
Theoretical Study on Ethylene Polymerization Catalyzed by Half-Titanocenes Bearing Different Ancillary Groups

 ,
,
Abstract
:
1. Introduction
2. Results and Discussion
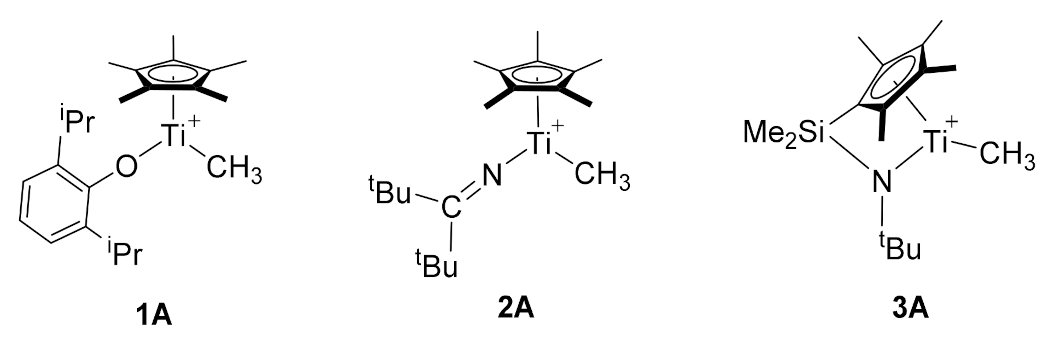
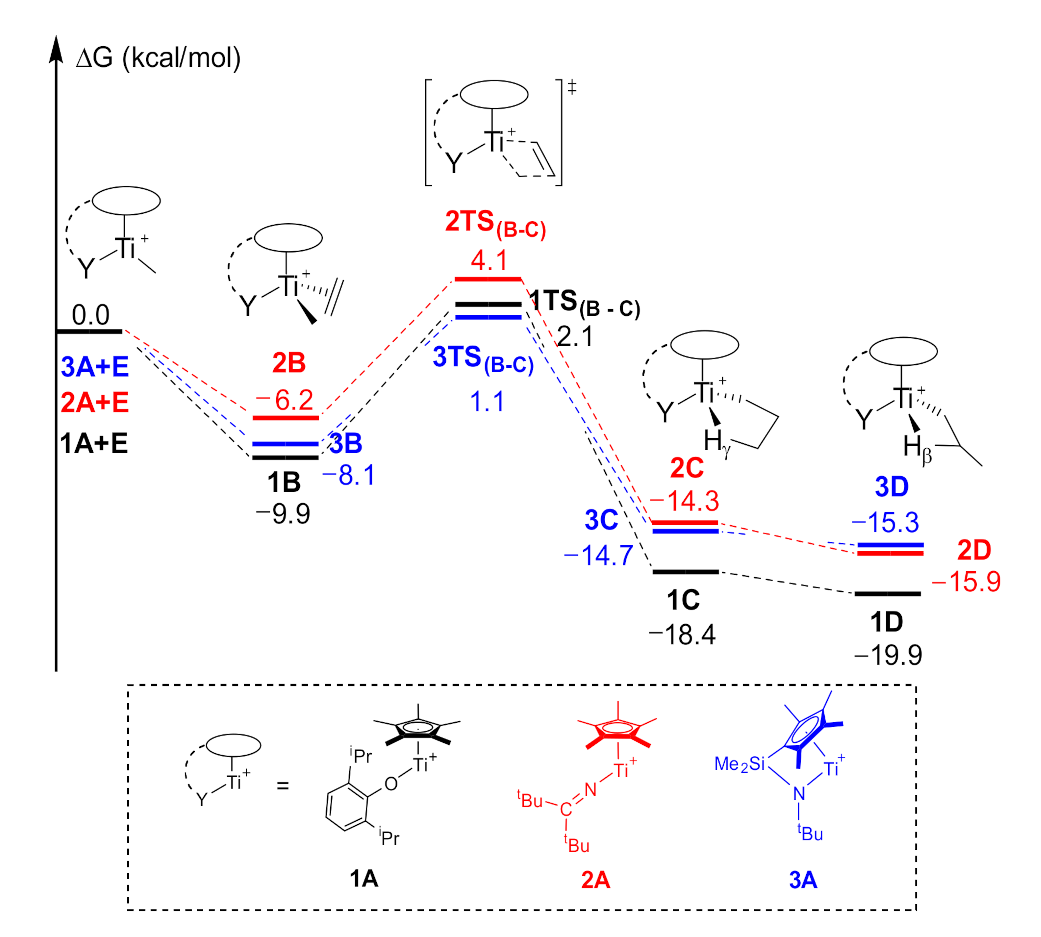
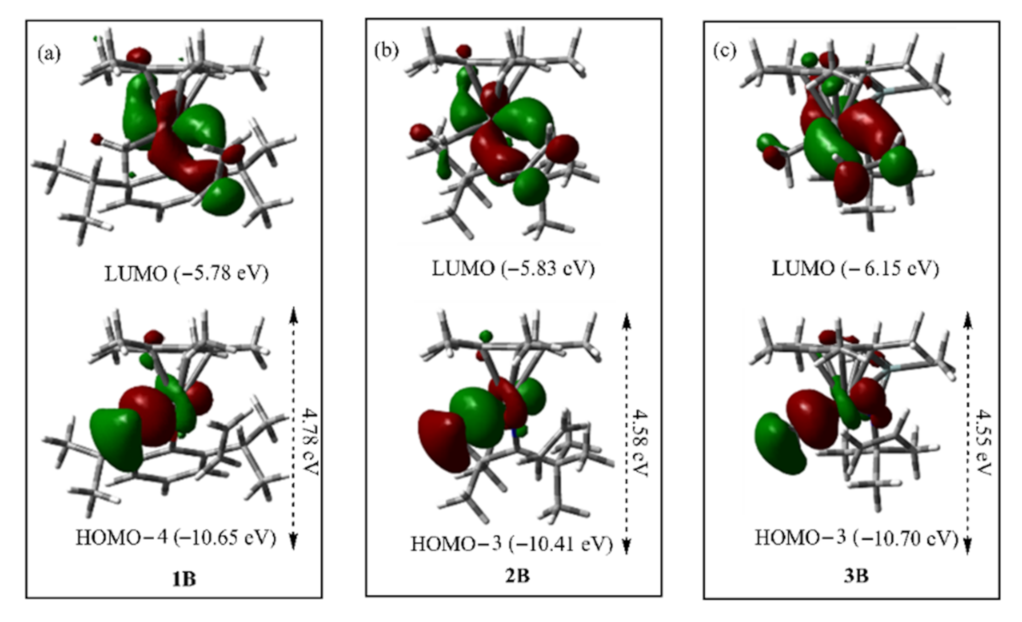
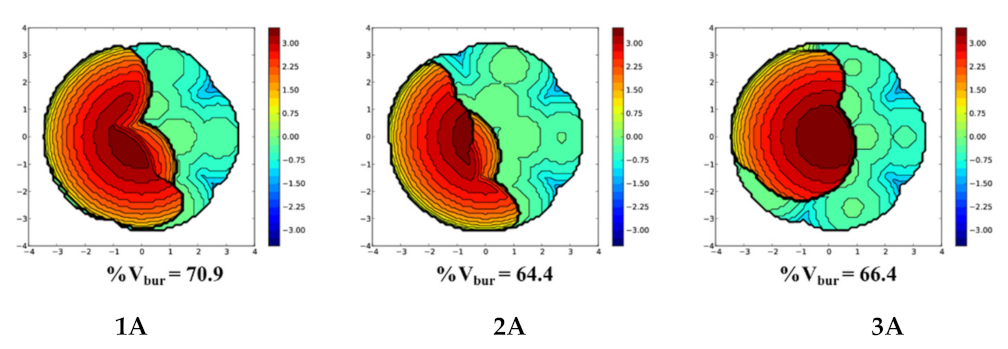
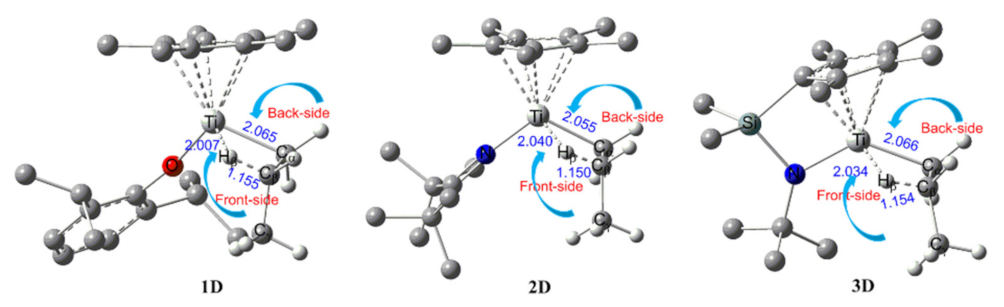
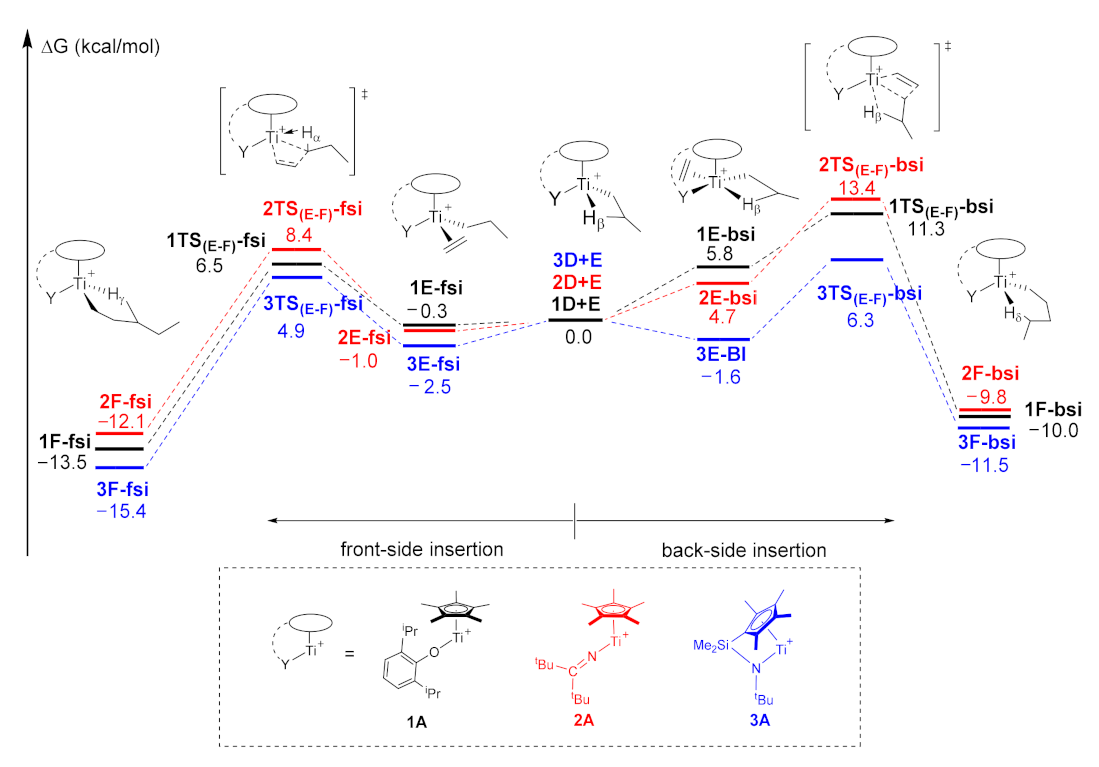
2.1. The Chain Initiation
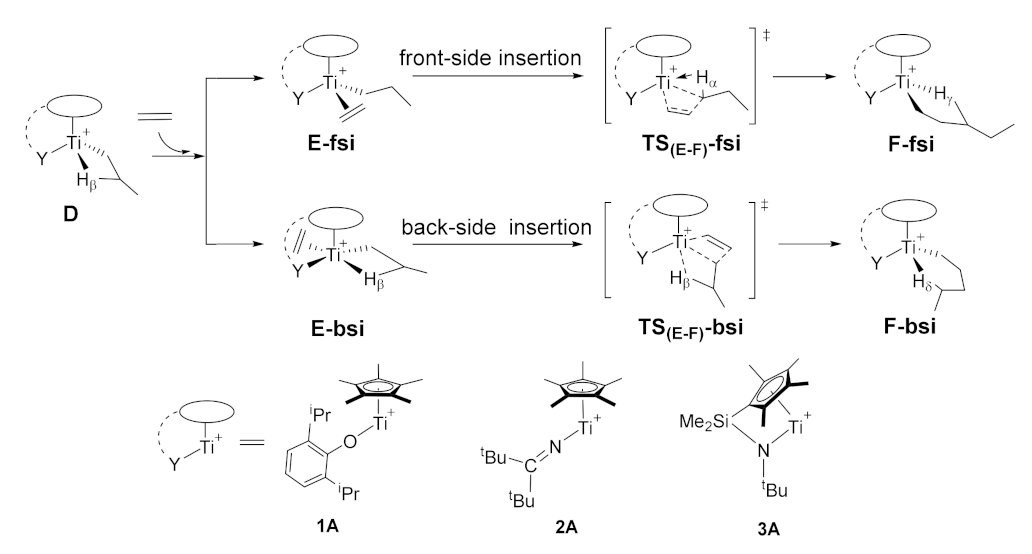
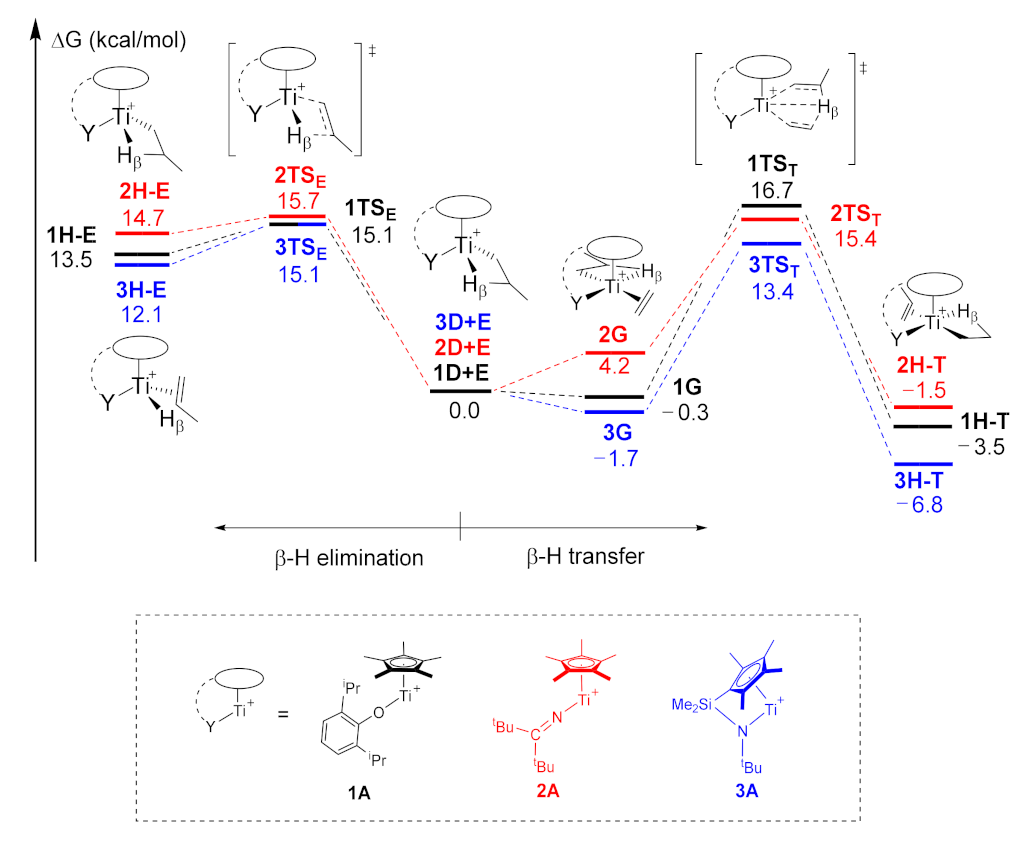
2.2. The Chain Propagation
2.3. The Chain Termination
3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sauter, D.; Taoufik, M.; Boisson, C. Polyolefins, a Success Story. Polymers 2017, 9, 185. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Brookhart, M. Exploring Ethylene/Polar Vinyl Monomer Copolymerizations Using Ni and Pd α-Diimine Catalysts. Acc. Chem. Res. 2018, 51, 1831–1839. [Google Scholar] [CrossRef]
- Ma, Z.; Yang, W.; Sun, W.-H. Recent Progress on Transition Metal (Fe, Co, Ni, Ti and V) Complex Catalysts in Olefin Polymerization with High Thermal Stability. Chin. J. Chem. 2017, 35, 531–540. [Google Scholar] [CrossRef]
- Zhang, S.; Wu, Y.-X. Controlled polymerization of olefin and its macromolecular engineering. Sci. Sin. Chim. 2018, 48, 590–600. [Google Scholar] [CrossRef]
- Baier, M.C.; Zuideveld, M.A.; Mecking, S. Post-Metallocenes in the Industrial Production of Polyolefins. Angew. Chem. Int. Ed. 2014, 53, 9722–9744. [Google Scholar] [CrossRef]
- Chen, C. Designing catalysts for olefin polymerization and copolymerization: Beyond electronic and steric tuning. Nat. Rev. Chem. 2018, 2, 6–14. [Google Scholar] [CrossRef]
