
Rapid Identification of New Biomarkers for the Classification of GM1 Type 2 Gangliosidosis Using an Unbiased 1H NMR-Linked Metabolomics Strategy


Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Approval
2.2. Collection of Human Blood Samples
2.3. Preparation of Human Blood Plasma Samples for 1H NMR Analysis
2.4. 1H NMR Analysis of Plasma Samples
2.5. Preprocessing of 1H NMR Biofluid Datasets
2.6. Statistical and Computational Intelligence Analysis Strategies Employed
2.7. Model Validation
2.8. QMSEA
2.9. MV Power Calculations
3. Results
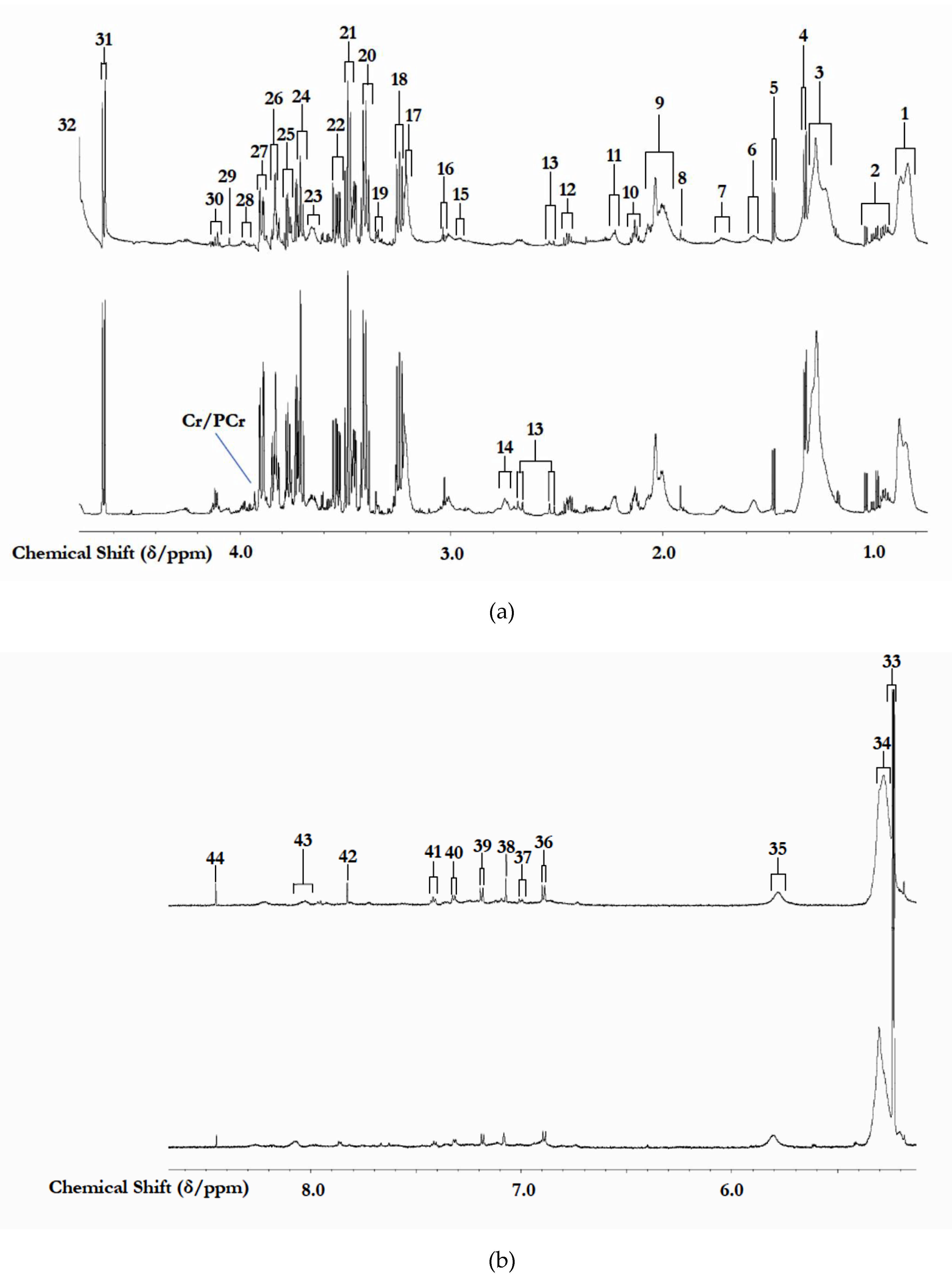
3.1. 1H NMR Analysis of Study Biofluids Collected
3.2. Univariate and Preliminary Metabolomics Analysis of Biofluid 1H NMR Profile Datasets
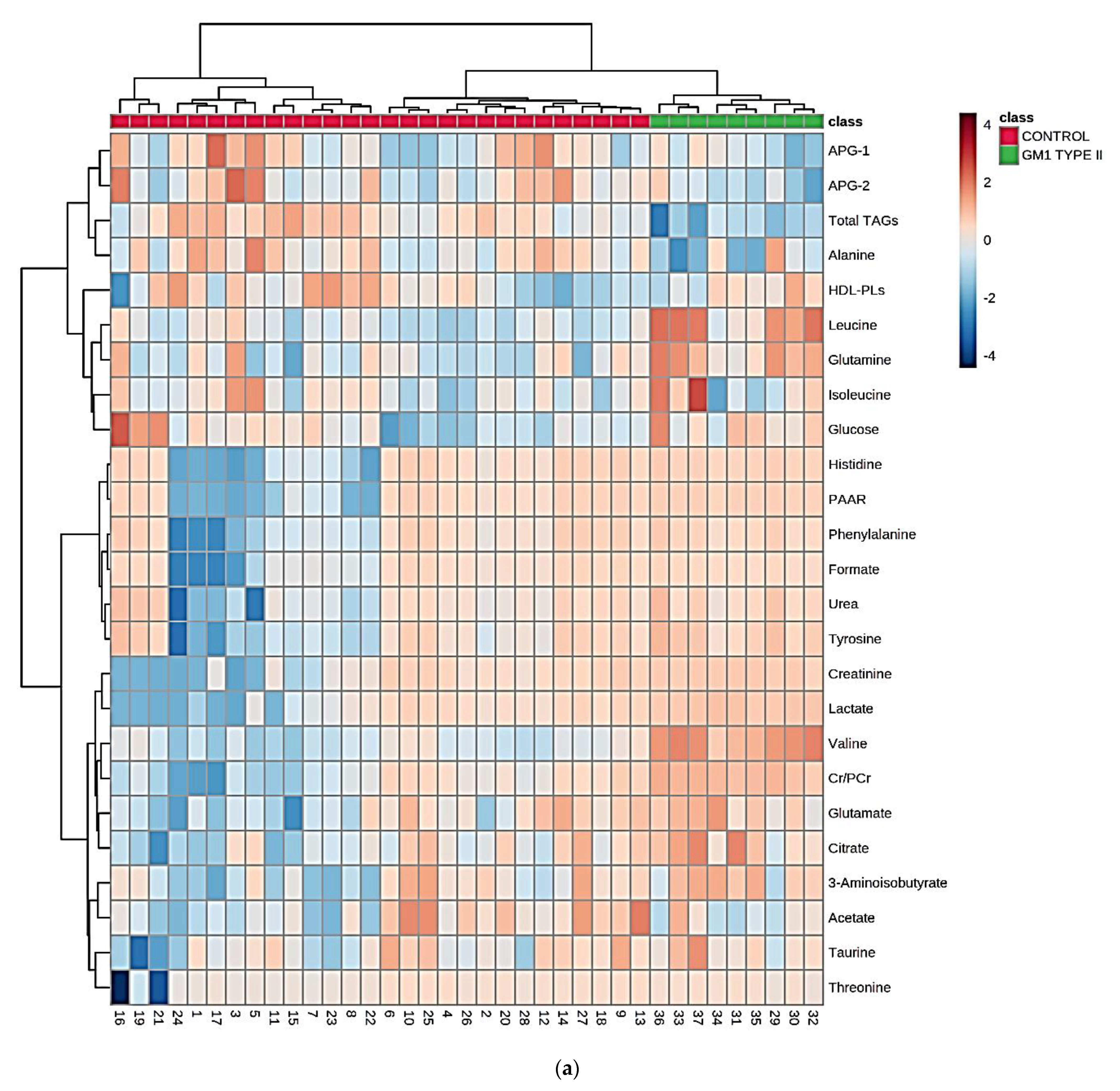
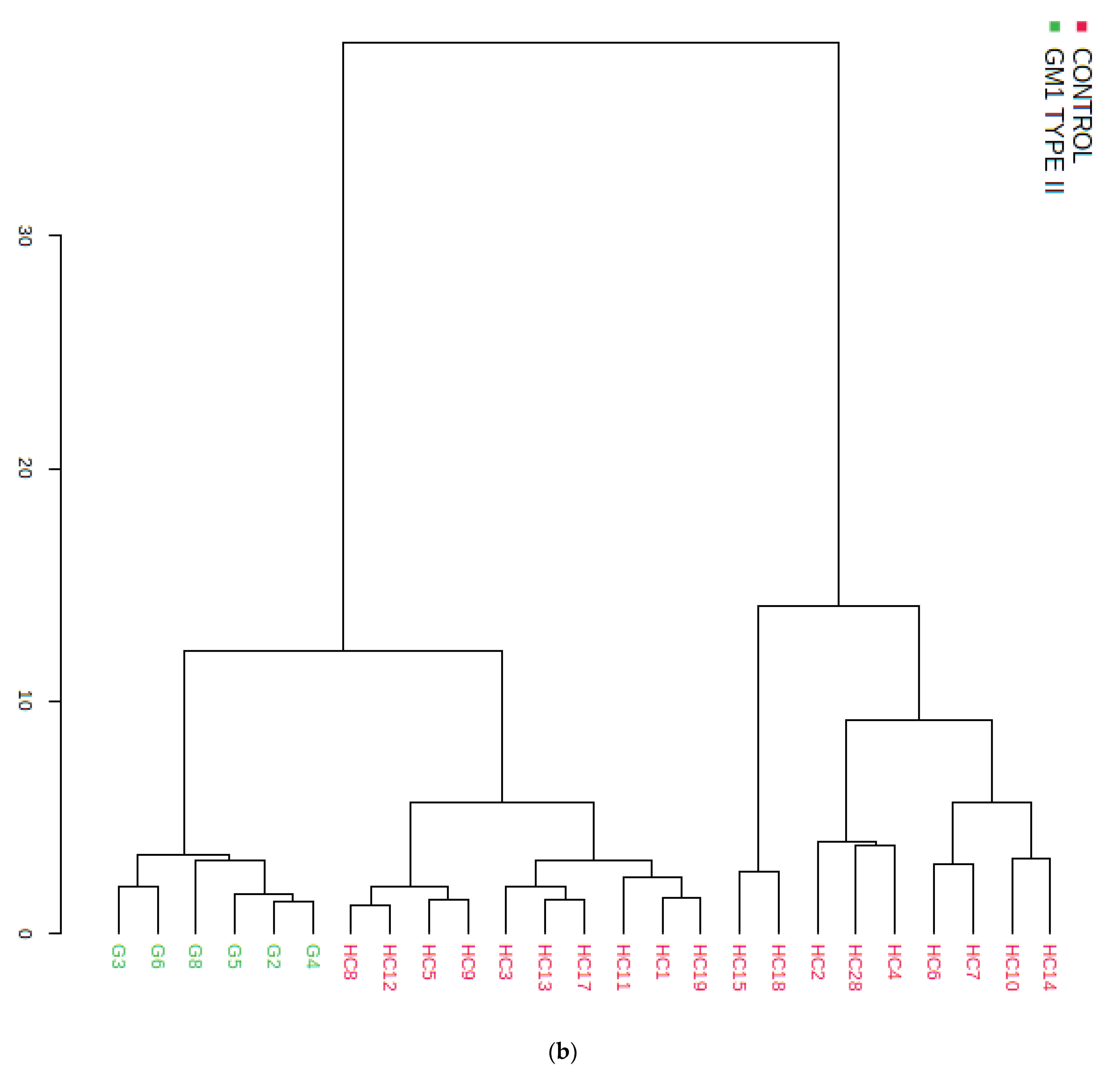
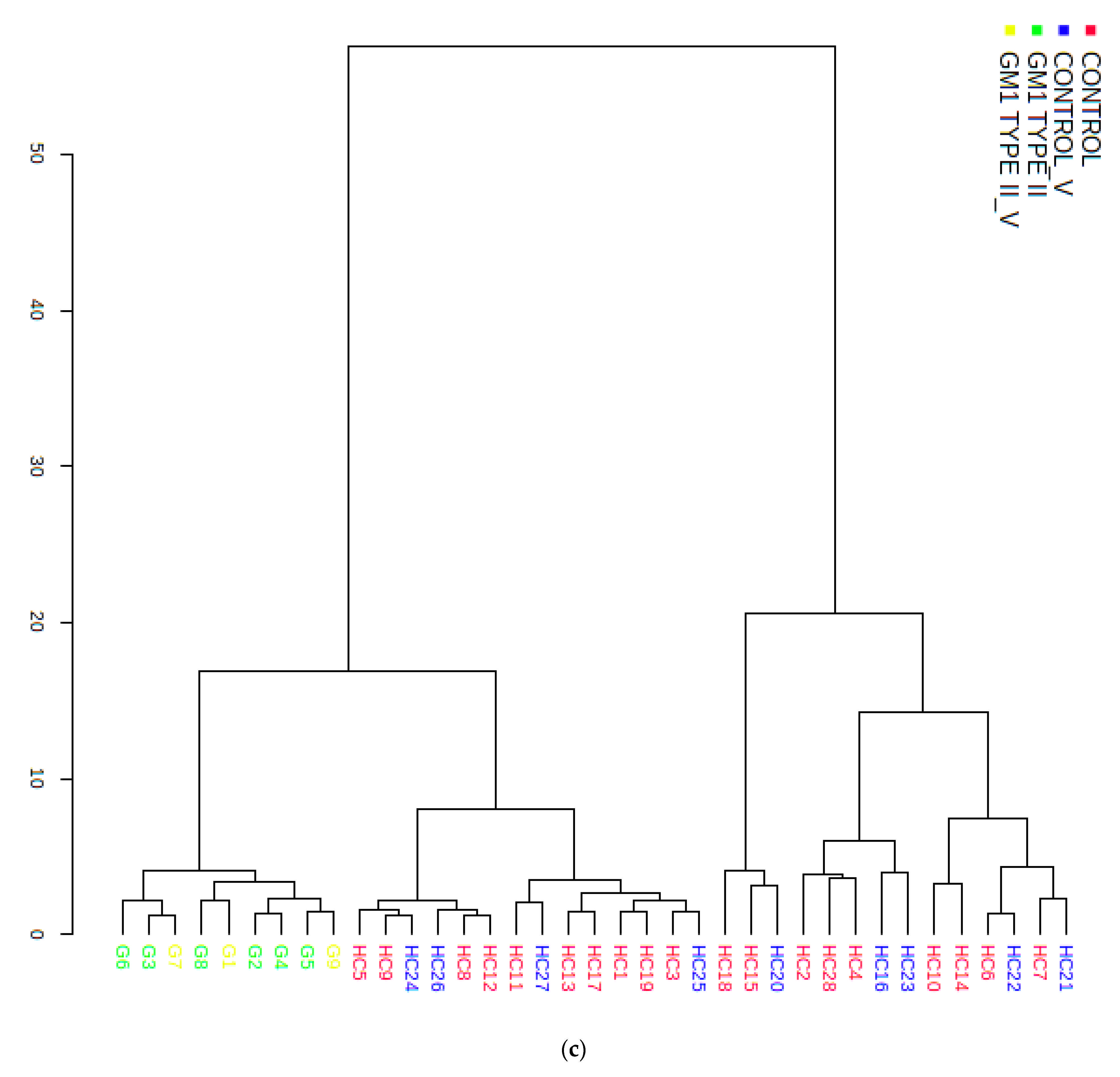
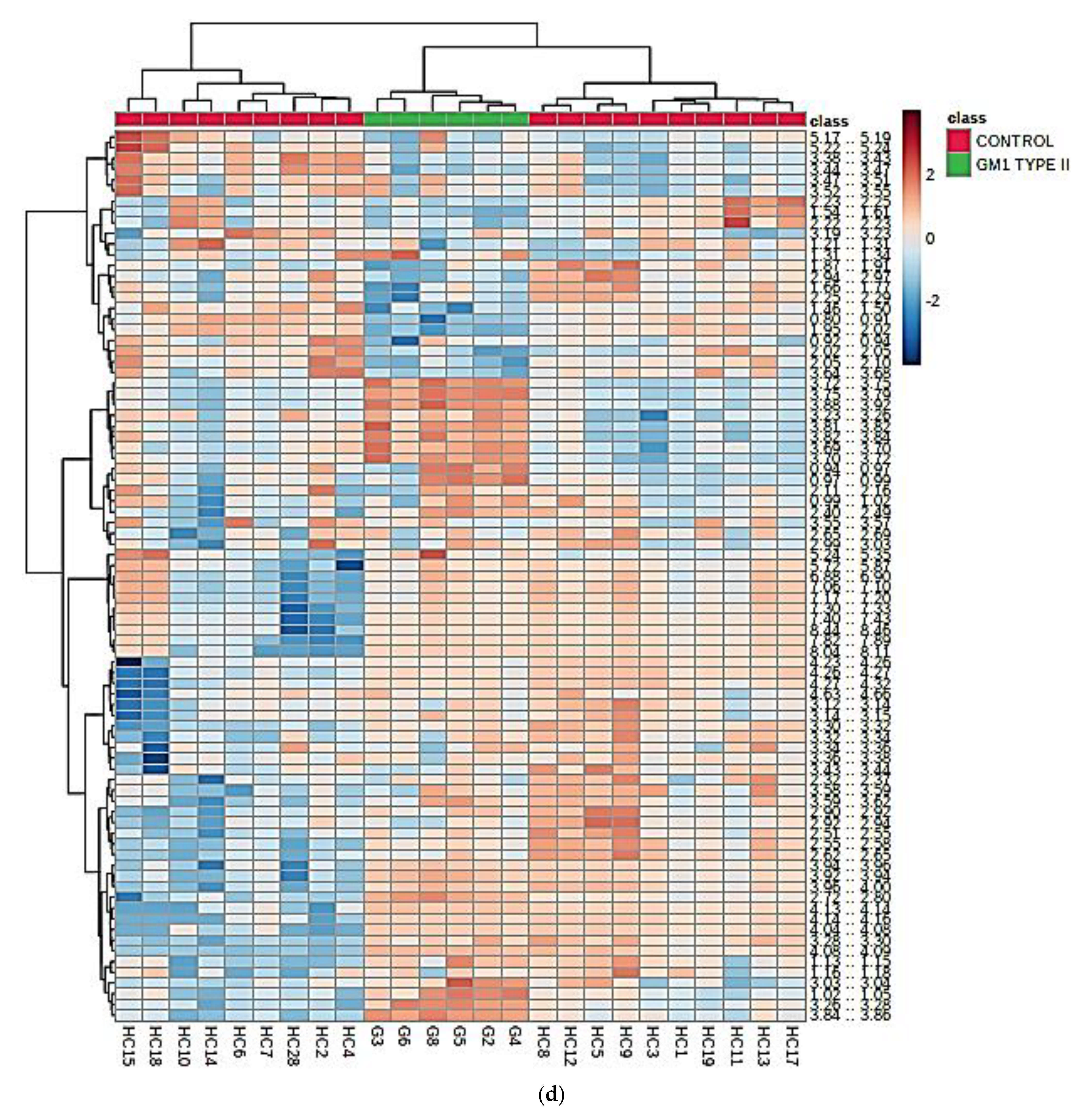
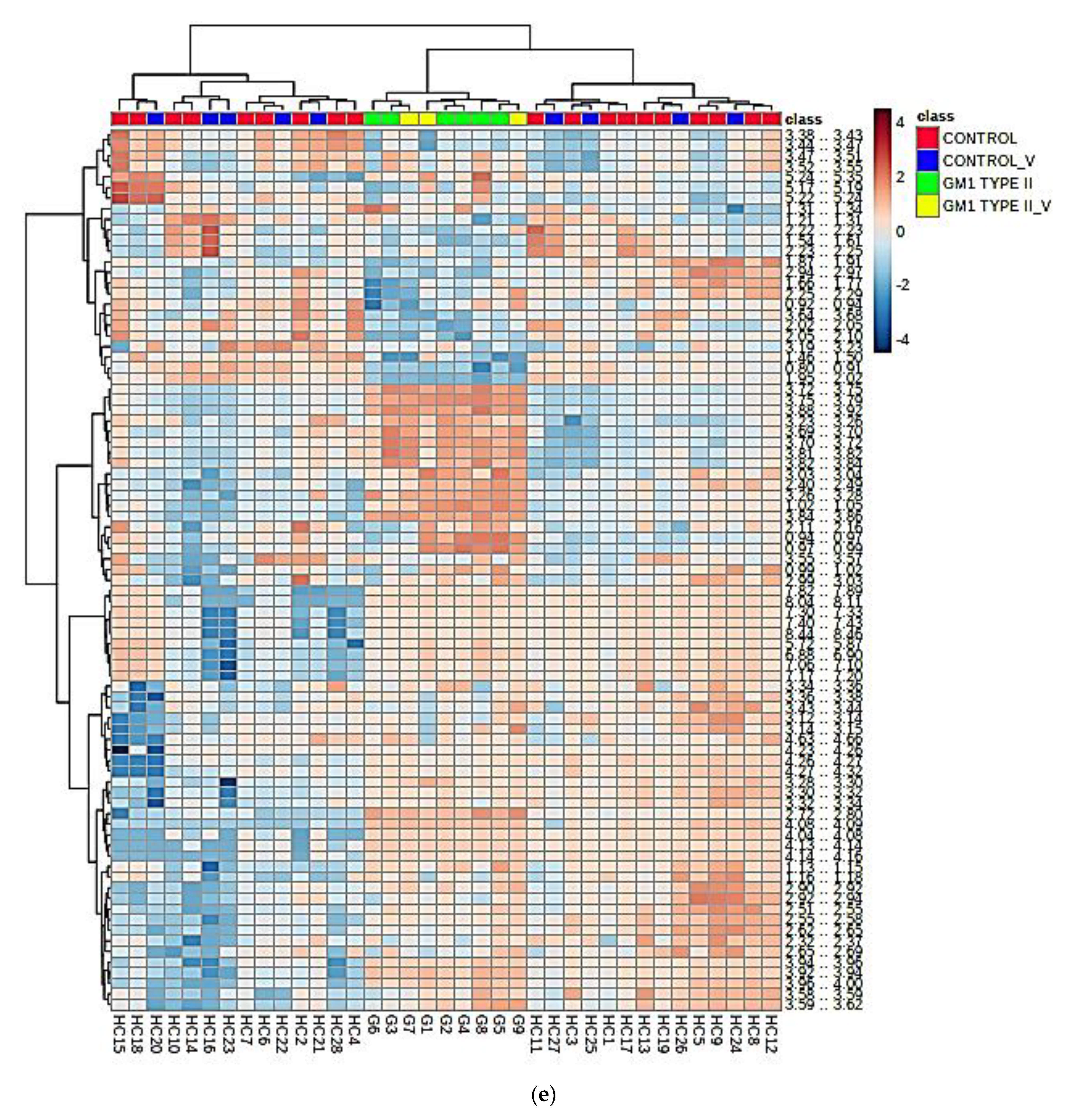
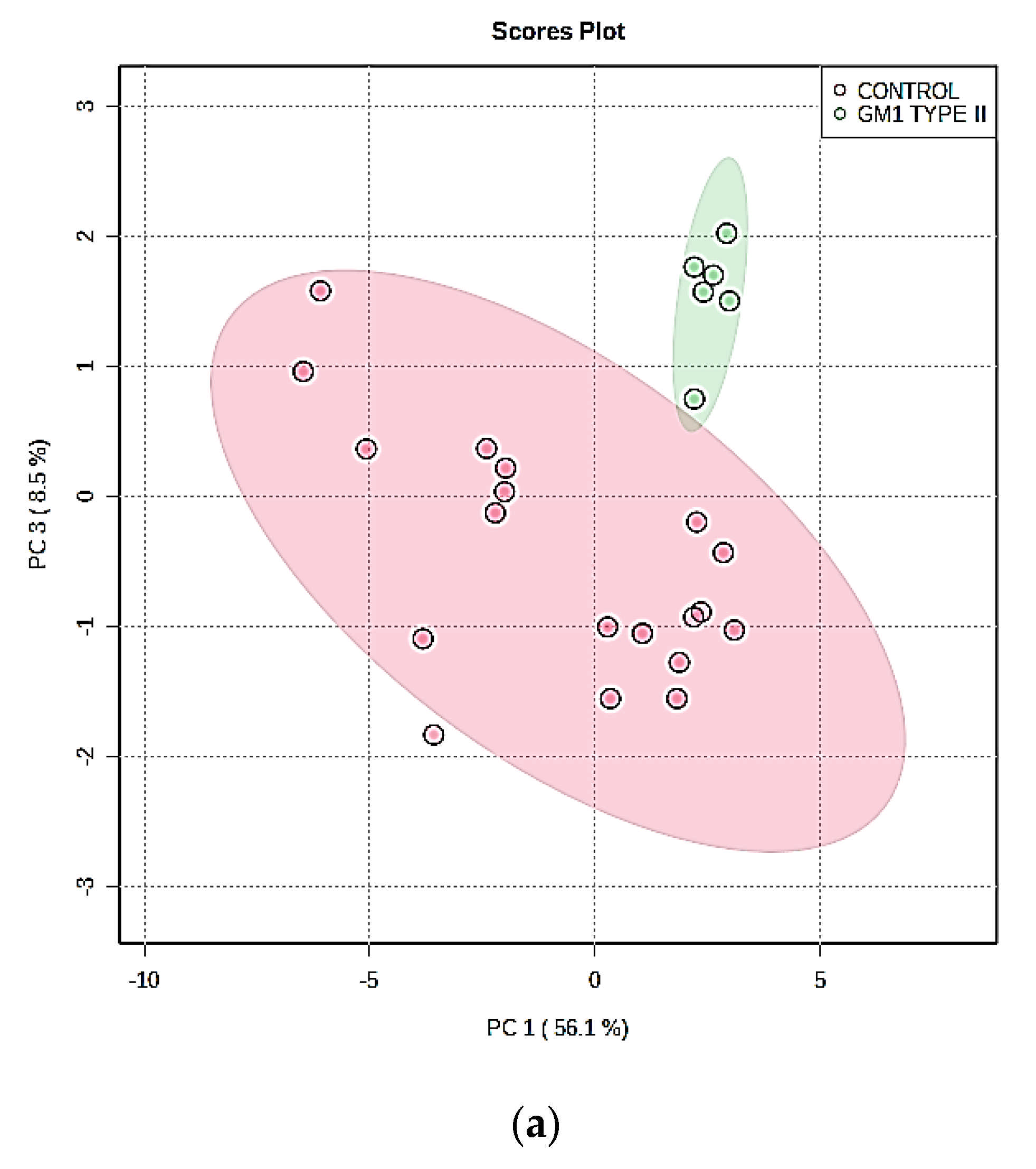
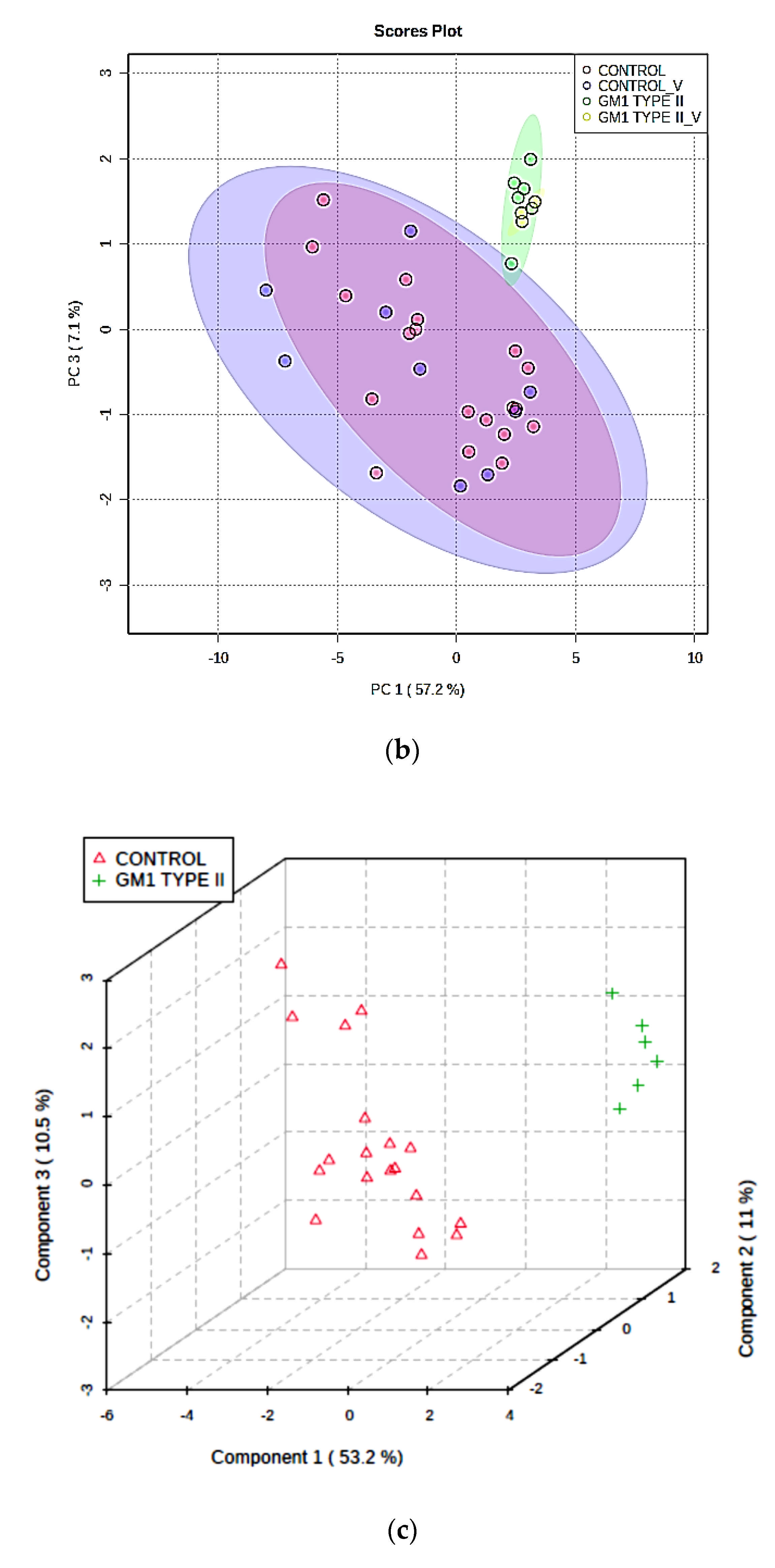
3.3. Multivariate Metabolomics Analysis of Biofluid 1H NMR Profile Datasets
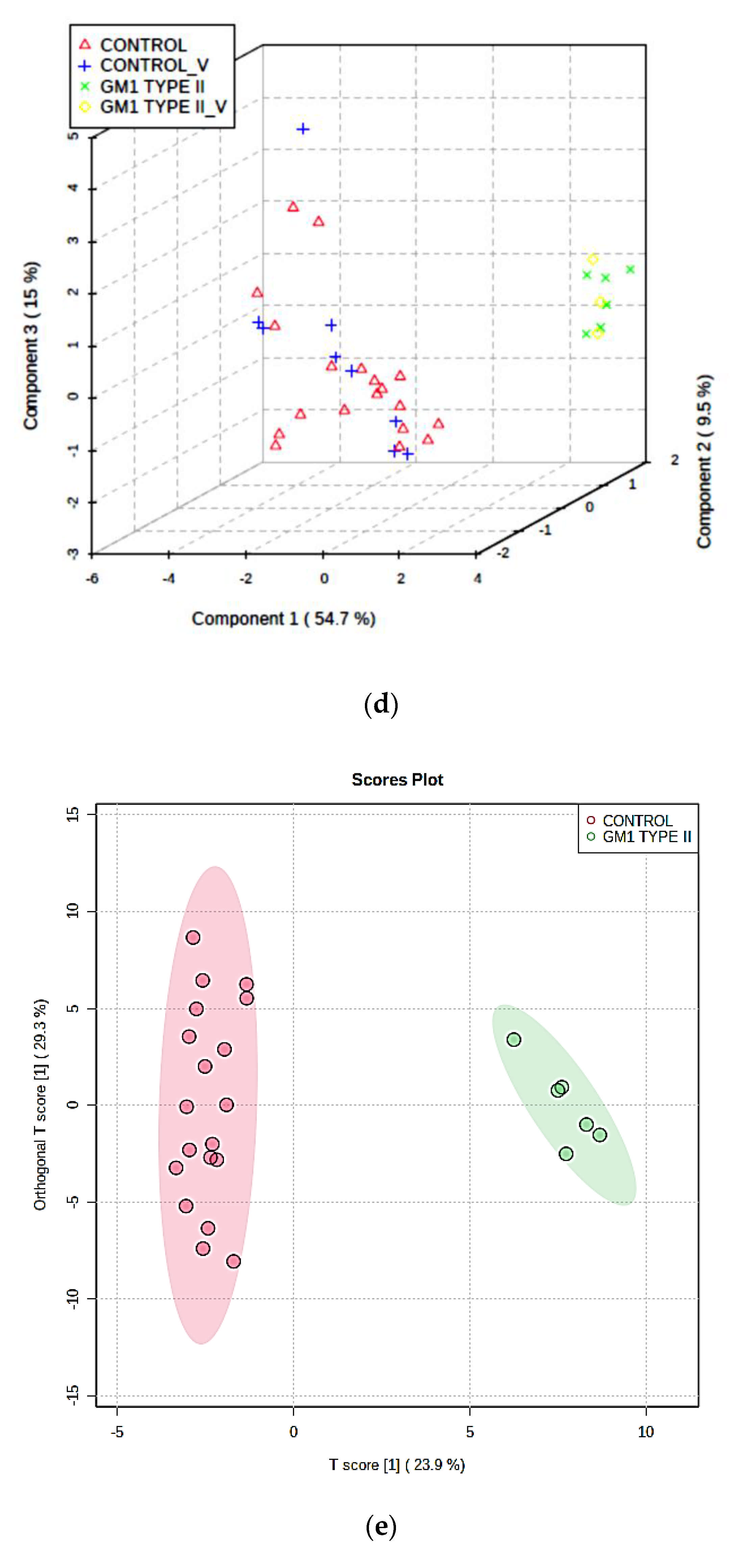
3.3.1. PCA, PLS-DA, and OPLS-DA
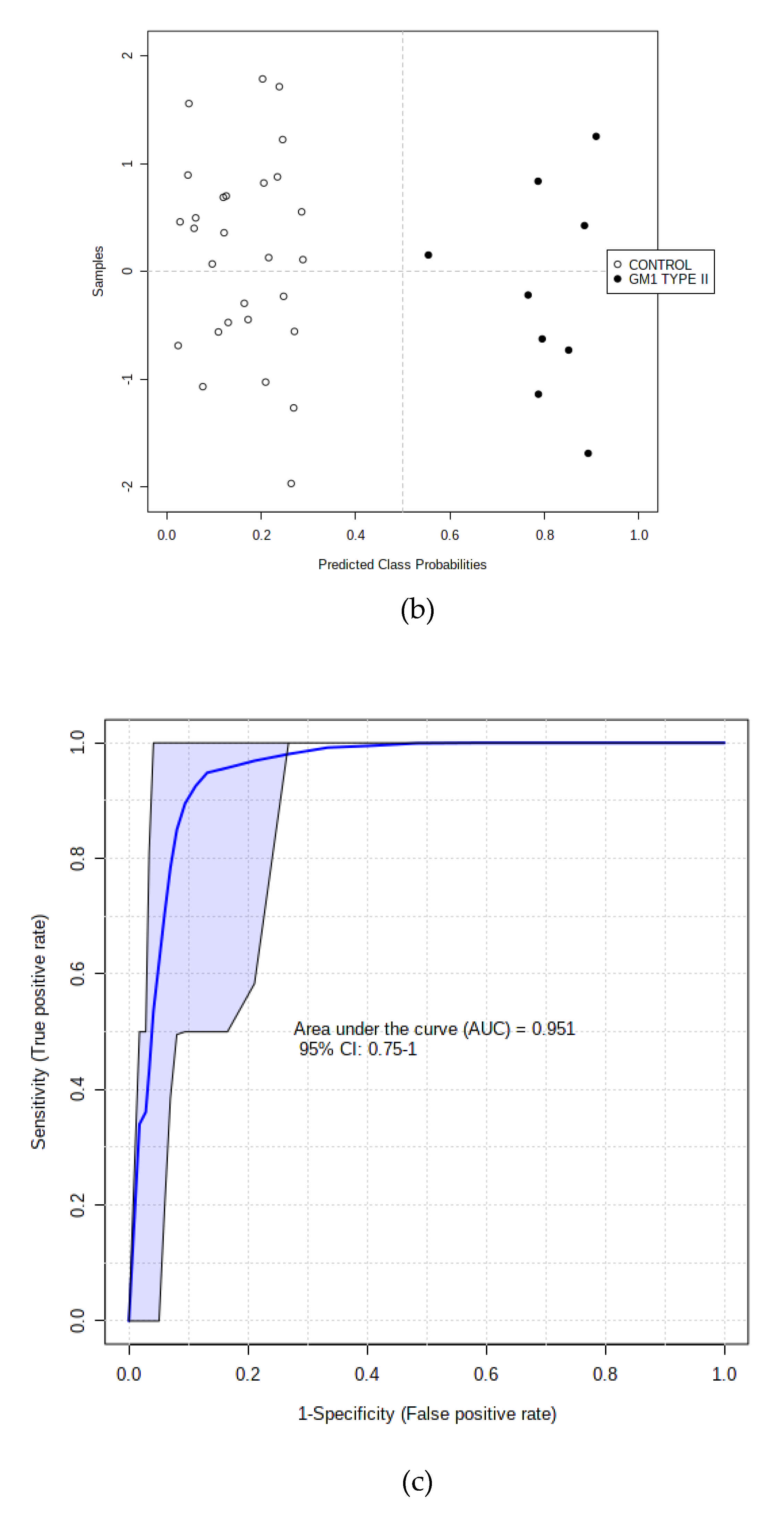
3.3.2. Supporting Model Validation and Predictive Accuracy Estimates
3.3.3. RF and SVM Classification Strategies
3.3.4. AUROC Testings of Complete and Hold-out Validation Datasets
3.4. Further Investigation of Potential Metabolic Influences Exerted by Participant Ages and Genders
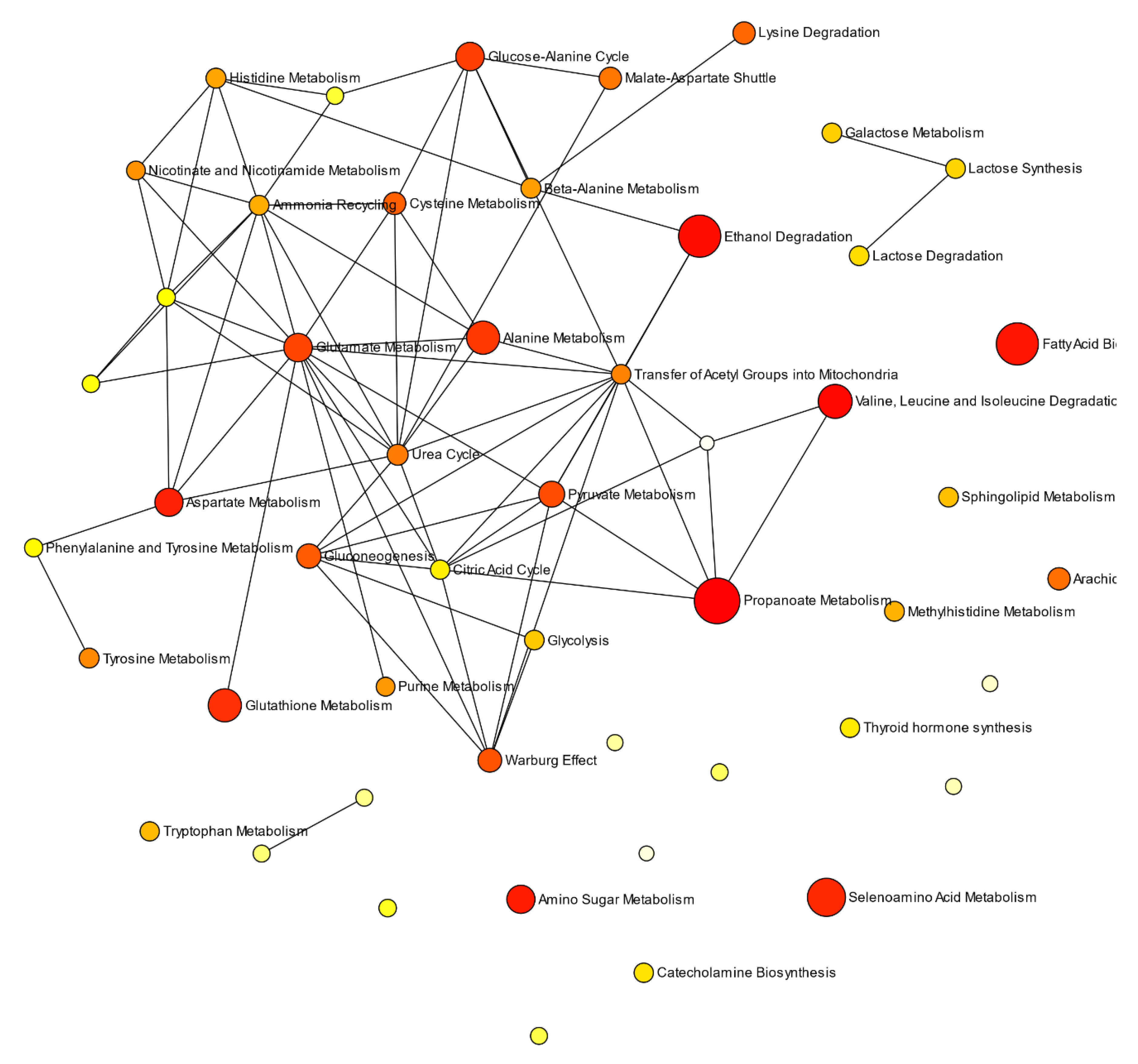
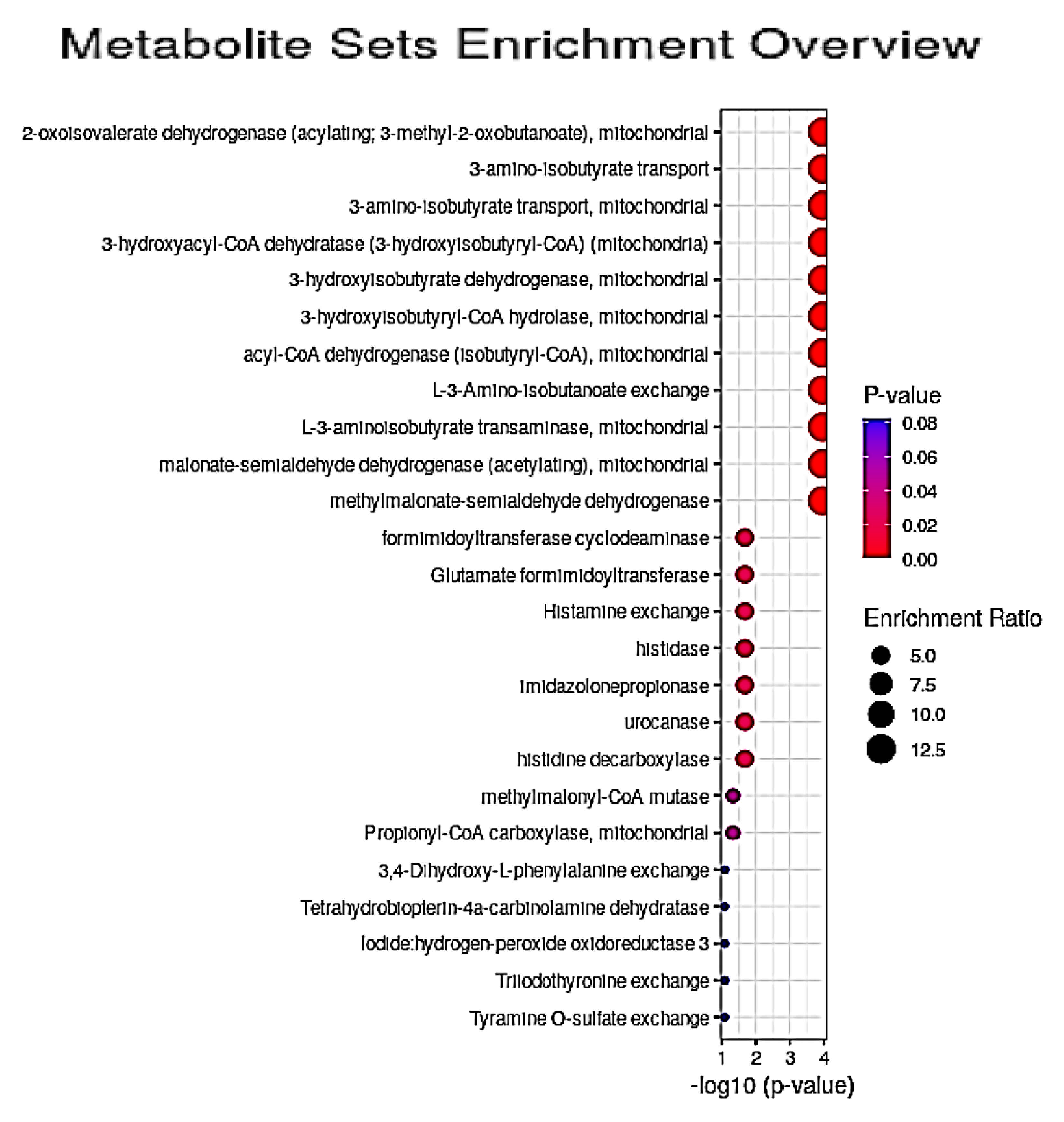
3.5. Quantitative Pathway Enrichment Analysis of Significant Biomolecules
4. Discussion
4.1. Key Discriminatory Biomarker Variables Identified by 1H NMR-Linked Metabolomics Analysis
4.2. Potential Pathological Significance of GM1T2-Mediated Alterations to Plasma Metabolic Profiles Indicated by QMSEA Investigations
4.2.1. Plasma Lipoprotein-Associated Lipids and FA Metabolism
4.2.2. Propionate (Propanoate) and N-Butyrate (Butanoate) Metabolism
4.2.3. BCAA Degradation
4.2.4. Ethanol Degradation
4.2.5. Seleno-Amino Acid and Glutathione (GSH) Metabolism
4.2.6. Aspartate Metabolism
4.2.7. Amino-Sugar Metabolism
4.2.8. Alanine Metabolism
4.3. QMSEA of GM1T2 Pathological Localisations Determined from Metabolic Disturbances Observed
4.3.1. Fibrobasts
4.3.2. Golgi Apparatus
4.3.3. Mitochondria
4.3.4. Spleen and Muscular/Skeletomuscular Systems
4.3.5. Consideration of Marginally Significant QMSEA Location-Based Metabolite Sets
4.4. Overview of Potential Contributions of Dysregulated Plasma Biomolecule Concentrations, Metabolic Pathways and Pinpointed Disorder Locations to GM1T2 Disease Pathogenesis
4.5. Comparative Evaluations with Metabolomics Datasets Acquired on Plasma/Serum Samples Collected from Patients with other LSDs
5. Potential Limitations of the Study
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brunetti-Pierri, N.; Scaglia, F. GM1 gangliosidosis: Review of clinical, molecular, and therapeutic aspects. Mol. Genet. Metab. 2008, 94, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Regier, D.S.; Proia, R.L.; D’Azzo, A.; Tifft, C.J. The GM1 and GM2 gangliosidoses: Natural history and progress toward therapy. Pediatr. Endocrinol. Rev. 2016, 13 (Suppl. 1), 663–673. [Google Scholar]
- Masingue, M.; Dufour, L.; Lenglet, T.; Saleille, L.; Goizet, C.; Ayrignac, X.; Ory-Magne, F.; Barth, M.; Lamari, F.; Mandia, D.; et al. Natural history of adult patients with GM2 gangliosidosis. Ann. Neurol. 2020, 87, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; O’Brien, J.S. Generalized gangliosidosis: Beta-galactosidase deficiency. Science 1968, 160, 1002–1004. [Google Scholar] [CrossRef] [PubMed]
- Sinigerska, I.; Chandler, D.; Vaghjiani, V.; Hassanova, I.; Gooding, R.; Morrone, A.; Kremensky, I.; Kalaydjieva, L. Founder mutation causing infantile GM1-gangliosidosis in the Gypsy population. Mol. Genet. Metab. 2006, 88, 93–95. [Google Scholar] [CrossRef]
- Sperb, F.; Vairo, F.; Burin, M.; Mayer, F.Q.; Matte, U.; Giugliani, R. Genotypic and phenotypic characterization of Brazilian patients with GM1 gangliosidosis. Gene 2013, 512, 113–116. [Google Scholar] [CrossRef] [Green Version]
- Ballabio, A.; Gieselmann, V. Lysosomal disorders: From storage to cellular damage. Biochim. Biophys. Acta 2009, 1793, 684–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisel, B.; Pavone, F.S.; Calamai, M. GM1 and GM2 gangliosides: Recent developments. Biomol. Concepts 2014, 5, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Hofer, D.; Paul, K.; Fantur, K.; Beck, M.; Bürger, F.; Caillaud, C.; Fumic, K.; Ledvinova, J.; Lugowska, A.; Michelakakis, H.; et al. GM1 gangliosidosis and Morquio B disease: Expression analysis of missense mutations affecting the catalytic site of acid beta-galactosidase. Hum. Mutat. 2009, 30, 1214–1221. [Google Scholar] [CrossRef]
- Caciotti, A.; Garman, S.C.; Rivera-Colón, Y.; Procopio, E.; Catarzi, S.; Ferri, L.; Guido, C.; Martelli, P.; Parini, R.; Antuzzi, D.; et al. GM1 gangliosidosis and Morquio B disease: An update on genetic alterations and clinical findings. Biochim. Biophys. Acta 2011, 1812, 782–790. [Google Scholar] [CrossRef]
