Rational Design of Chiral Selenium-π-Acid Catalysts


Institut für Organische und Biomolekulare Chemie, Universität Göttingen, Tammannstrasse 2, 37077 Göttingen, Germany
*
Author to whom correspondence should be addressed.
Catalysts 2019, 9(2), 153; https://0-doi-org.brum.beds.ac.uk/10.3390/catal9020153
Submission received: 16 January 2019
/
Revised: 25 January 2019
/
Accepted: 28 January 2019
/
Published: 3 February 2019
(This article belongs to the Special Issue Chalcogens in Catalysis: Synthesis and Biology)
Abstract
:A series of unprecedented chiral selenium-π-acid catalysts for the asymmetric, oxidative functionalization of alkenes has been developed. In total, eleven different chiral dihydrodiselenocine and (di-)alkoxyphenyl (di)selenide motifs have been synthesized in a concise, modal, and straightforward fashion. Commercially available, non-racemic alcohols have been predominantly used as chiral building blocks for the facile assembly of the selenium-π-acid catalysts. These species have been exemplarily applied to the enantioselective intermolecular imidation and intramolecular acyloxylation of two olefins using N-fluorobenzenesulfonimide (NFSI) and ambient air, respectively, as terminal oxidants. In part, the catalysts provide very good yields of up to 99% and enantiomeric ratios of up to 83.5:16.5 under aerobic conditions.
1. Introduction
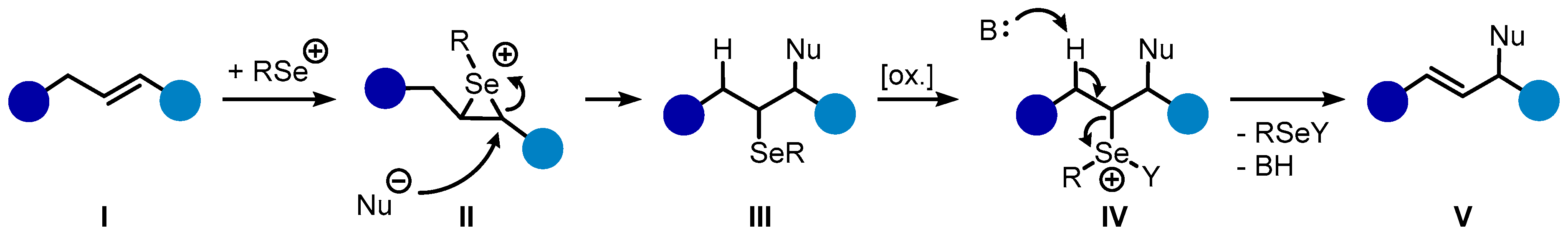
The oxidative introduction of stereochemical information into simple unsaturated hydrocarbons such as alkenes by means of catalytic processes represents a very expedient strategy in the realm of modern chemical synthesis [1]. The majority of protocols for the enantioselective oxidation of non-activated, non-aromatic C–C π-bonds are so far driven by transition metal-catalysts. Cognate organocatalytic procedures, by contrast, are comparatively rare and often require certain ancillary groups such as a carbonyl functionality in proximity to the C–C π-bond to operate effectively [2,3]. Notable exceptions to these methodological limitations are, for example, certain chalcogen Lewis-base and Lewis acid catalysts (chalcogen = S, Se) and certain halogen species for which asymmetric oxidations of electronically-unbiased olefins have been sporadically reported [4,5,6,7,8,9]. Among these transformations, those catalyzed by selenium-π-acids represent by far the smallest subcategory [10,11,12]. This circumstance is to some degree surprising since stoichiometric diastereoselective selenofunctionalizations of simple alkenes with chiral, non-racemic selenium electrophiles have been on record for quite some time [13,14,15,16,17]. In general, these reactions proceed through the stereospecific attack of a given selenium electrophile onto an alkene (I), resulting in the transient formation of seleniranium intermediate II (Scheme 1) [18,19]. Subsequent ring opening by a given nucleophile then leads to the formation of the respective addition product III in an anti-specific manner. Under oxidative conditions, a possible pathway for the removal of the selenium moiety is via elimination, thus giving enantioselective access to allylic functionalization products V. Efficient orchestration of all these steps within a single catalytic regime still constitutes a tremendous challenge.
Critical factors that play a leading role in the design of a generally applicable asymmetric selenium-π-acid catalysis concept are the conformational rigidity of the selenium species to provide sufficient stereoinduction and the catalyst compatibility with the terminal oxidants [7]. In this context, the purpose of this report is to delineate the rational design of a series of new chiral selenium-π-acid catalysts and to evaluate their catalytic potency in the enantioselective intermolecular imidation and intramolecular acyloxylation of two olefins using N-fluorobenzenesulfonimide (NFSI) and ambient air, respectively, as terminal oxidants. Notable features of the presented work are short and modal synthetic routes toward the selenium species, good catalyst compatibility with air as a terminal oxidant, and, in part, high catalytic activities (up to 99% yield).