- Matsugi, T.; Fujita, T. High-performance olefin polymerization catalysts discovered on the basis of a new catalyst design concept. Chem. Soc. Rev. 2008, 37, 1264–1277. [Google Scholar] [CrossRef]
- Zaccaria, F.; Budzelaar, P.H.M.; Zuccaccia, C.; Cipullo, R.; Macchioni, A.; Busico, V.; Ehm, C. Chain Transfer to Solvent and Monomer in Early Transition Metal Catalyzed Olefin Polymerization: Mechanisms and Implications for Catalysis. Catalysts 2021, 11, 215. [Google Scholar] [CrossRef]
- Shamiri, A.; Chakrabarti, M.H.; Jahan, S.; Hussain, M.A.; Kaminsky, W.; Aravind, P.V.; Yehye, W.A. The Influence of Ziegler-Natta and Metallocene Catalysts on Polyolefin Structure, Properties, and Processing Ability. Materials 2014, 7, 5069–5108. [Google Scholar] [CrossRef] [PubMed]
- Nomura, K. Half-Titanocenes Containing Anionic Ancillary Donor Ligands: Effective Catalyst Precursors for Ethylene/Styrene Copolymerization. Catalysts 2013, 3, 157–175. [Google Scholar] [CrossRef] [Green Version]
- Nomura, K.; Naga, N.; Miki, M.; Yanagi, K. Olefin Polymerization by (Cyclopentadienyl) (aryloxy) titanium(IV) Complexes−Cocatalyst Systems. Macromolecules 1998, 31, 7588–7597. [Google Scholar] [CrossRef]
- Nomura, K.; Naga, N.; Miki, M.; Yanagi, K.; Imai, A. Synthesis of Various Nonbridged Titanium(IV) Cyclopentadienyl−Aryloxy Complexes of the Type CpTi(OAr)X2 and Their Use in the Catalysis of Alkene Polymerization. Important Roles of Substituents on both Aryloxy and Cyclopentadienyl Groups. Organometallics 1998, 17, 2152–2154. [Google Scholar] [CrossRef]
- Nomura, K.; Fujita, K.; Fujiki, M. Olefin polymerization by (cyclopentadienyl)(ketimide)titanium(IV) complexes of the type, Cp’TiCl2(N = CtBu2)-methylaluminoxane (MAO) catalyst systems. J. Mol. Catal. A Chem. 2004, 220, 133–144. [Google Scholar] [CrossRef]