- Hofer, D.; Paul, K.; Fantur, K.; Beck, M.; Roubergue, A.; Vellodi, A.; Poorthuis, B.J.; Michelakakis, H.; Plecko, B.; Paschke, E. Phenotype determining alleles in GM1 gangliosidosis patients bearing novel GLB1 mutations. Clin. Genet. 2010, 78, 236–246. [Google Scholar] [CrossRef]
- Jarnes Utz, J.R.; Kim, S.; King, K.; Ziegler, R.; Schema, L.; Redtree, E.S.; Whitley, C.B. Infantile gangliosidoses: Mapping a timeline of clinical changes. Mol. Genet. Metab. 2017, 121, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Renaud, D.L. Leukoencephalopathies associated with macrocephaly. Semin. Neurol. 2012, 32, 51–54. [Google Scholar] [CrossRef]
- Ruiz-Rodado, V.; Luque-Baena, R.M.; te Vruchte, D.; Probert, F.; Lachmann, R.H.; Hendriksz, C.J.; Wraith, J.E.; Imrie, J.; Elizondo, D.; Sillence, D.; et al. 1H NMR-linked urinary metabolic profiling of Niemann-Pick class C1 (NPC1) disease: Identification of potential new biomarkers using correlated component regression (CCR) and genetic algorithm (GA) analysis strategies. Curr. Metab. 2014, 2, 88–121. [Google Scholar] [CrossRef]
- Ruiz-Rodado, V.; Nicoli, E.-R.; Probert, F.; Smith, D.A.; Morris, M.; Wassif, C.A.; Platt, F.M.; Grootveld, M. 1H NMR-linked metabolomics analysis of liver from a mouse model of NP-C1 disease. J. Proteome Res. 2016, 15, 3511–3527. [Google Scholar] [CrossRef]
- Quansah, E.; Ruiz-Rodado, V.; Grootveld, M.; Probert, F.; Zetterstrom, T.S.Z. 1H NMR-based metabolomics reveals neurochemical alterations in the brain of adolescent rats following acute methylphenidate administration. Neurochem. Internat. 2017, 108, 109–120. [Google Scholar] [CrossRef]
- Grootveld, M. Metabolic Profiling: Disease and Xenobiotics; Issues in Toxicology Series; Royal Society of Chemistry: Cambridge, UK, 2014; ISBN 1849731632. [Google Scholar]
- López-Rubio, E.; Elizondo, D.A.; Grootveld, M.; Jerez, J.M.; Luque-Baena, R.M. Computational Intelligence Techniques in Medicine; Special issue of Computational and Mathematical Methods in Medicine; Hindawi Publishing Corporation: London, UK, 2014. [Google Scholar]
- Available online: https://www.umassmed.edu/es/news/news-archives/2019/05/nih-administers-first-clinical-trial-treatment-for-gm1-gangliosidosis-a-lysosomal-storage-disorder/. (accessed on 24 November 2020).
- Probert, F.; Ruiz-Rodado, V.; Zhang, X.; te Vruchte, D.; Claridge, T.D.W.; Edgar, M.; Zonato-Tocchio, A.; Lachmann, R.H.; Platt, F.M.; Grootveld, M. Urinary excretion and metabolism of miglustat and valproate in patients with Niemann-Pick type C1 Disease: One- and two-Dimensional solution-state 1H NMR studies. J. Pharm. Biomed. Anal. 2016, 117, 276–288. [Google Scholar] [CrossRef] [PubMed]
- Percival, B.C.; Gibson, M.; Wilson, P.B.; Platt, F.M.; Grootveld, M. Metabolomic studies of lipid storage disorders, with special reference to Niemann-Pick type C disease: A critical review with future perspectives. Int. J. Mol. Sci. 2020, 21, 2533. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0—The human metabolome database for 2018. Nucleic Acids Res. 2018, 4, D608–D617. [Google Scholar]
- Ellul, S.; Wake, M.; Clifford, S.A.; Lange, K.; Würtz, P.; Juonala, M.; Dwyer, T.; Carlin, J.B.; Burgner, D.P.; Saffery, R. Metabolomics: Population epidemiology and concordance in Australian children aged 11–12 years and their parents. BMJ Open 2019, 9, 106–117. [Google Scholar] [CrossRef]
- van Vliet, L.J.; van Kempen, G.M.P. Mean and variance of ratio estimators used in fluorescence ratio imaging. Cytometry Part. A J. Quant. Cell Sci. 2000, 39, 300–305. [Google Scholar] [CrossRef]
- Chong, J.; Soufan, O.; Li, C.; Caraus, J.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [Green Version]
- Xia, J.; Wishart, D.S. MSEA: A web-based tool to identify biologically meaningful patterns in quantitative metabolomic data. Nucleic Acids Res. 2010, 38, W71–W77. [Google Scholar] [CrossRef] [Green Version]
- Elmonem, M.A.; Abdelazim, A.M. Novel biomarkers for lysosomal storage disorders: Metabolomic and proteomic approaches. Clinica Chimica Acta 2020, 509, 195–209. [Google Scholar] [CrossRef]
- Lee, J.S.; Choi, J.-M.; Lee, M.; Kim, S.Y.; Lee, S.; Lim, B.C.; Cheon, J.-E.; Kim, I.-O.; Kim, K.J.; Choi, M.; et al. Diagnostic challenge for the rare lysosomal storage disease: Late infantile GM1 gangliosidosis. Brain Dev. 2018, 40, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Kılıç, C.S.; Kasapkara, S.; Kılavuz, N.O.; Mungan, G.A. Biberoğlu Possible biomarker of neurocytolysis in infantile gangliosidoses: Aspartate transaminase. Metab. Brain. Dis. 2019, 34, 495–503. [Google Scholar] [CrossRef]
- Utz, J.R.J.; Crutcher, T.; Schneider, J.; Sorgen, P.; Whitley, C.B. Biomarkers of central nervous system inflammation in infantile and juvenile gangliosidoses. Mol. Genet. Metab. 2015, 114, 274–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaluarachchi, M.; Boulangé, C.L.; Karaman, I.; Lindon, J.C.; Ebbels, T.M.D.; Elliott, P.; Tracy, R.P.; Olson, N.C. A comparison of human serum and plasma metabolites using untargeted 1H NMR spectroscopy and UPLC-MS. Metabolomics 2018, 14, 32. [Google Scholar] [CrossRef]