2. Results and Discussion
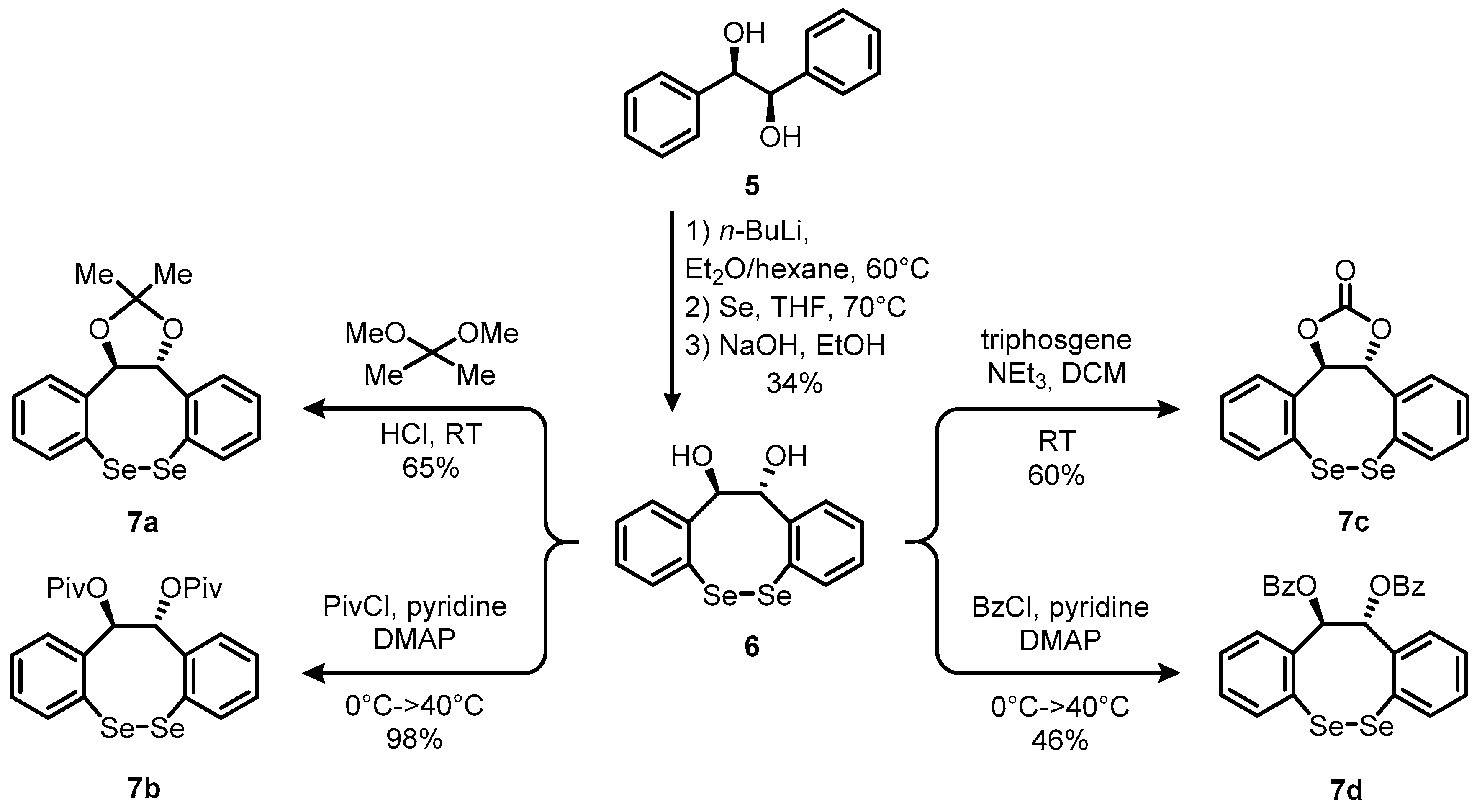
Our investigations began with a systematic study on the enantioselective allylic imidation of alkene 1 using N-fluorobenzenesulfonimide (2, NFSI) as both the terminal oxidant and source of the nitrogenous coupling partner. On the basis of our preliminary results on the racemic version of this reaction and reports from other groups [10,20], we speculated that conformationally rigidified chiral selenium-catalysts might be particularly suitable for the aspired asymmetric allylic alkene imidation [21]. Therefore, we initially tested the literature-known chiral diselenide 4 (Equation (1)) [22], which possesses a Lewis-basic side chain. Such selenium species were frequently reported to adopt significantly stabilized conformations by means of nonbonding interactions between a transiently-formed cationic selenium center and a heteroatom (e.g., oxygen or nitrogen) suitably juxtaposed in the side chain [23]. For the title reaction, benzyl pent-3-enoate (1) was chosen as a model olefin. Treatment of substrate 1 with 1.1 equivalent of NFSI in the presence of 5 mol% of catalyst 4 at ambient temperature did not result in any product formation (Equation (1)). Based on this observation, we speculated that the presence of Lewis-basic residues in the proximity of the selenium atom under the title conditions may result in the formation of catalytically inactive selenonium species [14,23]. We therefore turned our attention toward the design of complementary molecular architectures that would still display pronounced conformational rigidity. Thus, we envisioned the use of chiral dihydrodibenzodiselenocines 7 as prospective candidates (Scheme 2). Apart from the two-carbon tether, diselenocines 7 barely differ from (PhSe)2 in terms of structural features, which led us to believe that these compounds should perform equally well in the allylic imidation of alkenes. In addition, variation of the residues at the stereogenic positions should provide a versatile starting point for the customization of the selenium catalysts.
![Catalysts 09 00153 i001]()

To test our hypothesis, our campaign continued with the conversion of commercially-available (R,R)-hydrobenzoin (5) into a set of diselenocines 7a–d. Initial lithiation of (R,R)-hydrobenzoin (5) with 6.0 equivalents of n-butyl lithium, followed by quenching of the resulting polyanion with Se-powder furnished diselenocine core 6 in 34% yield (Scheme 2). As mentioned above, the diol unit offered the possibility to easily introduce different functional groups such as esters, ethers, or a cyclic carbonate into the backbone of the catalysts. Such structural modifications were anticipated to have a substantial effect on the stereochemical outcome of the title reaction. In a divergent manner, diselenocine 6 was accordingly converted into catalysts 7a–d in a single step, and in yields ranging from 46–98% using standard reaction conditions (Scheme 2).
Next, our investigations continued with the examination of chiral diselenocines 7a–d in the enantioselective imidation of alkene 1. Initially, diselenocine 7a, bearing an acetonide backbone, was used to determine any solvent effects (Table 1, entries 1–10). When the reaction was performed in hydrocarbonous solvents, such as cyclohexane and toluene (entries 1,2), product formation was only observed for toluene with an enantiomeric ratio (er) of 57:43 and a yield of 16%. Changing to ethereal solvents only led to a moderate improvement in terms of yield (up to 29%, entries 3–6). The er values were in all cases comparable to the result obtained for toluene, with a maximum er of 59.5:40.5 for THF (entry 6). Performing the title reaction in more polar solvents such as dichloromethane, nitromethane, and acetonitrile (entries 7–10) led to mixed results. While dichloromethane gave the best er value within this series (59: 41, entry 7), the yield turned out to be best in acetonitrile (50%, entry 10). At this stage, the remaining catalysts 7b–d were all tested in THF as the solvent of choice (entries 11–13). Catalysts 7b and 7d, which both possess an acyclic tether in their backbone, furnished target structure 3 in reasonable yields of 49% and 53%, respectively. However, the er values were quite similar to that of catalyst 7a. On the contrary, tetracyclic catalyst 7d provided access to allylic imide 3 in a good yield of 81% and an er of 75:25 (entry 13). Despite all our efforts, this last result could not be further improved for the allylic functionalization of alkenes using NFSI (2) as the terminal oxidant.