- Nomura, K.; Fujita, K.; Fujiki, M. Effects of cyclopentadienyl fragment in ethylene, 1-hexene, and styrene polymerizations catalyzed by half-titanocenes containing ketimide ligand of the type, Cp’TiCl2(N = CtBu2). Catal. Commun. 2004, 5, 413–417. [Google Scholar] [CrossRef]
- Stephan, D.W.; Stewart, J.C.; Guérin, F.; Spence, R.E.V.H.; Xu, W.; Harrison, D.G. Phosphinimides as a Steric Equivalent to Cyclopentadienyl: An Approach to Ethylene Polymerization Catalyst Design. Organometallics 1999, 18, 1116–1118. [Google Scholar] [CrossRef]
- Yue, N.; Hollink, E.; Guérin, F.; Stephan, D.W. Zirconium Phosphinimide Complexes: Synthesis, Structure, and Deactivation Pathways in Ethylene Polymerization Catalysis. Organometallics 2001, 20, 4424–4433. [Google Scholar] [CrossRef]
- Nomura, K.; Liu, J.; Padmanabhan, S.; Kitiyanan, B. Nonbridged half-metallocenes containing anionic ancillary donor ligands: New promising candidates as catalysts for precise olefin polymerization. J. Mol. Catal. A Chem. 2007, 267, 1–29. [Google Scholar] [CrossRef]
- Nomura, K. Half-titanocenes containing anionic ancillary donor ligands as promising new catalysts for precise olefin polymerization. Dalton Trans. 2009, 38, 8811–8823. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Nomura, K. Design of Efficient Molecular Catalysts for Synthesis of Cyclic Olefin Copolymers (COC) by Copolymerization of Ethylene and α-Olefins with Norbornene or Tetracyclododecene. Catalysts 2016, 6, 175. [Google Scholar] [CrossRef] [Green Version]