- Yu, Z.; Kastenmuller, G.; He, Y.; Belcredi, P.; Moller, G.; Prehn, C.; Mendes, J.; Wahl, S.; Roemisch-Margl, W.; Ceglarek, U.; et al. Differences between human plasma and serum metabolite profiles. PLoS ONE 2011, 6, e21230. [Google Scholar] [CrossRef] [PubMed]
- Duarte, J.F.; Goodfellow, B.J.; Barros, A.; Jones, J.G.; Barosa, C.; Diogo, L.; Garcia, P.; Gil, A.M. Metabolic characterisation of plasma in juveniles with glycogen storage disease type 1a (GSD1a) by high resolution 1H NMR spectroscopy. NMR Biomed. 2007, 20, 401–412. [Google Scholar] [CrossRef] [Green Version]
- Garver, W.S.; Jelinek, D.; Meaney, F.J.; Flynn, J.; Pettit, K.M.; Shepherd, G.; Heidenreich, R.A.; Vockley, C.M.; Castro, G.; Francis, G.A. The National Niemann-Pick Type C1 Disease Database: Correlation of lipid profiles, mutations, and biochemical phenotypes. J. Lipid Res. 2010, 51, 406–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Y.; Choi, B.K.; Chan, T.; Vance, J.E.; Greer, W.L.; Heidenreich, R.A.; Garver, W.S.; Francis, G.A. Impaired ABCA1-dependent lipid efflux and hypoalphalipoproteinemia in human Niemann-Pick type C disease. J. Biol. Chem. 2003, 278, 32569–32577. [Google Scholar]
- De Meyer, T.; Sinnaeve, D.; Van Gasse, B.; Rietzschel, E.R.; De Buyzere, M.L.; Langlois, M. Evaluation of standard and advanced preprocessing methods for the univariate analysis of blood serum 1H-NMR spectra. Anal. Bioanal. Chem. 2010, 398, 1781–1790. [Google Scholar] [CrossRef] [PubMed]
- Probert, F.; Rice, P.; Scudamore, C.L.; Wells, S.; Williams, R.; Hough, T.A.; Cox, I.J. 1H NMR metabolic profiling of plasma reveals additional phenotypes in knockout mouse models. J. Proteome Res. 2015, 14, 2036–2045. [Google Scholar] [CrossRef]
- Thorpe, C.; Kim, J.J. Structure and mechanism of action of the acyl-CoA dehydrogenases. FASEB J. 1995, 9, 718–725. [Google Scholar] [CrossRef]
- Ou, L.; Przybilla, M.J.; Whitley, C.B. Metabolomics profiling reveals profound metabolic impairments in mice and patients with Sandhoff disease. Mol. Genet. Metab. 2019, 126, 151–156. [Google Scholar] [CrossRef]
- Solem, E. The absolute configuration of beta-aminoisobutyric acid formed by degradation of thymine in man. Clin. Chim. Acta 1974, 53, 183–190. [Google Scholar] [CrossRef]
- Kalhan, S.C.; Guo, L.; Edmison, J.; Dasarathy, S.; McCullough, R.J.; Hanson, R.J.; Milburn, M. Plasma metabolomic profile in nonalcoholic fatty liver disease. Metabolism 2011, 60, 404–413. [Google Scholar] [CrossRef] [Green Version]
- Caciotti, A.; Bardelli, T.; Cunningham, J.; D’Azzo, A.; Zammarchi, E.; Morrone, A. Modulating action of the new polymorphism L436F detected in the GLB1 gene of a type-II GM1 gangliosidosis patient. Hum. Genet. 2003, 113, 44–50. [Google Scholar] [CrossRef]
- Suhre, K.; Wallaschofski, H.; Raffler, J.; Nele, F.; Haring, R.; Michael, K.; Wasner, C.; Krebs, A.; Kronenberg, F.; Chang, D.; et al. A genome-wide association study of metabolic traits in human urine. Nat. Genet. 2011, 43, 565–569. [Google Scholar] [CrossRef]
- Kittel, A.; Müller, F.; König, J.; Mieth, M.; Sticht, H.; Zolk, O.; Kralj, A.; Heinrich, M.R.; Fromm, M.F.; Maas, R. Alanine-glyoxylate aminotransferase 2 (AGXT2) polymorphisms have considerable impact on methylarginine and β-aminoisobutyrate metabolism in healthy volunteers. PLoS ONE 2014, 9, e88544. [Google Scholar] [CrossRef]
- Kennedy, B.E.; Hundert, A.S.; Goguen, D.; Weaver, I.C.G.; Karten, B. Presymptomatic Alterations in Amino Acid Metabolism and DNA Methylation in the Cerebellum of a Murine Model of Niemann-Pick Type C Disease. Am. J. Pathol. 2016, 186, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Nie, Y.; Chen, S.; Xie, C.; Fan, Q.; Wang, Z.; Long, B.; Yan, G.; Zhong, Q.; Yan, X. Leucine reduces reactive oxygen species levels via an energy metabolism switch by activation of the mTOR-HIF-1α pathway in porcine intestinal epithelial cells. Int. J. Biochem. Cell Biol. 2017, 89, 42–56. [Google Scholar] [CrossRef]
- Liu, R.; Li, H.; Fan, W.; Jin, Q.; Chao, T.; Wu, Y.; Huang, J.; Hao, L.; Yang, X. Leucine supplementation differently modulates branched-chain amino acid catabolism, mitochondrial function and metabolic profiles at the different stage of insulin resistance in rats on high-fat diet. Nutrients 2017, 9, 565. [Google Scholar]
- Kaya, E.; Smith, D.A.; Smith, C.; Morris, L.; Bremova-Ertl, T.; Cortina-Borja, M.; Fineran, P.; Morten, K.J.; Poulton, J.; Boland, B.; et al. Acetyl-leucine slows disease progression in lysosomal storage disorders. Brain Commun. 2021, 3. [Google Scholar] [CrossRef]
- Donohue, T.M., Jr.; Zetterman, R.K.; Tuma, D.J. Effect of chronic ethanol administration on protein catabolism in rat liver. Alcohol Clin. Exp. Res. 1989, 13, 49–57. [Google Scholar] [CrossRef]
- Thomes, P.G.; Ehlers, R.A.; Trambly, C.S.; Clemens, D.L.; Fox, H.S.; Tuma, D.J.; Donohue, T.M. Multilevel regulation of autophagosome content by ethanol oxidation in HepG2 cells. Autophagy 2013, 9, 63–73. [Google Scholar] [CrossRef] [Green Version]