Considering the fact that the use of NFSI (2) as the terminal oxidant only led to moderate enantiomeric ratios, we turned our attention to a more generalized enantioselective selenocatalytic oxidation concept that—in the long run—potentially allowed for a broader scope of coupling partners to simple alkenes. Recently, our group disclosed a light-driven dual catalytic protocol that enabled for the first time the use of ambient air as the terminal oxidant in selenium-π-acid-catalyzed allylic alkene functionalizations [24,25,26,27,28]. A key asset of this strategy is the circumstance that various nucleophilic reaction partners, such as carboxylic acids, alcohols, and hydrogen phosphates, are compatible with aerobic conditions. This aspect is particularly noteworthy in the context of intermolecular processes. In the majority of traditional methods that rely on selenium-π-acid catalysis, the use of oxidants is required that simultaneously function as the source of the nucleophilic coupling partner for the alkenes (e.g., chloride nucleophile from N-chlorosuccinimide or benzenesulfonimide nucleophile from NFSI (2)) [21,29,30,31,32,33,34,35]. Against this background, we wondered whether this dual photoredox/selenium-π- acid catalysis concept could be rendered enantioselective through the use of chiral selenium species.
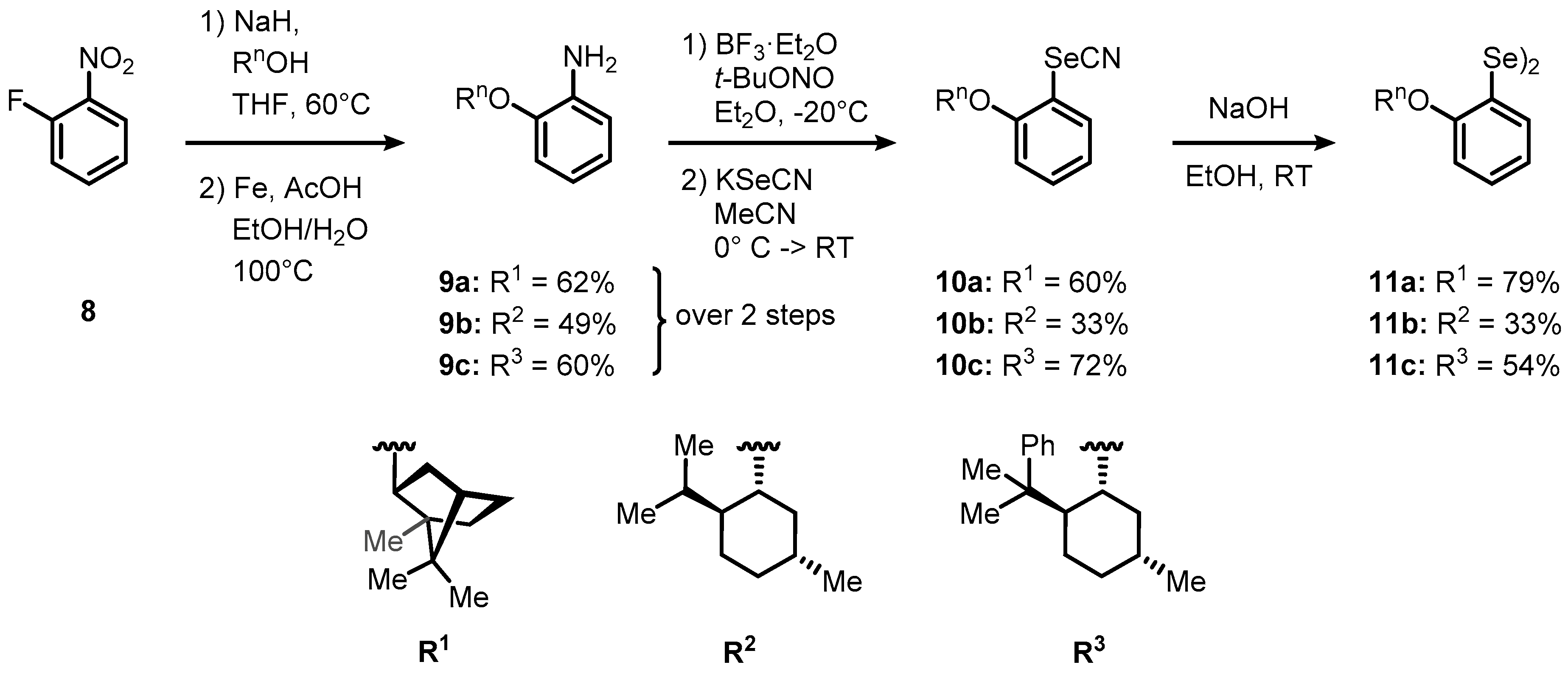
At the outset of this part of the investigation, we devised two main synthetic routes toward an extended series of C1- and C2-symmetric chiral, non-racemic diselenides (Scheme 3). For the first set of C1-symmetric catalysts, commercially available 2-fluoronitrobenzene (8) was reacted with 2.5 equivalents of a given chiral sodium alkoxide, derived from sodium hydride and the corresponding alcohol (i.e., (–)-menthol, (–)-8-phenylmenthol, and (–)-borneol) to furnish the respective aryl ethers in yields ranging from 76–99%. (Scheme 3). Subsequently, the nitro group was selectively reduced by the aid of elemental iron in acidic media, [36] furnishing anilines 9a–c in 61–75% isolated yield. The selenium group was next introduced through a Sandmeyer-type reaction sequence, in which anilines 9a–c were initially converted into their corresponding diazonium salts and then treated with 1.0 equivalent of potassium selenocyanate. Eventually, transient aryl selenocyanates 10a–c were solvolyzed in basic ethanol under aerobic atmosphere to furnish the target diselenides 11a–c in 11–47% yield over three steps.