- Nomura, K.; Liu, J. Half-titanocenes for precise olefin polymerisation: Effects of ligand substituents and some mechanistic aspects. Dalton Trans. 2011, 40, 7666–7682. [Google Scholar] [CrossRef]
- Nomura, K.; Yamada, J.; Wei, W.; Liu, J. Effect of ketimide ligand for ethylene polymerization and ethylene/norbornene copolymerization catalyzed by (cyclopentadienyl) (ketimide) titanium complexes-MAO catalyst systems: Structural analysis for Cp*TiCl2(N = CPh2). J. Organomet. Chem. 2007, 692, 4675–4682. [Google Scholar] [CrossRef]
- Kim, T.J.; Kim, S.K.; Kim, B.J.; Hahn, J.S.; Ok, M.A.; Song, J.H.; Shin, D.H.; Ko, J.; Cheong, M.; Kim, J.; et al. Half-Metallocene Titanium(IV) Phenyl Phenoxide for High Temperature Olefin Polymerization: Ortho-Substituent Effect at Ancillary o-Phenoxy Ligand for Enhanced Catalytic Performance. Macromolecules 2009, 42, 6932–6943. [Google Scholar] [CrossRef]
- Deng, L.; Margl, P.; Ziegler, T. A Density Functional Study of Nickel(II) Diimide Catalyzed Polymerization of Ethylene. J. Am. Chem. Soc. 1997, 119, 1094–1100. [Google Scholar] [CrossRef]
- Xu, Z.; Vanka, K.; Ziegler, T. Influence of the Counterion MeB(C6F5)3- and Solvent Effects on Ethylene Polymerization Catalyzed by [(CpSiMe2NR)TiMe]+: A Combined Density Functional Theory and Molecular Mechanism Study. Organometallics 2004, 23, 104–116. [Google Scholar] [CrossRef]
- Kang, X.; Zhou, G.; Wang, X.; Qu, J.; Hou, Z.; Luo, Y. Alkyl Effects on the Chain Initiation Efficiency of Olefin Polymerization by Cationic Half-Sandwich Scandium Catalysts: A DFT Study. Organometallics 2016, 35, 913–920. [Google Scholar] [CrossRef]
- Luo, G.; Luo, Y.; Hou, Z.; Qu, J. Intermetallic Cooperation in Olefin Polymerization Catalyzed by a Binuclear Samarocene Hydride: A Theoretical Study. Organometallics 2016, 35, 778–784. [Google Scholar] [CrossRef]