- Kharbanda, K.K.; McVicker, D.L.; Zetterman, R.K.; Donohue, T.M., Jr. Ethanol consumption reduces the proteolytic capacity and protease activities of hepatic lysosomes. Biochim. Biophys. Acta. 1995, 1245, 421–429. [Google Scholar] [CrossRef]
- Kharbanda, K.K.; McVicker, D.L.; Zetterman, R.K.; Donohue, T.M., Jr. Ethanol consumption alters trafficking of lysosomal enzymes and affects the processing of procathepsin L in rat liver. Biochim. Biophys. Acta. 1996, 1291, 45–52. [Google Scholar] [CrossRef]
- Ding, W.X.; Li, M.; Chen, X.; Ni, H.M.; Lin, C.W.; Gao, W.; Lu, B.; Stolz, D.B.; Clemens, D.L.; Yin, X.-M. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology 2010, 139, 1740–1752. [Google Scholar] [CrossRef] [Green Version]
- Grootveld, M.; Percival, B.C.; Leenders, J.; Wilson, P. Potential adverse public health effects afforded by the ingestion of dietary lipid oxidation product toxins: Significance of fried food sources. Nutrients 2020, 12, 974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labunskyy, V.M.; Hatfield, D.L.; Gladyshev, V.N. Selenoproteins: Molecular pathways and physiological roles. Physiol. Rev. 2014, 94, 739–777. [Google Scholar] [CrossRef] [Green Version]
- Halliwell, B.; Grootveld, M. The measurement of free radical reactions in humans. FEBS Lett. 1987, 231, 9–14. [Google Scholar] [CrossRef] [Green Version]
- Ursini, F.; Maiorino, M. Glutathione Peroxidases. In Encyclopedia of Biological Chemistry, 2nd ed.; Lennarz, W.J., Lane, M.D., Eds.; Academic Press: Cambridge, MA, USA, 2013; pp. 399–404. ISBN 9780123786319. [Google Scholar] [CrossRef]
- Nakamura, H.; Nakamura, K.; Yodoi, J. Redox regulation of cellular activation. Ann. Rev. Immunol. 1997, 15, 351–369. [Google Scholar] [CrossRef]
- Lushchak, V.I. Glutathione homeostasis and functions: Potential targets for medical interventions. J. Amino Acids 2012. [Google Scholar] [CrossRef] [Green Version]
- Vázquez, M.C.; Balboa, E.; Alvarez, A.R.; Zanlungo, S. Oxidative Stress: A Pathogenic Mechanism for Niemann-Pick Type C Disease. Oxidative Med. Cell. Longev. 2012, 2012, 205713. [Google Scholar] [CrossRef] [Green Version]
- Jeyakumar, M.; Thomas, R.; Elliot-Smith, E.; Smith, D.A.; Van der Spoel, A.C.; D’Azzo, A.; Perry, V.H.; Butters, T.D.; Dwek, R.A.; Platt, F.M. Central nervous system inflammation is a hallmark of pathogenesis in mouse models of GM1 and GM2 gangliosidosis. Brain 2003, 126, 974–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, R.A.; Burk, R.F. Species, tissue and subcellular distribution of mon Se-dependent glutathione peroxidase activity. J. Nutr. 1978, 108, 211–215. [Google Scholar] [CrossRef]
- Paglia, G.; Stocchero, M.; Cacciatore, S.; Lai, S.; Angel, P.; Alam, M.T.; Keller, M.; Ralser, M.; Astarita, G. Unbiased metabolomic investigation of Alzheimer’s disease brain points to dysregulation of mitochondrial aspartate metabolism. J. Proteome Res. 2016, 15, 608–618. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, Y.; Oshima, A.; Namba, E. disease. In The Metabolic and Molecular Bases of Inherited Disease; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3775–3809. [Google Scholar]
- Allende, M.L.; Proia, R.L. Lubricating cell signaling pathways with gangliosides. Curr. Opin. Struct. Biol. 2002, 12, 587–592. [Google Scholar] [CrossRef]
- Regina Todeschini, A.; Hakomori, S.I. Functional role of glycosphingolipids and gangliosides in control of cell adhesion, motility, and growth, through glycosynaptic microdomains. Biochim. Biophys. Acta 2008, 1780, 421–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grootveld, M.; Claxson, A.W.D.; Chander, C.L.; Haycock, P.; Blake, D.R.; Hawkes, G.E. High resolution proton NMR investigations of rat blood plasma. Assisgnment of resonances for the molecularly mobile carbohydrate side-chains of ‘acute-phase’ glycoproteins. FEBS Lett. 1993, 322, 266–276. [Google Scholar] [CrossRef] [Green Version]
- Engelke, U.F.H.; Liebrand-van Sambeek, M.L.F.; de Jong, J.G.N.; Leroy, J.G.; va Morava, E.; Smeitink, J.A.M.; Wevers, R.A. N-Acetylated metabolites in urine: Proton nuclear magnetic resonance spectroscopic study on patients with inborn errors of metabolism. Clin. Chem. 2004, 50, 58–66. [Google Scholar] [CrossRef] [Green Version]
- Krawczyk, H.; Gradowska, W. Characterisation of the 1H and 13C NMR spectra of N-acetylaspartylglutamate and its detection in urine from patients with Canavan disease. J. Pharm. Biomed. Anal. 2003, 31, 455–463. [Google Scholar] [CrossRef]
- Mathews, C.K.; Van Holde, K.E.; Ahern, K.G. Biochemistry, 3rd ed.; Benjamin/Cummings Publishing: San Francisco, CA, USA, 2000; ISBN 0805330666. [Google Scholar]
- Bröer, S.; Bröer, A.; Hansen, J.T.; Bubb, W.A.; Balcar, V.J.; Nasrallah, F.A.; Garner, B.; Rae, C. Alanine metabolism, transport, and cycling in the brain. J. Neurochem. 2007, 102, 1758–1770. [Google Scholar] [CrossRef]
- Condori, J.; Acosta, W.; Ayala, J.; Katta, V.; Flory, A.; Martin, R.; Radin, J.; Cramer, C.L.; Radin, D.N. Enzyme replacement for GM1-gangliosidosis: Uptake, lysosomal activation, and cellular disease correction using a novel β-galactosidase:RTB lectin fusion. Molec. Genet. Metab. 2016, 117, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Cavallini, L.; Venerando, R.; Miotto, G.; Alexandre, A. Ganglioside GM1 protection from apoptosis of rat heart fibroblasts. Arch. Biochem. Biophys. 1999, 370, 156–162. [Google Scholar] [CrossRef]