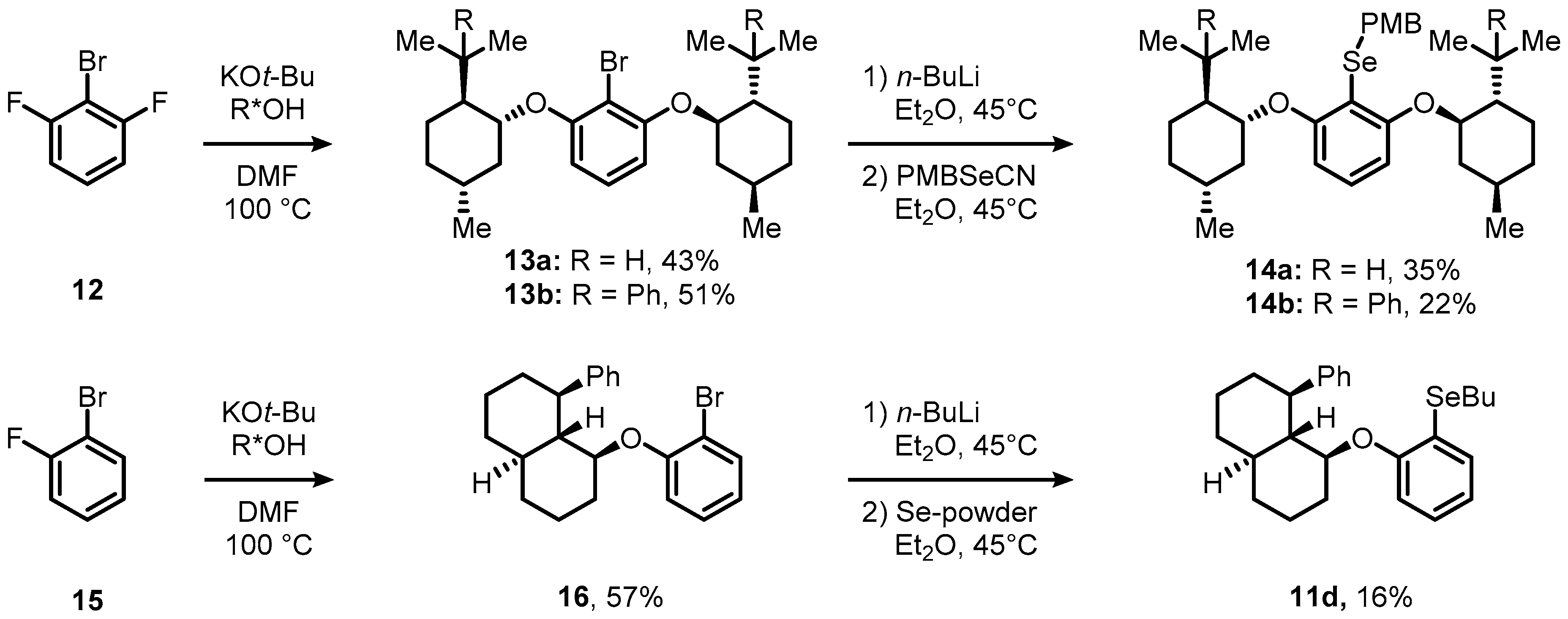
When we attempted to apply the reaction sequence depicted in Scheme 3 to the synthesis of C2-symmetric, 2,6-dialkoxyaryldiselenides, all our efforts unfortunately met with failure. We noticed that the corresponding 2,6-dialkoxybenzenediazonium salts were too stable to react with potassium selenocyanate. Thus, we used 2-bromo-1,3-difluorobenzene (12) as an alternative starting point. Substrate 12 was consequently treated either with (–)-menthol or (–)-8-phenylmenthol in the presence of 2.15 equivalents of potassium tert-butoxide in DMF at elevated temperatures to provide access to aryl ethers 13a and 13b in 43 and 51% yield, respectively (Scheme 4). Subsequent halogen-metal-exchange using n-butyl lithium and capture of the resulting aryl lithium species with para-methoxybenzylselenocyanate (PMBSeCN) provided the desired selenides 14a and 14b in yields of 22 and 35%, respectively. Since the SNAr displacement of the fluoride substituents from fluorinated bromobenzenes with sterically-demanding alkoxides proved reliable, we synthesized C1-symmetric selenide 11d by the same strategy starting from 1-bromo-2-fluorobenzene (15, Scheme 4, bottom). Treatment of the requisite decalinol with 1.15 equivalents of tert-butoxide in the presence of arene 15 in DMF led to aryl ether 16 in 57% yield. Conversion of compound 16 with 1.1 equivalents of n-butyl lithium and 3 equivalents of selenium powder resulted in the formation of arylbutyl selenide 11d in 16% yield. As shown below, compound 11d could be directly used as a reactive pre-catalyst without the necessity of removing the butyl group.
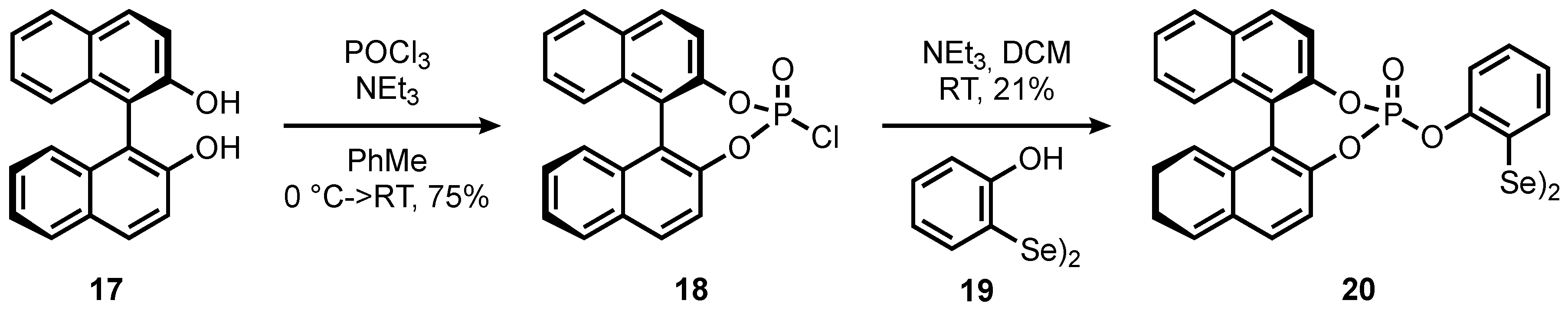
Eventually, we also wanted to incorporate a chiral, non-racemic phosphoric ester motif into the arene skeleton of the selenium-π-acid catalyst. Since a broad variety of chiral phosphoric acid derivatives are commercially available, we felt that such a catalytic motif may bear the advantage of facile structural diversification. Thus, to exemplarily probe the synthetic feasibility of such a catalyst, 2-hydroxyphenyldiselenide (19) was synthesized in 30% in a single step from 2-bromophenol (Scheme 5) [14]. The resulting diselenide was then reacted with (R)-BINOL chlorophosphoric diester (18) [37] to give corresponding chiral catalyst 20 in 21% isolated yield.