- Liu, K.; Liu, Z.; Cheng, R.; He, X.; Liu, B. Mechanistic study on the effects of co-catalyst on ethylene polymerization over supported vanadocene catalyst. Mol. Catal. 2020, 486, 110852. [Google Scholar] [CrossRef]
- Parveen, R.; Cundari, T.R.; Younker, J.M.; Rodriguez, G.; McCullough, L. DFT and QSAR Studies of Ethylene Polymerization by Zirconocene Catalysts. ACS Catal. 2019, 9, 9339–9349. [Google Scholar] [CrossRef]
- Parveen, R.; Cundari, T.R.; Younker, J.M.; Rodriguez, G. Computational Assessment of Counterion Effect of Borate Anions on Ethylene Polymerization by Zirconocene and Hafnocene Catalysts. Organometallics 2020, 39, 2068–2079. [Google Scholar] [CrossRef]
- Ustynyuk, L.Y.; Bulychev, B.M. Activation effect of metal chlorides in post-metallocene catalytic systems for ethylene polymerization: A DFT study. J. Organomet. Chem. 2015, 793, 160–170. [Google Scholar] [CrossRef]
- Lanza, G.; Fragalà, I.L.; Marks, T.J. Highly Electrophilic Olefin Polymerization Catalysts. Counteranion and Solvent Effects on Constrained Geometry Catalyst Ion Pair Structure and Reactivity. J. Am. Chem. Soc. 1998, 120, 8257–8258. [Google Scholar] [CrossRef]
- Lanza, G.; Fragalà, I.L.; Marks, T.J. Ligand Substituent, Anion, and Solvation Effects on Ion Pair Structure, Thermodynamic Stability, and Structural Mobility in “Constrained Geometry” Olefin Polymerization Catalysts: an Ab Initio Quantum Chemical Investigation. J. Am. Chem. Soc. 2000, 122, 12764–12777. [Google Scholar] [CrossRef]