- Meran, S.; Luo, D.D.; Simpson, R.; Martin, J.; Wells, A.; Steadman, R.; Phillips, A.O. Hyaluronan facilitates transforming growth factor-β1-dependent proliferation via CD44 and epidermal growth factor receptor interaction. J. Biol. Chem. 2011, 286, 17618–17630. [Google Scholar] [CrossRef] [Green Version]
- Momoi, T.; Furuya, T.; Suzuki, Y.; Sato, H.; Yamaguchi, N. In vitro establishment of human fibroblasts of lysosomal diseases, GM1-gangliosidosis and Sandhoff disease, by transformation with origin-minus SV40 DNA. Biosci. Rep. 1985, 5, 267–273. [Google Scholar] [CrossRef]
- Maccioni, H.; Daniotti, J.; Martina, J. Organization of ganglioside synthesis in the Golgi apparatus. Biochim. Biophys. Acta 1999, 1437, 101–118. [Google Scholar] [CrossRef]
- Bisel, B.; Calamai, M.; Vanzi, F.; Pavone, F.S. Decoupling polarization of the Golgi apparatus and GM1 in the plasma membrane. PLoS ONE 2013, 8, e80446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yusuf, H.K.M.; Pohlentz, G.; Sandhoff, K. Ganglioside biosynthesis in Golgi apparatus: New perspectives on its mechanism. J. Neurosci. Res. 1984, 12, 161–178. [Google Scholar] [CrossRef]
- Demine, S.; Michel, S.; Vannuvel, K.; Wanet, A.; Renard, P.; Arnould, T. Macroautophagy and cell responses related to mitochondrial dysfunction, lipid metabolism and unconventional secretion of proteins. Cells 2012, 1, 168–203. [Google Scholar] [CrossRef] [Green Version]
- de la Mata, M.; Cotán, D.; Villanueva-Paz, M.; de Lavera, I.; Álvarez-Córdoba, M.; Luzón-Hidalgo, R.; Suárez-Rivero, J.M.; Tiscornia, G.; Oropesa-Ávila, M. Mitochondrial dysfunction in lysosomal storage disorders. Diseases 2016, 4, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, R.; Annunziata, I.; Patterson, A.; Moshiach, S.; Gomero, E.; Opferman, J.; Forte, M.; d’Azzo, A. Gm1-ganglioside accumulation at the mitochondria-associated er membranes links er stress to Ca(2+)-dependent mitochondrial apoptosis. Mol. Cell. 2009, 36, 500–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takamura, A.; Higaki, K.; Kajimaki, K.; Otsuka, S.; Ninomiya, H.; Matsuda, J.; Ohno, K.; Suzuki, Y.; Nanba, E. Enhanced autophagy and mitochondrial aberrations in murine g(m1)-gangliosidosis. Biochem. Biophys. Res. Commun. 2008, 367, 616–622. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative stress in neurodegenerative diseases: From molecular mechanisms to clinical applications. Oxid. Med. Cell. Longev. 2017, 4, 1–11. [Google Scholar] [CrossRef]
- Levine, B.; Yuan, J. Autophagy in cell death: An innocent convict? J. Clin. Invest. 2005, 115, 2679–2688. [Google Scholar] [CrossRef]
- Huang, J.; Philbert, M.A. Distribution of glutathione and glutathione-related enzyme systems in mitochondria and cytosol of cultured cerebellar astrocytes and granule cells. Brain Res. 1995, 680, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Kiselyov, K.; Muallem, S. Mitochondrial Ca2+ homeostasis in lysosomal storage diseases. Cell Calcium. 2008, 44, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Heur, M.; Duanmu, M.; Grabowski, G.A.; Hui, D.Y.; Witte, D.P.; Mishra, J. Lysosomal acid lipase-deficient mice: Depletion of white and brown fat, severe hepatosplenomegaly, and shortened life span. J. Lipid Res. 2001, 42, 489–500. [Google Scholar] [CrossRef]
- Ou, L.; Przybilla, M.J.; Whitley, C.B. Proteomic analysis of mucopolysaccharidosis I mouse brain with two-dimensional polyacrylamide gel electrophoresis. Mol. Genet. Metab. 2017, 120, 101–110. [Google Scholar] [CrossRef] [Green Version]
- Hutchesson, A.; Preece, M.A.; Gray, G.; Green, A. Measurement of lactate in cerebrospinal fluid in investigation of inherited metabolic disease. Clin. Chem. 1997, 43, 158–161. [Google Scholar] [CrossRef] [Green Version]
- Phypers, B.; Pierce, J.M.T. Lactate physiology in health and disease. Contin. Educ. Anaesthes. Crit. Care Pain 2006, 6, 128–132. [Google Scholar] [CrossRef]
- Xiang, Z.; Yuan, M.; Hassen, G.W.; Gampel, M.; Bergold, P.J. Lactate induced excitotoxicity in hippocampal slice cultures. Experiment. Neurol. 2004, 186, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Kannebley, J.S.; Silveira-Moriyama, L.; Bastos, L.O.D.; Steiner, C.E. Clinical findings and natural history in ten unrelated families with juvenile and adult GM1 gangliosidosis. In JIMD Reports; Zschocke, J., Baumgartner, M., Morava, E., Patterson, M., Rahman, S., Peters, V., Eds.; Springer: Berlin/Heidelberg, Germany, 2015; Volume 24. [Google Scholar] [CrossRef] [Green Version]
- Gografe, S.I.; Sanberg, P.R.; Chamizo, W.; Monforte, H.; Garbuzova-Davis, S. Novel pathologic findings associated with urinary retention in a mouse model of mucopolysaccharidosis type IIIB. Comp. Med. 2009, 59, 139–146. [Google Scholar]
- Koyama, K.; Moda, Y.; Sone, A.; Tanaka, H.; Hino, Y. Neurogenic bladder in Hunter’s syndrome. J. Med. Genet. 1994, 31, 257–258. [Google Scholar] [CrossRef] [Green Version]