With a proper set of chiral selenium-π-acid catalysts in hand, we next examined their efficiency in the asymmetric allylic functionalization of an alkene under dual photoredox catalytic conditions (Table 2 and Table 3). As a benchmark reaction, we chose the asymmetric aerobic lactonization of 5-phenylpent-3-enoic acid (21). From previous investigations in our laboratories, we knew that 2,4,6-tris(4-anisyl)pyrylium tetrafluoroborate (TAPT, E1/2*,red = 1.74 V vs SCE) and 10-(3,5-Dimethoxyphenyl)-9-mesityl-1,3,6,8-tetramethoxyacridin-10-ium tetrafluoroborate (DMTA, E1/2*,red = 1.65 V vs SCE) were both compatible with (PhSe)2 [38,39]. We had also discovered that mono- and dialkoxy-substituted diaryl diselenides usually display oxidation potentials in the range of 0.98–1.29 V vs. SCE in acetonitrile and that the applied redox potentials during catalysis (e.g., by a suitable photoredox catalyst or by electrochemical means) need to be about 200 mV higher than the oxidation potential of the respective diselenide catalysts [40]. Consequently, we used this data to further establish our target protocol. To evaluate the effectivity of our aerobic conditions, we used chiral selenium-catalyst 23 as a reference system (Table 3), which was recently reported by Maruoka et al. and found to be highly effective in the asymmetric lactonization of enoic acids in the presence of NFSI (2) as the terminal oxidant [10]. In order to determine operable reaction conditions, we used C2-symmetric catalyst 14b as a starting point (Table 2). Conversion of 5-phenylpent-3-enoic acid (21) in the presence of 10 mol% of selenide 14b and 5 mol% of TAPT in acetonitrile under an aerial atmosphere and 465 nm irradiation (ca. 35 °C internal temp.) for 20 h led to an isolated yield of 24% and an er value of 74:26 (entry 1). Under these conditions, the starting material was not fully consumed. When we increased the reaction temperature to 50 °C and the reaction time to 96 h, we could increase the yield to 99%; however, this was at the expense of stereoinduction (er = 62:38, entry 2). Use of a modified TAPT catalyst through anion exchange (i.e., replacement of BF4– with BPh4– [TAPTP]) under otherwise unaltered reaction conditions (cf. entry 1), led to diminished yields of only 10–13% and low stereoinduction (er = 56:44; entries 3,4). These results already indicate that the counterion of the photoredox catalyst has a marked impact on the stereochemical outcome of the title reaction. Changing to DMAT as the photocatalyst and toluene as the solvent also resulted in a significant decrease of the er to 54:46 (entry 5). Other photocatalysts, such as rhodamine G and Ru(bpz)3(PF6)2, in acetonitrile at 35 and 45 °C, respectively, either led to no (entry 6) or very low yield (19%, er = 52:48, entry 7). As a result of these initial experiments, we decided to use acetonitrile and TAPT as the standard solvent and photocatalyst for the screening of the remaining (di)selenide catalysts (Table 3).
Our endeavors continued with the evaluation of the C2-symmetric catalysts 14a, 7a, and 7c. Employment of catalyst 14a resulted in a slightly better reaction outcome in terms of yield (40%) and stereoinduction (er = 77.5:22.5) compared to its phenylated congener 14b. With regard to diselenocine catalysts 7a and 7c, the former gave a notably better result (44% yield, er = 39:61) under dual photoredox/selenium-π-acid catalysis conditions than the latter (11% yield, er = 50:50). This observation is in contrast to the performance of these catalysts in the asymmetric allylic imidation using NFSI as the terminal oxidant (cf. Table 1, entries 10,13), which clearly underscores the intricate interplay of factors governing the stereoinduction under this multi-catalytic regime.
Eventually, we also evaluated C1-symmetric catalysts 11a–d and 20 under the title conditions. Catalysts 11b and 11c, which are the respective mono-substituted analogs of diselenides 14a and 14b, gave comparable yields (68% and 70%, respectively) but differed significantly in terms of er values (74.5:25.5 and 59.5:40.5, respectively). Catalyst 11c was therefore more efficient than all of the C2-symmetric catalysts. Borneol- and BINOL phosphate-substituted catalysts 11a and 20 also provided lactone 22 in very good yields of 81% and 78%, respectively, and with full conversion, albeit with poor stereoselectivity (50:50 and 55:45, respectively). When selenide 11d was used, the er reached a reasonable level of 22.5:77.5 together with a yield of 40%. In combination with the result obtained with catalyst 11c, these data seem to suggest that an arene ring in the proximity of the catalytically active selenium center leads to an increase of stereoinduction. A potential rationale for these observations could be a cation-π-interaction during the transient formation of a seleniranium intermediate—the stereodetermining event in these reactions. Conceptually related π-π-interactions have already been considered as being operative in other enantioselective transformations, [41,42,43,44] but such a non-bonding interaction has not been exploited in the realm of asymmetric aerobic selenium-π-acid catalysis before. However, further experimental and computational data will be required to either corroborate or refute this hypothesis.
Finally, we tested catalysts 4 and 23 [10]. While the former diselenide 4 suffered from rapid degradation under photoredox conditions, the latter performed particularly well at 0 °C over the course of 88 h in terms of enantiomeric ratio (83.5:16.5), albeit with a moderate yield of 44% [45]. In general, our data are in line with our initial hypothesis that conformationally rigid selenium-π-acid catalysts—either through a properly stiffened carbon backbone or by non-bonding interactions— lead to noticeable stereoinduction in the allylic oxidation of alkenes. Obviously, at this stage, significant further improvement will be required in order to obtain synthetically useful enantiomeric ratios. However, the C2-symmetric diselenocine motifs (e.g., structures 7a and 7c) and those catalysts possessing an arene ring as a potential source of cation-π-interactions (e.g., diselenides 11c and 11d) will serve in the future as highly promising scaffolds for the design of new and more efficient selenium-π-acids in our group.
3. Materials and Methods
Experimental preparation procedures of all novel compounds, copies of NMR and IR-spectra as well as HPLC chromatograms are demonstrated in the supplementary materials.