- Lanza, G.; Fragalà, I.L.; Marks, T.J. Energetic, Structural, and Dynamic Aspects of Ethylene Polymerization Mediated by Homogeneous Single-Site “Constrained Geometry Catalysts” in the Presence of Cocatalyst and Solvation: An Investigation at the ab Initio Quantum Chemical Level. Organometallics 2002, 21, 5594–5612. [Google Scholar] [CrossRef]
- Pan, Y.; Zhao, A.; Li, Y.; Li, W.; So, Y.-M.; Yan, X.; He, G. Bis(oxazoline)-Derived N-Heterocyclic Carbene Ligated Rare-Earth Metal Complexes: Synthesis, Structure, and Polymerization Performance. Dalton Trans. 2018, 47, 13815–13823. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Li, W.; Wei, N.-N.; So, Y.-M.; Li, Y.; Jiang, K.; He, G. Anilido-Oxazoline-Ligated Rare-Earth Metal Complexes: Synthesis, Characterization and Highly cis-1,4-Selective Polymerization of Isoprene. Dalton Trans. 2019, 48, 3583–3592. [Google Scholar] [CrossRef]
- Li, W.; Jiang, X.; So, Y.-M.; He, G.; Pan, Y. Lutetium and Yttrium Complexes Supported by Anilido-oxazoline Ligand for Polymerization of 1,3-Conjugated Dienes and ε-Caprolactone. New J. Chem. 2020, 44, 121–128. [Google Scholar] [CrossRef]
- Pan, Y.; Xu, X.; Wei, N.-N.; Hao, C.; Zhu, X.; He, G. DFT study on 1,7-octadiene polymerization catalyzed by a non-bridged half-titanocene system. RSC Adv. 2016, 6, 69939–69946. [Google Scholar] [CrossRef]
- Xu, X.; He, G.; Wei, N.-N.; Hao, C.; Pan, Y. Selective Insertion in Copolymerization of Ethylene and Styrene Catalyzed by Half-Titanocene System Bearing Ketimide Ligand: A Theoretical Study. Chin. J. Chem. 2017, 35, 1731–1738. [Google Scholar] [CrossRef]
- Cossee, P. Ziegler-Natta catalysis I. Mechanism of polymerization of α-olefins with Ziegler-Natta catalysts. J. Catal. 1964, 3, 80–88. [Google Scholar] [CrossRef]