- McNamara, E.R.; Sullivan, J.; Nagaraj, S.K.; Wiener, J.S.; Kishnani, P.S. Neurogenic bladder dysfunction presenting as urinary retention in neuronopathic Gaucher disease. JIMD Rep. 2015, 15, 67–70. [Google Scholar] [CrossRef] [Green Version]
- Rosenbloom, B.E.; Weinreb, N.J.; Zimran, A.; Kacena, K.A.; Charrow, J.; Ward, E. Gaucher disease and cancer incidence: A study from the Gaucher Registry. Blood 2005, 105, 4569–4572. [Google Scholar] [CrossRef]
- Perera, R.; Stoykova, S.; Nicolay, B.; Perera, R.M.; Stoykova, S.; Nicolay, B.N.; Ross, K.N.; Fitamant, J.; Boukhali, M.; Lengrand, J.; et al. Transcriptional control of autophagy–lysosome function drives pancreatic cancer metabolism. Nature 2015, 524, 361–365. [Google Scholar] [CrossRef]
- Gray-Edwards, H.L.; Regier, D.S.; Shirley, J.L.; Randle, A.N.; Salibi, N.; Thomas, S.E.; Latour, Y.L.; Johnston, J.; Golas, G.; Maguire, A.S.; et al. Novel biomarkers of human GM1 gangliosidosis reflect the clinical efficacy of gene therapy in a feline model. Mol. Ther. 2017, 25, 892–903. [Google Scholar] [CrossRef] [Green Version]
- McCurdy, V.J.; Rockwell, H.E.; Arthur, J.R.; Bradbury, A.M.; Johnson, A.K.; Randle, A.N.; Brunson, B.L.; Hwang, M.; Gray-Edwards, H.L.; Morrison, N.E.; et al. Widespread correction of central nervous system disease after intracranial gene therapy in a feline model of Sandhoff disease. Gene Ther. 2015, 22, 181–189. [Google Scholar] [CrossRef] [Green Version]
- Aronson, S.M.; Saifer, A.; Kanof, A.; Volk, B.W. Progression of amaurotic family idiocy as reflected by serum and cerebrospinal fluid changes. Am. J. Med. 1958, 24, 390–401. [Google Scholar] [CrossRef]
- Satoh, H.; Yamato, O.; Asano, T.; Yonemura, M.; Yamauchi, T.; Hasegawa, D.; Orima, H.; Arai, T.; Yamasaki, M.; Maede, Y. Cerebrospinal fluid biomarkers showing neurodegeneration in dogs with GM1 gangliosidosis: Possible use for assessment of a therapeutic regimen. Brain Res. 2007, 1133, 200–208. [Google Scholar] [CrossRef] [Green Version]
- Lending, M.; Slobody, L.B.; Mestern, J. Cerebrospinal fluid glutamic oxalacetic transaminase and lactic dehydrogenase activities in children with neurologic disorders. J. Pediatrics 1964, 65, 415–421. [Google Scholar] [CrossRef]
- Wroblewski, F.; Decker, B.; Wroblewski, R. The clinical implications of spinal fluid LDH activity. N. Engl. J. Med. 1958, 258, 635–639. [Google Scholar] [CrossRef]
- Sinha, M.K.; Prasad, M.; Haque, S.S.; Agrawal, R.; Keshari, J.R.; Kumar, U.; Kumar, A.; Aziz, T.; Kumar, A. Evaluation of lactate dehydrogenase activity in cerebrospinal fluid in different etiology of meningitis. Curr. Trends Biomed. Eng. Biosci. 2017, 1, 41–43. [Google Scholar]
- Wyss, M.; Kaddurah-Daouk, R. Creatine and creatinine metabolism. Physiol. Rev. 2000, 80, 1107–1213. [Google Scholar] [CrossRef]
- Chaleckis, R.; Murakami, I.; Takada, J.; Kondoh, H.; Yanagida, M. Individual variability in human blood metabolites identifies age-related differences. Proc. Natl. Acad. Sci. USA 2016, 113, 4252–4259. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, W.J. Creatine and phosphocreatine in the human red cell. Br. J. Haematol. 1968, 15, 389–400. [Google Scholar] [CrossRef]
- Kaplan, L.A.; Pesce, A.J. Muscle disease. In Clinical Chemistry; Mosby Company: St. Louis, MO, USA, 1984; Volume 243, pp. 550–593. [Google Scholar]
- Tiao, J.Y.; Semmens, J.B.; Masarei, J.R.; Lawrence-Brown, M.M. The effect of age on serum creatinine levels in an aging population: Relevance to vascular surgery. Cardiovasc Surg. 2002, 10, 445–451. [Google Scholar] [CrossRef]
- Thongprayoon, C.; Cheungpasitporn, W.; Kashani, K. Serum creatinine level, a surrogate of muscle mass, predicts mortality in critically ill patients. J. Thorac. Dis. 2016, 8, E305–E311. [Google Scholar] [CrossRef] [Green Version]
- Nair, S.; O’Brien, S.V.; Hayden, K.; Pandya, B.; Lisboa, P.J.G.; Hardy, K.J.; Wilding, J.P.H. Effect of a cooked meat meal on serum creatinine and estimated glomerular filtration rate in diabetes-related kidney disease. Diabet. Care 2014, 37, 483–487. [Google Scholar] [CrossRef] [Green Version]
- Herder, V.; Kummrow, M.; Leeb, T.; Sewell, A.C.; Hansmann, F.; Lehmbecker, A.; Wohlsein, P.; Baumgärtner, W. Polycystic kidneys and GM2 gangliosidosis-like disease in neonatal springboks (Antidorcas marsupialis). Vet. Pathol. 2015, 52, 543–552. [Google Scholar] [CrossRef]
- Grantham, J.J.; Torres, V.E.; Chapman, A.B.; Guay-Woodford, L.M.; Bae, K.T.; King, B.F., Jr.; Wetzel, L.H.; Baumgarten, D.A.; Kenney, P.J.; Harris, P.C.; et al. Volume progression in polycystic kidney disease. N. Engl. J. Med. 2006, 354, 2122–2130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horiuchi, M.; Takeda, T.; Takanashi, H.; Ozaki-Masuzawa, Y.; Taguchi, Y.; Toyoshima, Y.; Otani, L.; Kato, H.; Sone-Yonezawa, M.; Hakuno, F.; et al. Branched-chain amino acid supplementation restores reduced insulinotropic activity of a low-protein diet through the vagus nerve in rats. Nutr. Metab. 2017, 15. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, R.; Hassanpour, N.; Liu, L.-P.; Hassoun, S. Pathway-activity likelihood analysis and metabolite annotation for untargeted metabolomics using probabilistic modeling. Metabolites 2020, 10, 183. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Resonance Code | Chemical Shift (δ/ppm) | Multiplicity | Assignment | Median STN Value * |