4. Conclusion
In summary, we have delineated the rational design of eleven new chiral selenium-π-acid catalysts and analyzed their performance—together with some literature-known (di)selenides—in the asymmetric allylic imidation of benzyl pent-3-enoate (1), and the aerobic lactonization of 5-phenylpent-3-enoic acid (21). The majority of catalysts tested have shown moderate to very good activity in terms of yield (up to 99%) in either of the two model transformations. With regard to stereoinduction, catalysts 11c, 11d, 14b, and 23 displayed particularly promising enantiomeric ratios of up to 77.5:22.5. Even though selenide 23 provided the highest er of 83.5:16.5 under dual photoredox catalytic conditions, this catalyst severely suffered from prolonged reaction times (88 h) and moderate yields (44%) [10]. The syntheses of the newly introduced catalyst motifs are for the most part short (2–5 steps) and predominantly involve commercially available chiral building blocks. Furthermore, our results seem to indicate that cation-π-interactions possibly play a significant role in the stereoinduction. To the best of our knowledge, this type of non-bonding interaction has not been exploited in the realm of asymmetric selenium-π-acid catalysis. Thus, future efforts within our group will be in part concerned with the systematic study of reaction and catalyst parameters (e.g., solvent polarity, counterion effects, steric and electronic features of the π-interacting arene unit within the catalyst) to eventually establish highly potent asymmetric protocols for the functionalization of simple alkenes by selenium-π-acid catalysis.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2073-4344/9/2/153/s1, Experimental preparation procedures of all novel compounds, copies of NMR and IR-spectra as well as HPLC chromatograms.
Author Contributions
Conceptualization, A.B.; experimental investigation, F.K., S.O., R.Y.N.W.; writing—original draft preparation, A.B., F.K.; writing—review and editing, A.B., F.K., S.O.; supervision, A.B.; project administration, A.B.; funding acquisition, A.B.
Funding
This research was funded by the German Research Foundation (Emmy Noether-Fellowship to A.B. (BR 4907/1), the Fonds der Chemischen Industrie (PhD Fellowship to S.O.), and the Dr. Otto Röhm Gedächtnisstiftung. The authors thank Göttingen University for financial support.
Acknowledgments
We thank Lutz Ackermann (University of Göttingen) for generous infrastructural support. We also thank Katharina Rode, Christian Schlawis, Thorben Kühl, Yue Wang, Mingyue Zhao for assisting in the synthesis of some starting materials and catalysts.
Conflicts of Interest
The authors declare no conflict of interest.
References and Notes
- Comprehensive Asymmetric Catalysis; Jacobsen, E.N.; Pfaltz, A.; Yamamoto, H. (Eds.) Springer: Berlin, Germany, 1999. [Google Scholar]
- Torres, R.R. Stereoselective Organocatalysis—Bond Formation Methodologies and Activation Modes; John Wiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Dalko, P.I. Comprehensive Enantioselective Organocatalysis: Catalysts, Reactions, and Applications; WileyalystVerlag GmbH & Co. KGaA: Weinheim, Germany, 2013. [Google Scholar]
- Denmark, S.E.; Kuester, W.E.; Burk, M.T. Catalytic, Asymmetric Halofunctionalization of Alkenes—A Critical Perspective. Angew. Chem. Int. Ed. 2012, 51, 10938–10953. [Google Scholar] [CrossRef]
- Tan, C.K.; Yeung, Y.-Y. Recent Advances in Stereoselective Bromofunctionalization of Alkenes using N-Bromoamide Reagents. Chem. Commun. 2013, 49, 7985–7996. [Google Scholar] [CrossRef] [PubMed]
- Breder, A.; Ortgies, S. Recent Developments in Sulfur- and Selenium-Catalyzed Oxidative and Isohypsic Functionalization Reactions of Alkenes. Tetrahedron Lett. 2015, 56, 2843–2852. [Google Scholar] [CrossRef]
- Ortgies, S.; Breder, A. Oxidative Alkene Functionalizations via Selenium-π-Acid Catalysis. ACS Catal. 2017, 7, 5828–5840. [Google Scholar] [CrossRef]
- Romero, R.M.; Wöste, T.H.; Muñiz, K. Vicinal Difunctionalization of Alkenes with Iodine(III) Reagents and Catalysts. Chem. Asian J. 2014, 9, 972–983. [Google Scholar] [CrossRef] [PubMed]
- Haubenreisser, S.; Wöste, T.H.; Martínez, C.; Ishihara, K.; Muñiz, K. Structurally Defined Molecular Hypervalent Iodine Catalysts for Intermolecular Enantioselective Reactions. Angew. Chem. Int. Ed. 2016, 55, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, Y.; Hashimoto, T.; Maruoka, K. A Chiral Electrophilic Selenium Catalyst for Highly Enantioselective Oxidative Cyclization. J. Am. Chem. Soc. 2016, 138, 5206–5209. [Google Scholar] [CrossRef]
- Wirth, T.; Häuptli, S.; Leuenberger, M. Catalytic Asymmetric Oxyselenenylation–Elimination Reactions Using Chiral Selenium Compounds. Tetrahedron 1998, 9, 547–550. [Google Scholar] [CrossRef]
- Fujita, K.; Iwaoka, M.; Tomoda, S. Synthesis of Diaryl Diselenides Having Chiral Pyrrolidine Rings with C2 Symmetry. Their Application to the Asymmetric Methoxyselenenylation of trans-β-Methylstyrenes. Chem. Lett. 1994, 23, 923–926. [Google Scholar] [CrossRef]
- Tiecco, M.; Testaferri, L.; Santi, C.; Tomassini, C.; Marini, F.; Bagnoli, L.; Temperini, A. Asymmetric Azidoselenenylation of Alkenes: A Key Step for the Synthesis of Enantiomerically Enriched Nitrogen-Containing Compounds. Angew. Chem. Int. Ed. 2003, 42, 3131–3133. [Google Scholar] [CrossRef]
- Zhao, L.; Li, Z.; Wirth, T. Asymmetric Methoxyselenenylations with Chiral Selenium Electrophiles. Eur. J. Org. Chem. 2011, 2011, 7080–7082. [Google Scholar] [CrossRef]
- Browne, D.; Wirth, T. New Developments with Chiral Electrophilic Selenium Reagents. Curr. Org. Chem. 2006, 10, 1893–1903. [Google Scholar] [CrossRef]
- Höltzle, G.; Jenny, W. Zur Kenntnis der Sulfen und Selenensäuren und ihrer Derivate: Additions- und Substitutionsreaktionen organischer Selenverbindungen mit unpolaren und polaren Äthylenen. Helv. Chim. Acta 1958, 73, 593–603. [Google Scholar] [CrossRef]
- Deziel, R.; Goulet, S.; Grenier, L.; Bordeleau, J.; Bernier, J. Asymmetric Selenomethoxylation of Olefins Involving a Chiral C2 Symmetrical Electrophilic Organoselenium Reagent. J. Org. Chem. 1993, 58, 3619–3621. [Google Scholar] [CrossRef]
