, 1–52. [Google Scholar]
- Hu, X.-M.; Rønne, M.H.; Pedersen, S.U.; Skrydstrup, T.; Daasbjerg, K. Enhanced Catalytic Activity of Cobalt Porphyrin in CO2 Electroreduction upon Immobilization on Carbon Materials. Angew. Chem. Int. Ed. 2017, 56, 6468–6472. [Google Scholar] [CrossRef]
- Pasternack, R.F.; Francesconi, L.; Raff, D.; Spiro, E. Aggregation of nickel(II), copper(II), and zinc(II) derivatives of water-soluble porphyrins. Inorg. Chem. 1973, 12, 2606–2611. [Google Scholar] [CrossRef]
- Ow, Y.P.; Green, D.R.; Hao, Z.; Mak, T.W. Cytochrome c: Functions beyond respiration. Nat. Rev. Mol. Cell Biol. 2008, 9, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zheng, J.; Wang, J.; Xu, J.; Hayat, T.; Alharbi, N.S. Direct electrochemistry of cytochrome c immobilized on one dimensional Au nanoparticles functionalized magnetic N-doped carbon nanotubes and its application for the detection of H2O2. Sens. Actuators B Chem. 2019, 282, 85–95. [Google Scholar] [CrossRef]
- Dong, Y.; Ji, X.; Laaksonen, A.; Cao, W.; He, H.; Lu, X. Excellent Protein Immobilization and Stability on Heterogeneous C–TiO2 Hybrid Nanostructures: A Single Protein AFM Study. Langmuir 2020, 36, 9323–9332. [Google Scholar] [CrossRef] [PubMed]
- Ciornii, D.; Kölsch, A.; Zouni, A.; Lisdat, F. A precursor-approach in constructing 3D ITO electrodes for the improved performance of photosystem I-cyt c photobioelectrodes. Nanoscale 2019, 11, 15862–15870. [Google Scholar] [CrossRef]
- Hou, K.; Puzzo, D.; Helander, M.G.; Lo, S.S.; Bonifacio, L.D.; Wang, W.; Lu, Z.-H.; Scholes, G.D.; Ozin, G.A. Dye-Anchored Mesoporous Antimony-Doped Tin Oxide Electrochemiluminescence Cell. Adv. Mater. 2009, 21, 2492–2496. [Google Scholar] [CrossRef]
- Carey, A.M.; Zhang, H.; Liu, M.; Sharaf, D.; Akram, N.; Yan, H.; Lin, S.; Woodbury, N.W.; Seo, D.K. Enhancing Photocurrent Generation in Photosynthetic Reaction Center-Based Photoelectrochemical Cells with Biomimetic DNA Antenna. ChemSusChem 2017, 10, 4457–4460. [Google Scholar] [CrossRef] [Green Version]
- Kwan, P.; Schmitt, D.; Volosin, A.M.; McIntosh, C.L.; Seo, D.-K.; Jones, A.K. Spectroelectrochemistry of cytochrome c and azurin immobilized in nanoporous antimony-doped tin oxide. Chem. Commun. 2011, 47, 12367–12369. [Google Scholar] [CrossRef]
- Day, N.U.; Wamser, C.C. Poly-tetrakis-5,10,15,20-(4-aminophenyl)porphyrin Films as Two-Electron Oxygen Reduction Photoelectrocatalysts for the Production of H2O2. J. Phys. Chem. C 2017, 121, 11076–11082. [Google Scholar] [CrossRef]
- Wadsworth, B.L.; Khusnutdinova, D.; Urbine, J.M.; Reyes, A.S.; Moore, G.F. Expanding the Redox Range of Surface-Immobilized Metallocomplexes Using Molecular Interfaces. ACS Appl. Mater. Interfaces 2020, 12, 3903–3911. [Google Scholar] [CrossRef]
- Wadsworth, B.L.; Beiler, A.M.; Khusnutdinova, D.; Reyes Cruz, E.A.; Moore, G.F. Interplay between Light Flux, Quantum Efficiency, and Turnover Frequency in Molecular-Modified Photoelectrosynthetic Assemblies. J. Am. Chem. Soc. 2019, 141, 15932–15941. [Google Scholar] [CrossRef]