|---|---|---|---|---|
| 1 | 0.92 | broad | Very-low-density-lipoprotein (vLDL)/low-density-lipoprotein (LDL) TAG-terminal-CH3 functions | 413 |
| 2 | 0.926 and 0.997 | t and d | Isoleucine-CH3 | 82 and 118 |
| 2 | 0.949 | t | Leucine-CH3 | 108 |
| 2 | 0.98 and 1.03 | 2 × d | Valine-CH3′s | 226 and 253 |
| 3 | 1.10–1.30 | broad | vLDL/LDL-bulk-chain-(-CH2-)n | 599 |
| 4 | 1.32 | d | Lactate-CH3 | 501 |
| 5 | 1.48 | d | Alanine-CH3 | 382 |
| 6 | 1.55 | broad | Lipoprotein TAG-CH2CH2CO | 157 |
| 7 | 1.74 | broad m | Arginine-γ-CH2/Lysine-δ-CH2 protein residues | 94 |
| 8 | 1.92 | s | Acetate-CH3 | 198 |
| 9 | 2.03 | broad | Acute-phase glycoprotein-carbohydrate side-chain N-acetyl-sugar-CH3 I (APG-I)/TAG-CH2-CH = CH- | 281 |
| 9 | 2.07 | broad | Acute-phase glycoprotein-carbohydrate side-chain N-acetyl-sugar-CH3 II (APG-II)/TAG-CH2-CH = CH- | 386 |
| 10 | 2.12 | m | Glutamine-β-CH2 | 122 |
| 11 | 2.21 | m | Lipoprotein TAG-CH2-CO2-/Acetone-CH3 | 111 |
| 12 | 2.43 | m | Glutamine-γ-CH2 | 169 |
| 13 | 2.53 | ½ dd (AB coupling pattern) | Citrate-CH2A/B | 107 |
| 14 | 2.75 | d | Lipoprotein TAG-CH = CH-CH2-CH = CH/Citrate-CH2A/B | 89 |
| 15 | 2.97 | broad | Albumin lysine residue-ε-CH2 | 82 |
| 16 | 3.02 | t/s | Free lysine-ε-CH2/Creatine- and Creatinine-N-CH3 | 151 |
| 17 | 3.19 | broad | High-density-lipoprotein phospholipid choline head-group-N(CH)3+ | 459 |
| 18 | 3.22 | m | Glucose-C2H | 478 |
| 19 | 3.39 | m | Taurine-CH2SO32- | 279 |
| 20 | 3.41 | m | Glucose-C2-6H | 463 |
| 21 | 3.43 | m | Glucose-β-C2/5-α-C3/5-H | 473 |
| 22 | 3.57 | m | Glucose-β-C2/5-α-C3/5-H | 539 |
| 23 | 3.62 | m | Unassigned | 133 |
| 24 | 3.71 | m | Glucose-α-C3/6- β-C6-H | 434 |
| 25 | 3.78 | m | Glucose-α-C3/6- β-C6-H | 451 |
| 26 | 3.82 | m | Glucose-α-C4/6- β-C4/6-H | 444 |
| 27 | 3.89 | m | Glucose-α-C4/6- β-C4/6-H | 421 |
| Cr/PCr | 3.94 | s | Creatine/Phosphocreatine-CH2 | 151 |
| 28 | 3.99 | ms | Tyrosine-/Histidine-/Phenylalanine-α-CH’s | 80 |
| 29 | 4.05 | s | Creatinine-CH2 | 133 |
| 30 | 4.14 | q | Lactate-CH | 135 |
| 31 | 4.63 | d | β-Glucose-CH | 594 |
| 32 | 4.80 | s | Residual H2O/HO2H | n/a |
| 33 | 5.25 | d | α-Glucose-CH | 341 |
| 34 | 5.27 | broad | Lipoprotein TAG-CH = CH- | 555 |
| 35 | 5.80 | broad | Urea-CO-NH2 | 49 |
| 36 | 6.89 | m | Tyrosine-CH | 57 |
| 37 | 7.01 | m | Phenylalanine-CH | 34 |
| 38 | 7.08 | s | Histidine-CH | 83 |
| 39 | 7.19 | m | Tyrosine-CH | 63 |
| 40 | 7.30 | m | Phenylalanine-CH | 50 |
| 41 | 7.41 | m | Phenylalanine-CH | 30 |
| 42 | 7.80 | s | Histidine-CH | 79 |
| 43 | 8.07 | broad | Protein aromatic amino acid residue(s) | 45 |
| 44 | 8.45 | s | Formate-CH | 63 |
| Metabolite | Regulation Status (↑/↓) * | Fold-Change ** | Age-Related Fold-Change (HC data) *** | WT p Value | Bonferroni-Corrected WT p-Value |
|---|---|---|---|---|---|
| Total TAGs | ↓ | 0.75 | 0.808 (total TAGs)0.842 (total FAs) | 4.50 × 10−5 | 1.12 × 10−3 |
| Isoleucine | ↑ | 1.04 | 0.947 | ns | ns |
| Leucine | ↑ | 1.37 | 0.935 | 4.67 × 10−4 | 1.17 × 10−2 |
| Valine | ↑ | 2.10 | 0.958 | <10−6 | <2.50 × 10−5 |
| 3-AIB | ↑ | 1.21 | na | 9.02 × 10−3 | ns |
| Alanine | ↓ | 0.87 | 0.973 | 4.37 × 10−2 | ns |
| Acetate | ↓ | 0.96 | 0.882 | ns | ns |
| APG-I/TAGs | ↓ | 0.91 | 0.930 | ns | ns |
| APG-II/TAGs | ↓ | 0.93 | na | 3.41 × 10−2 | ns |
| Glutamine | ↑ | 1.20 | 1.052 | 1.16 × 10−4 | 2.90 × 10−3 |
| Glutamate | ↑ | 1.14 | 1.308**** | 4.70 × 10−4 | 1.17 × 10−2 |
| Citrate | ↑ | 1.16 | 1.153 | 1.16 × 10−3 | 2.90 × 10−2 |
| HDL-PLs | ↑ | 1.03 | 0.859 (total PGs)/0.870 (total cholines) | ns | ns |
| α-/β-Glucose | ↑ | 1.14 | 0.998 | 2.91 × 10−2 | ns |
| Taurine | ↑ | 1.13 | 1.073 **** | 3.58 × 10−2 | ns |
| Cr/PCr | ↑ | 1.48 | 0.998 (Cr only) **** | 2.80 × 10−3 | ns |
| Cn | ↑ | 2.65 | 0.727 | 2.18 × 10−5 | 5.45 × 10−4 |
| Lactate | ↑ | 4.19 | 1.103 | 3.27 × 10−6 | 8.17 × 10−5 |
| Threonine | ↑ | 1.16 | 1.225 **** | ns | ns |
| Urea | ↑ | 1.66 | 0.473 **** | 1.55 × 10−3 | 3.87 × 10−2 |
| Tyrosine | ↑ | 1.62 | 1.000 | 7.41 × 10−5 | 1.85 × 10−3 |
| Phenylalanine | ↑ | 1.52 | 0.918 | 1.26 × 10−3 | 3.15 × 10−2 |
| Histidine | ↑ | 1.91 | 1.000 | 2.57 × 10−4 | 6.42 × 10−3 |
| PAAR | ↑ | 1.96 | na | 2.78 × 10−4 | 6.95 × 10−3 |
| Formate | ↑ | 1.54 | na | 4.05 × 10−3 | ns |
| Metabolite | Ile-N Fold-Change ± 95% CIs | Age-Related Ile-N Fold-Change (HC data) | Ile-N WT p Value | Ile-N Bonferroni-Corrected WT p-Value |
|---|---|---|---|---|
| Total TAGs | 0.74 ± 0.22 | 0.85 | 5.53 × 10−3 | ns |
| Isoleucine | na | na | n/a | n/a |
| Leucine | 1.32 ± 0.04 | 0.99 | 2.21 × 10−5 | 5.30 × 10−4 |
| Valine | 2.00 ± 0.05 | 1.01 | <10−6 | <2.5 × 10−4 |
| 3-AIB | 1.18 ± 0.16 | na | ns | ns |
| Alanine | 0.86 ± 0.22 | 1.03 | 0.079 | ns |
| Acetate | 0.92 ± 0.16 | 0.96 | ns | ns |
| APG-I/TAGs | 0.88 ± 0.27 | 0.98 | 4.62 × 10−2 | ns |
| APG-II/TAGs | 0.89 ± 0.11 | na | 3.53 × 10−2 | ns |
| Glutamine | 1.16 ± 0.09 | 1.11 | 6.83 × 10−3 | ns |
| Glutamate | 1.11 ± 0.39 | na | ns | ns |
| Citrate | 1.12 ± 0.09 | 1.22 | 0.057 | ns |