- Denmark, S.E.; Collins, W.R. Lewis Base Activation of Lewis Acids: Development of a Lewis Base Catalyzed Selenolactonization. Org. Lett. 2007, 9, 3801–3804. [Google Scholar] [CrossRef] [PubMed]
- Denmark, S.E.; Edwards, M.G. On the Mechanism of the Selenolactonization Reaction with Selenenyl Halides. J. Org. Chem. 2006, 71, 7293–7306. [Google Scholar] [CrossRef] [PubMed]
- Wirth, T. Organoselenium Chemistry in Stereoselective Reactions. Angew. Chem. Int. Ed. 2000, 39, 3740–3749. [Google Scholar] [CrossRef]
- Trenner, J.; Depken, C.; Weber, T.; Breder, A. Direct Oxidative Allylic and Vinylic Amination of Alkenes through Selenium Catalysis. Angew. Chem. Int. Ed. 2013, 52, 8952–8956. [Google Scholar] [CrossRef] [PubMed]
- Braga, A.L.; Silva, S.J.N.; Lüdtke, D.S.; Drekener, R.L.; Silveira, C.C.; Rocha, J.B.T.; Wessjohann, L.A. Chiral Diselenide Ligands for the Asymmetric Copper-Catalyzed Conjugate Cddition of Grignard Reagents to Enones. Tetrahedron Lett. 2002, 43, 7329–7331. [Google Scholar] [CrossRef]
- Mukherjee, A.J.; Zade, S.S.; Singh, H.B.; Sunoj, R.B. Organoselenium Chemistry: Role of Intramolecular Interactions. Chem. Rev. 2010, 110, 4357–4416. [Google Scholar] [CrossRef]
- Rode, K.; Palomba, M.; Ortgies, S.; Rieger, R.; Breder, A. Aerobic Allylation of Alcohols with Non-Activated Alkenes Enabled by Light-Driven Selenium-π-Acid Catalysis. Synthesis 2018, 50, 3875–3885. [Google Scholar] [CrossRef]
- Ortgies, S.; Depken, C.; Breder, A. Oxidative Allylic Esterification of Alkenes by Cooperative Selenium-Catalysis Using Air as the Sole Oxidant. Org. Lett. 2016, 18, 2856–2859. [Google Scholar] [CrossRef] [PubMed]
- Depken, C.; Krätzschmar, F.; Rieger, R.; Rode, K.; Breder, A. Photocatalytic Aerobic Phosphatation of Alkenes. Angew. Chem. Int. Ed. 2018, 57, 2459–2463. [Google Scholar] [CrossRef] [PubMed]
- Ortgies, S.; Rieger, R.; Rode, K.; Koszinowski, K.; Kind, J.; Thiele, C.M.; Rehbein, J.; Breder, A. Mechanistic and Synthetic Investigations on the Dual Selenium-π-Acid/Photoredox Catalysis in the Context of the Aerobic Dehydrogenative Lactonization of Alkenoic Acids. ACS Catal. 2017, 7, 7578–7586. [Google Scholar] [CrossRef]
- Leisering, S.; Riaño, I.; Depken, C.; Gross, L.J.; Weber, M.; Lentz, D.; Zimmer, R.; Stark, C.B.W.; Breder, A.; Christmann, M. Synthesis of (+)-Greek Tobacco Lactone via a Diastereoablative Epoxidation and a Selenium-Catalyzed Oxidative Cyclization. Org. Lett. 2017, 19, 1478–1481. [Google Scholar] [CrossRef] [PubMed]
- Hori, T.; Sharpless, K.B. Conversion of Allylic Phenylselenides to the Rearranged Allylic Chlorides by N-Chlorosuccinimide. Mechanism of Selenium-Catalyzed Allylic Chlorination of beta-Pinene. J. Org. Chem. 1979, 44, 4208–4210. [Google Scholar] [CrossRef]
- Tunge, J.A.; Mellegaard, S.R. Selective Selenocatalytic Allylic Chlorination. Org. Lett. 2004, 6, 1205–1207. [Google Scholar] [CrossRef] [PubMed]
- Mellegaard, S.R.; Tunge, J.A. Selenium-Catalyzed Halolactonization: Nucleophilic Activation of Electrophilic Halogenating Reagents. J. Org. Chem. 2004, 69, 8979–8981. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Guo, R.; Zhao, X. Organoselenium-Catalyzed Regioselective C-H Pyridination of 1,3-Dienes and Alkenes. Angew. Chem. Int. Ed. 2017, 56, 3201–3205. [Google Scholar] [CrossRef]
- Iwaoka, M.; Tomoda, S. Catalytic Conversion of Alkenes into Allylic Ethers and Esters using Diselenides having Internal Tertiary Amines. J. Chem. Soc. Chem. Commun. 1992, 1165–1167. [Google Scholar] [CrossRef]
- Cresswell, A.J.; Eey, S.T.-C.; Denmark, S.E. Catalytic, Stereospecific syn-Dichlorination of Alkenes. Nat. Chem. 2015, 7, 146–152. [Google Scholar] [CrossRef]
- Niyomura, O.; Cox, M.; Wirth, T. Electrochemical Generation and Catalytic Use of Selenium Electrophiles. Synlett 2006, 2, 251–254. [Google Scholar] [CrossRef]
- Altermann, S.M.; Richardson, R.D.; Page, T.K.; Schmidt, R.K.; Holland, E.; Mohammed, U.; Paradine, S.M.; French, A.N.; Richter, C.; Bahar, A.M.; et al. Catalytic Enantioselective α-Oxysulfonylation of Ketones Mediated by Iodoarenes. Eur. J. Org. Chem. 2008, 2008, 5315–5328. [Google Scholar] [CrossRef]
- Kattuboina, A.; Li, G. Chiral N-Phosphonyl Imine Chemistry: New Reagents and their Applications for Asymmetric Reactions. Tetrahedron Lett. 2008, 49, 1573–1577. [Google Scholar] [CrossRef]
- Martiny, M.; Steckhan, E.; Esch, T. Cycloaddition Reactions Initiated by Photochemically Excited Pyrylium Salts. Chem. Ber. 1993, 126, 1671–1682. [Google Scholar] [CrossRef]
- Joshi-Pangu, A.; Lévesque, F.; Roth, H.G.; Oliver, S.F.; Campeau, L.-C.; Nicewicz, D.; DiRocco, D.A. Acridinium-Based Photocatalysts: A Sustainable Option in Photoredox Catalysis. J. Org. Chem. 2016, 81, 7244–7249. [Google Scholar] [CrossRef]
- Wilken, M.; Ortgies, S.; Breder, A.; Siewert, I. Mechanistic Studies on the Anodic Functionalization of Alkenes Catalyzed by Diselenides. ACS Catal. 2018, 8, 10901–10912. [Google Scholar] [CrossRef]
- Cardoso do Vale, M.L.; Rodríguez-Borges, H.E.; Caamaño, O.; Fernández, F.; García-Mera, X. The Use of (−)-8-Phenylisoneomenthol and (−)-8-Phenylmenthol in the Enantioselective synthesis of 3-functionalized 2-azabicyclo[2.2.1]heptane derivatives via aza-Diels–Alder reaction. Tetrahedron 2006, 62, 9475–9482. [Google Scholar] [CrossRef]
- Whitesell, J.K.; Lawrence, R.M.; Chen, H.H. Auxiliary Structure and Asymmetric Induction in the Ene Reactions of Chiral Glyoxylates. J. Org. Chem. 1986, 51, 4779–4784. [Google Scholar] [CrossRef]
- Corey, E.J.; Ensley, H.E. Preparation of an Optically Active Prostaglandin Intermediate via Asymmetric Induction. J. Am. Chem. Soc. 1975, 97, 6908–6909. [Google Scholar] [CrossRef]
- Letzel, M.C.; Schäfer, H.J.; Fröhlich, R. Diastereoselective Anodic Hetero- and Homo-Coupling of Menthol-, 8-Methylmenthol- and 8-Phenylmenthol-2-Alkylmalonates. Beilstein J. Org. Chem. 2017, 13, 33–42. [Google Scholar] [CrossRef]
- Maruoka et al. reported an e.r. of 97.5:2.5 for the same substrate when NFSI was used as the terminal oxidant at room temperature in toluene (cf. Ref. [10]).