- Arlman, E.J. Ziegler-Natta catalysis II. Surface structure of layer-lattice transition metal chlorides. J. Catal. 1964, 3, 89–98. [Google Scholar] [CrossRef]
- Niu, S.; Hall, M.B. Theoretical Studies on Reactions of Transition-Metal Complexes. Chem. Rev. 2000, 100, 353–405. [Google Scholar] [CrossRef] [PubMed]
- Nomura, K.; Mitsudome, T.; Igarashi, A.; Nagai, G.; Tsutsumi, K.; Ina, T.; Omiya, T.; Takaya, H.; Yamazoe, S. Synthesis of (Adamantylimido) vanadium(V) Dimethyl Complex Containing (2-Anilidomethyl) pyridine Ligand and Selected Reactions: Exploring the Oxidation State of the Catalytically Active Species in Ethylene Dimerization. Organometallics 2017, 36, 530–542. [Google Scholar] [CrossRef] [Green Version]
- Falivene, L.; Cao, Z.; Petta, A.; Serra, L.; Poater, A.; Oliva, R.; Scarano, V.; Cavallo, L. Towards the online computer-aided design of catalytic pockets. Nat. Chem. 2019, 11, 872–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitaura, K.; Morokuma, K. A new energy decomposition scheme for molecular interactions within the Hartree-Fock approximation. Int. J. Quantum Chem. 1976, 10, 325–340. [Google Scholar] [CrossRef]
- Kawamura-Kuribayashi, H.; Miyatake, T. A theoretical study of the ligand effect of thiobisphenoxytitanium complex catalyst on the catalytic activity for ethylene polymerization. J. Organomet. Chem. 2003, 674, 73–85. [Google Scholar] [CrossRef]
- Margl, P.; Deng, L.; Ziegler, T. A Unified View of Ethylene Polymerization by d0 and d0fn Transition Metals. 3. Termination of the Growing Polymer Chain. J. Am. Chem. Soc. 1999, 121, 154–162. [Google Scholar] [CrossRef]