- Frasca, S.; von Graberg, T.; Feng, J.-J.; Thomas, A.; Smarsly, B.M.; Weidinger, I.M.; Scheller, F.W.; Hildebrandt, P.; Wollenberger, U. Mesoporous Indium Tin Oxide as a Novel Platform for Bioelectronics. ChemCatChem 2010, 2, 839–845. [Google Scholar] [CrossRef]
- Graf, M.; García, R.G.; Wätzig, H. Protein adsorption in fused-silica and polyacrylamide-coated capillaries. Electrophoresis 2005, 26, 2409–2417. [Google Scholar] [CrossRef]
- Kleingardner, J.G.; Kandemir, B.; Bren, K.L. Hydrogen Evolution from Neutral Water under Aerobic Conditions Catalyzed by Cobalt Microperoxidase-11. J. Am. Chem. Soc. 2014, 136, 4–7. [Google Scholar] [CrossRef]
- Primus, J.L.; Boersma, M.G.; Mandon, D.; Boeren, S.; Veeger, C.; Weiss, R.; Rietjens, I.M. The effect of iron to manganese substitution on microperoxidase 8 catalysed peroxidase and cytochrome P450 type of catalysis. J. Biol. Inorg. Chem. 1999, 4, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Dizicheh, Z.B. Design of Redox Proteins as Catalysts for Fuel Production; Arizona State University: Phoenix, AZ, USA, 2019. [Google Scholar]
- Mersch, D.; Lee, C.-Y.; Zhang, J.Z.; Brinkert, K.; Fontecilla-Camps, J.C.; Rutherford, A.W.; Reisner, E. Wiring of Photosystem II to Hydrogenase for Photoelectrochemical Water Splitting. J. Am. Chem. Soc. 2015, 137, 8541–8549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Mandal, S.; Carey, A.-M.; Liu, M.; Chen, S.; Seo, D.-K.; Yan, H.; Woodbury, N. Interfacing Photosystem I Reaction Centers with a Porous Antimony-Doped Tin Oxide Electrode to Perform Light Driven Redox Chemistry. Biophys. J. 2019, 116, 443a. [Google Scholar] [CrossRef] [Green Version]
- Mieritz, D.; Liang, R.; Zhang, H.; Carey, A.-M.; Chen, S.; Volosin, A.; Lin, S.; Woodbury, N.; Seo, D.-K. Thickness-Dependent Bioelectrochemical and Energy Applications of Thickness-Controlled Meso-Macroporous Antimony-Doped Tin Oxide. Coatings 2018, 8, 128. [Google Scholar] [CrossRef] [Green Version]
- Call, A.; Casadevall, C.; Romero-Rivera, A.; Martin-Diaconescu, V.; Sommer, D.J.; Osuna, S.; Ghirlanda, G.; Lloret-Fillol, J. Improved Electro- and Photocatalytic Water Reduction by Confined Cobalt Catalysts in Streptavidin. ACS Catal. 2019, 9, 5837–5846. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casadevall, C.; Zhang, H.; Chen, S.; Sommer, D.J.; Seo, D.-K.; Ghirlanda, G. Photoelectrochemical Water Oxidation by Cobalt Cytochrome C Integrated-ATO Photoanode. Catalysts 2021, 11, 626. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11050626
Casadevall C, Zhang H, Chen S, Sommer DJ, Seo D-K, Ghirlanda G. Photoelectrochemical Water Oxidation by Cobalt Cytochrome C Integrated-ATO Photoanode. Catalysts. 2021; 11(5):626. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11050626
Chicago/Turabian StyleCasadevall, Carla, Haojie Zhang, Shaojiang Chen, Dayn J. Sommer, Dong-Kyun Seo, and Giovanna Ghirlanda. 2021. "Photoelectrochemical Water Oxidation by Cobalt Cytochrome C Integrated-ATO Photoanode" Catalysts 11, no. 5: 626. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11050626