Scheme 1.
A simplified mechanistic pathway for the allylic functionalization of alkenes by selenium electrophiles.
Scheme 1.
A simplified mechanistic pathway for the allylic functionalization of alkenes by selenium electrophiles.

Scheme 2.
The synthetic route toward chiral, non-racemic dihydrodibenzodiselenocines 7a–d.

Scheme 3.
The synthetic route toward chiral, non-racemic diselenides 11a–c.

Scheme 4.
Synthetic route toward chiral, non-racemic diselenides 11d and 14a,b.

Scheme 5.
The synthetic route toward chiral, non-racemic diselenide 20.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Solvent and catalyst screening for the enantioselective imidation.

| Entry | Se-Catalyst | Solvent | er | Yield [%] |
|---|---|---|---|---|
| 1 | 7a | CyH | - | 0% |
| 2 | 7a | PhMe | 57:43 | 16% |
| 3 | 7a | Et2O | 57:43 | 27% |
| 4 | 7a | 1,4-dioxane | 57.5:42.5 | 17% |
| 5 | 7a | MTBE | 58:42 | 28% |
| 6 | 7a | THF | 59.5:40.5 | 29% |
| 7 | 7a | DCM | 59:41 | 18% |
| 8 | 7a | MeNO2 | 54:46 | 16% |
| 9 | 7a | THF/MeCN | 53.5:46.5 | 37% |
| 10 | 7a | MeCN | 51.5:48.5 | 50% |
| 11 | 7b | THF | 54:46 | 49% |
| 12 | 7d | THF | 58:42 | 52% |
| 13 | 7c | THF | 75:25 | 81% |
Table 2.
Screening for proper reaction conditions for the enantioselective aerobic lactonization of 5-phenylpent-3-enoic acid (21).
Table 2.
Screening for proper reaction conditions for the enantioselective aerobic lactonization of 5-phenylpent-3-enoic acid (21).

| Entry | Photocatalyst | Solvent | T | t | Yield | er4 |
|---|---|---|---|---|---|---|
| 1 | TAPT | MeCN | 35 °C | 20 h | 24% 1 | 74:26 |
| 2 | TAPT | MeCN | 50 °C | 96 h | 99% 2 | 62:38 |
| 3 | TAPTP | MeCN | 35 °C | 17 h | 13% 2 | 56:44 |
| 4 | TAPTP 3 | MeCN | 35 °C | 18 h | 10% 2 | 56:44 |
| 5 | DMAT | PhMe | 35 °C | 16 h | 33% 1 | 54:46 |
| 6 | rhodamine G | MeCN | 35 °C | 16 h | 0% | nd |
| 7 | Ru(bpz)3(PF6)2 | MeCN | 45 °C | 16 h | 19% 2 | 52:48 |
Reactions were carried out in a 0.1 m solution in MeCN using 5 mol% of diselenide catalyst 14b and 5 mol% of the corresponding photocatalyst under air with LED irradiation (465 nm). 1 Incomplete conversion; 2 Complete conversion; TAPTP = 2,4,6-tris(4-anisyl)pyrylium tetraphenylborate. 3 Reaction was performed with pre-dried solvent. 4 The depicted configuration of 22 refers to the major enantiomer and is in accordance with the literature [10].
Table 3.
Screening of selenium-π-acid catalysts in the enantioselective aerobic lactonization of 5-phenylpent-3-enoic acid (21).
Table 3.
Screening of selenium-π-acid catalysts in the enantioselective aerobic lactonization of 5-phenylpent-3-enoic acid (21).

 |
Reactions were carried out in a 0.1 m solution in MeCN using in each case 5 mol% of diselenide catalyst or 10 mol% of the monoselenide catalyst and 5 mol% of TAPT under air with LED irradiation (465 nm; cf. Table 2, entry 1). The depicted configuration of 22 refers to the major enantiomer and is in accordance with the literature [10]. aWith this catalyst, the enantiomer of compound 22 was predominantly formed.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Krätzschmar, F.; Ortgies, S.; Willing, R.Y.N.; Breder, A. Rational Design of Chiral Selenium-π-Acid Catalysts. Catalysts 2019, 9, 153. https://0-doi-org.brum.beds.ac.uk/10.3390/catal9020153
AMA Style
Krätzschmar F, Ortgies S, Willing RYN, Breder A. Rational Design of Chiral Selenium-π-Acid Catalysts. Catalysts. 2019; 9(2):153. https://0-doi-org.brum.beds.ac.uk/10.3390/catal9020153
Chicago/Turabian StyleKrätzschmar, Felix, Stefan Ortgies, Robert Y. N. Willing, and Alexander Breder. 2019. "Rational Design of Chiral Selenium-π-Acid Catalysts" Catalysts 9, no. 2: 153. https://0-doi-org.brum.beds.ac.uk/10.3390/catal9020153
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.