- Devlin, F.J.; Finley, J.W.; Stephens, P.J.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields: A Comparison of Local, Nonlocal, and Hybrid Density Functionals. J. Phys. Chem. 1995, 99, 16883–16902. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Fukui, K. The path of chemical reactions-the IRC approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Active Species | Insertion Direction | Transition State | ∆Eint (kcal/mol) | ∆Edef (I) (kcal/mol) | ∆Edef (II) (kcal/mol) | ∆Edef (kcal/mol) 1 |
|---|---|---|---|---|---|---|---|
| 1 | 1A | front-side | 1TS(E–F)-fsi | −37.0 | 21.5 | 5.8 | 27.3 |
| 2 | 1A | back-side | 1TS(E–F)-bsi | −40.3 | 20.1 | 12.4 | 32.5 |
| 3 | 2A | front-side | 2TS(E–F)-fsi | −36.0 | 21.0 | 8.6 | 29.6 |
| 4 | 2A | back-side | 2TS(E–F)-bsi | −37.5 | 20.0 | 14.6 | 34.6 |
| 5 | 3A | front-side | 3TS(E–F)-fsi | −36.3 | 18.5 | 8.0 | 26.5 |
| 6 | 3A | back-side | 3TS(E–F)-bsi | −42.7 | 16.4 | 15.7 | 32.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Lai, X.; Xu, X.; So, Y.-M.; Du, Y.; Zhang, Z.; Pan, Y. Theoretical Study on Ethylene Polymerization Catalyzed by Half-Titanocenes Bearing Different Ancillary Groups. Catalysts 2021, 11, 1392. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11111392
Li Y, Lai X, Xu X, So Y-M, Du Y, Zhang Z, Pan Y. Theoretical Study on Ethylene Polymerization Catalyzed by Half-Titanocenes Bearing Different Ancillary Groups. Catalysts. 2021; 11(11):1392. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11111392
Chicago/Turabian StyleLi, Yang, Xiaoling Lai, Xiaowei Xu, Yat-Ming So, Yijing Du, Zhengze Zhang, and Yu Pan. 2021. "Theoretical Study on Ethylene Polymerization Catalyzed by Half-Titanocenes Bearing Different Ancillary Groups" Catalysts 11, no. 11: 1392. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11111392