| HDL-PLs | 1.01 ± 0.19 | 0.89 (total PGs)/0.92 (total cholines) | ns | ns |
| α-/β-Glucose | 1.06 ± 0.10 | 1.05 | ns | ns |
| Taurine | 1.08 ± 0.11 | na | ns | ns |
| Cr/PCr | 2.59 ± 0.10 | na | <10−6 | <2.50 × 10−4 |
| Cn | 2.50 ± 0.11 | 0.77 | 3.67 × 10−5 | 8.81 × 10−4 |
| Lactate | 3.98 ± 0.40 | 1.16 | 4.98 × 10−6 | 1.19 × 10−4 |
| Threonine | 1.11 ± 0.33 | na | ns | ns |
| Urea | 1.54 ± 0.21 | na | 2.37 × 10−3 | 0.057 |
| Tyrosine | 1.53 ± 0.14 | 1.06 | 2.00 × 10−4 | 9.60 x 10−3 |
| Phenylalanine | 1.43 ± 0.19 | 0.97 | 2.45 × 10−3 | 0.059 |
| Histidine | 1.78 ± 0.19 | 1.06 | 4.06 × 10−4 | 9.74 × 10−3 |
| PAAR | 2.11 ± 0.17 | na | 2.10 × 10−4 | 5.04 × 10−3 |
| PC | PC1 | PC2 | PC3 | PC4 | PC5 |
|---|---|---|---|---|---|
| Eigenvalue | 9.69 | 3.61 | 2.91 | 2.40 | 1.05 |
| Total TAGs | −0.50 (0.25) | −0.79 (0.62) | nc | nc | nc |
| Isoleucine | nc | 0.70 (0.49) | nc | nc | nc |
| Leucine | nc | 0.94 (0.88) | nc | nc | nc |
| Valine | 0.47 (0.22) | 0.72 (0.53) | nc | nc | nc |
| 3-AIB | 0.53 (0.28) | nc | nc | nc | 0.69 (0.47) |
| Alanine | nc | nc | nc | 0.40 (0.16) | −0.62 (0.38) |
| Acetate | 0.45 (0.20) | nc | 0.48 (0.23) | nc | 0.52 (0.28) |
| APG-1 | nc | nc | nc | 0.81 (0.65) | nc |
| APG-2 | nc | nc | nc | 0.87 (0.76) | nc |
| Glutamine | nc | 0.88 (0.78) | nc | nc | nc |
| Glutamate | 0.53 (0.28) | nc | 0.60 (0.36) | nc | nc |
| Citrate | nc | 0.42 (0.18) | 0.60 (0.36) | nc | 0.45 (0.20) |
| HDL-PLs | −0.42 (0.17) | nc | nc | −0.83 (0.69) | nc |
| Taurine | nc | nc | 0.74 (0.55) | nc | nc |
| Cr/PCr | 0.65 (0.43) | nc | 0.60 (0.36) | nc | nc |
| Cn | 0.50 (0.25) | nc | 0.75 (0.57) | nc | nc |
| Lactate | 0.47 (0.22) | nc | 0.75 (0.57) | nc | nc |
| Threonine | nc | nc | 0.87 (0.76) | nc | nc |
| Glucose | nc | 0.56 (0.32) | −0.69 (0.47) | nc | nc |
| Urea | 0.92 (0.85) | nc | nc | nc | nc |
| Tyrosine | 0.93 (0.87) | nc | nc | nc | nc |
| Phenylalanine | 0.95 (0.90) | nc | nc | nc | nc |
| Histidine | 0.93 (0.87) | nc | nc | nc | nc |
| PAAR | 0.92 (0.84) | nc | nc | nc | nc |
| Formate | 0.93 (0.86) | nc | nc | nc | nc |
| GM1T2 Disease | HC | Total | |
|---|---|---|---|
| Training Set (n = 19) | 89.35 ± 1.92% | 99.91 ± 0.09% | 97.95 ± 0.31% |
| Test Set (n = 9) | 83.22 ± 3.17% | 99.06 ± 0.53% | 94.66 ± 0.95% |
| Mean Validation Set Classification Success Score (n = 9) | 8.75/9.00 (97.22%) | ||
| Pathway | Total Metabolites | Hits | Q Statistic | FDR-Corrected p-value |
|---|---|---|---|---|
| Propanoate Metabolism | 42 | 2 | 59.66 | 7.12 × 10−12 |
| Valine, Leucine and Isoleucine Degradation | 60 | 4 | 37.97 | 1.33 × 10−9 |
| Ethanol Degradation | 19 | 1 | 53.21 | 3.79 × 10−6 |
| Fatty Acid Biosynthesis | 35 | 1 | 53.21 | 3.79 × 10−6 |
| Amino-sugar Metabolism | 33 | 3 | 27.23 | 1.90 × 10−5 |
| Aspartate Metabolism | 35 | 3 | 27.23 | 1.90 × 10−5 |
| Seleno-Amino Acid Metabolism | 28 | 1 | 45.64 | 3.21 × 10−5 |
| Glutathione Metabolism | 21 | 2 | 36.41 | 4.25 × 10−5 |
| Alanine Metabolism | 17 | 2 | 36.41 | 4.25 × 10−5 |
| Glucose-Alanine Cycle | 13 | 3 | 27.72 | 0.00017 |
| Glutamate Metabolism | 49 | 3 | 27.79 | 0.00030 |
| Pyruvate Metabolism | 48 | 2 | 22.98 | 0.008 |
| Warburg Effect | 58 | 5 | 19.02 | 0.014 |
| Gluconeogenesis | 35 | 2 | 20.28 | 0.016 |
| Cysteine Metabolism | 26 | 1 | 16.29 | 0.037 |
| Lysine Degradation | 30 | 1 | 16.29 | 0.037 |
| Arachidonic Acid Metabolism | 69 | 1 | 16.29 | 0.037 |
| Malate-Aspartate Shuttle | 10 | 1 | 16.29 | 0.037 |
| Urea Cycle | 29 | 4 | 13.69 | 0.037 |
| Transfer of Acetyl Groups into Mitochondria | 22 | 2 | 10.95 | 0.049 |
| Site | Total Metabolites Involved | Number of Hits | FDR-Adjusted p Value | Metabolites Featured (Up-/Down Regulation in GM1T2 Disease Observed) |
|---|---|---|---|---|
| Fibroblasts | 183 | 1 | 1.31 × 10−12 | Valine↑ |
| Golgi Apparatus | 14 | 1 | 3.04 × 10−6 | Acetate↓ |
| Mitochondria | 98 | 5 | 4.74 × 10−3 | Valine↑/Acetate↓/Urea↑/ Glutamine↑/Leucine↑ |
| Spleen | 170 | 3 | 3.33 × 10−2 | Glutamate↑/Creatinine↑/ Glutamine↑ |
| Skeletal Muscle | 123 | 5 | 3.33 × 10−2 | Glutamate↑/Creatinine↑/ Taurine↑/Glutamine↑/Leucine↑ |
| Muscle | 160 | 6 | 3.33 × 10−2 | Glutamate↑/Creatinine↑/ Glucose↑/Taurine↑/Leucine↑ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Percival, B.C.; Latour, Y.L.; Tifft, C.J.; Grootveld, M. Rapid Identification of New Biomarkers for the Classification of GM1 Type 2 Gangliosidosis Using an Unbiased 1H NMR-Linked Metabolomics Strategy. Cells 2021, 10, 572. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10030572
Percival BC, Latour YL, Tifft CJ, Grootveld M. Rapid Identification of New Biomarkers for the Classification of GM1 Type 2 Gangliosidosis Using an Unbiased 1H NMR-Linked Metabolomics Strategy. Cells. 2021; 10(3):572. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10030572
Chicago/Turabian StylePercival, Benita C., Yvonne L. Latour, Cynthia J. Tifft, and Martin Grootveld. 2021. "Rapid Identification of New Biomarkers for the Classification of GM1 Type 2 Gangliosidosis Using an Unbiased 1H NMR-Linked Metabolomics Strategy" Cells 10, no. 3: 572. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10030572