
Transcriptional Profiles of Cell Fate Transitions Reveal Early Drivers of Neuronal Apoptosis and Survival

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Design
2.2. RNA Isolation and Microarray Hybridization
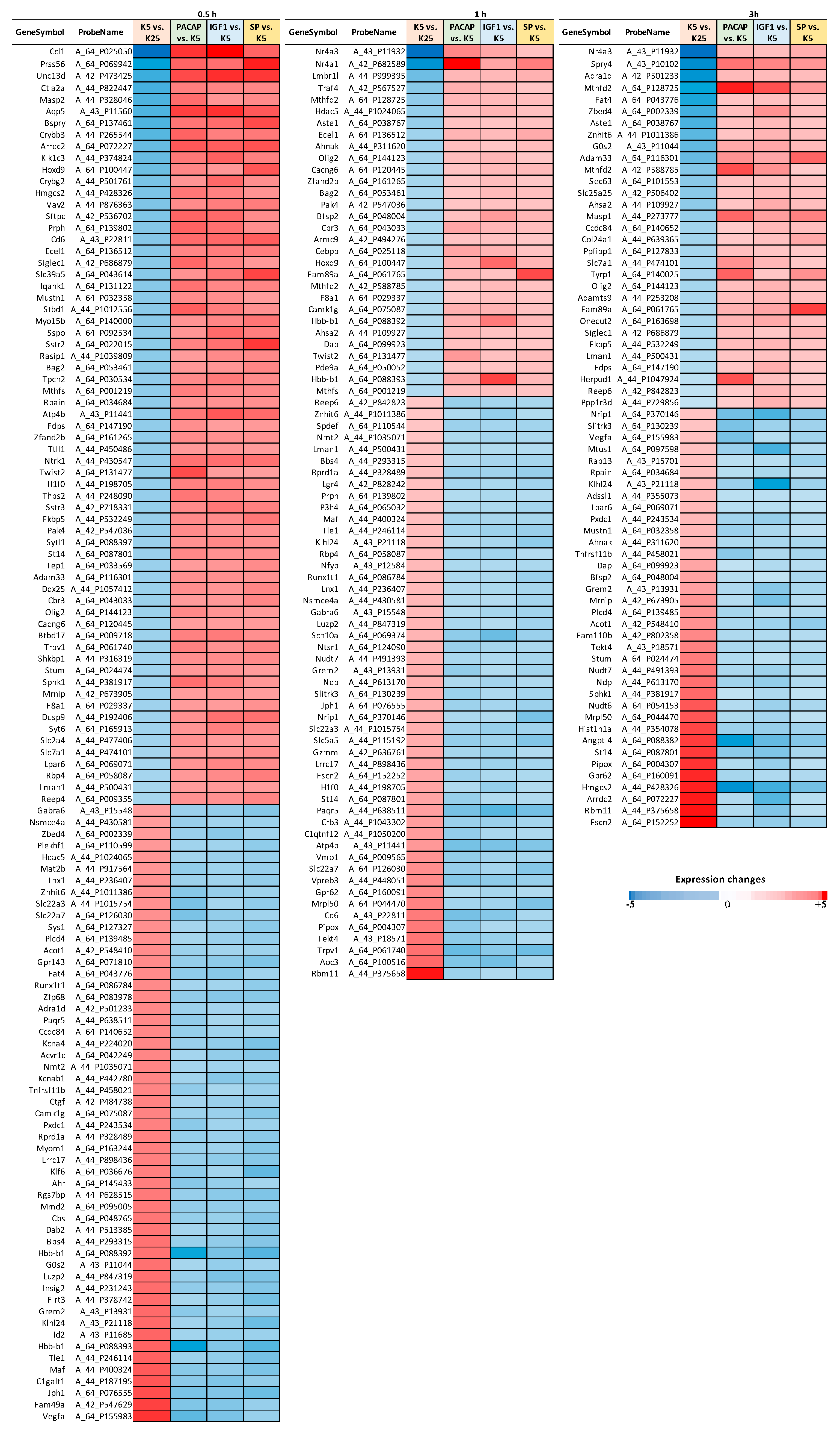
2.3. Time-Point Differential Gene Expression Analysis
2.4. Protein–Protein Interaction (PPI) Network Functional Enrichment Analysis
2.5. Upstream Transcriptional Regulator Analysis
2.6. Disease Biomarker Enrichment and Selection of Potential Drug Targets
3. Results
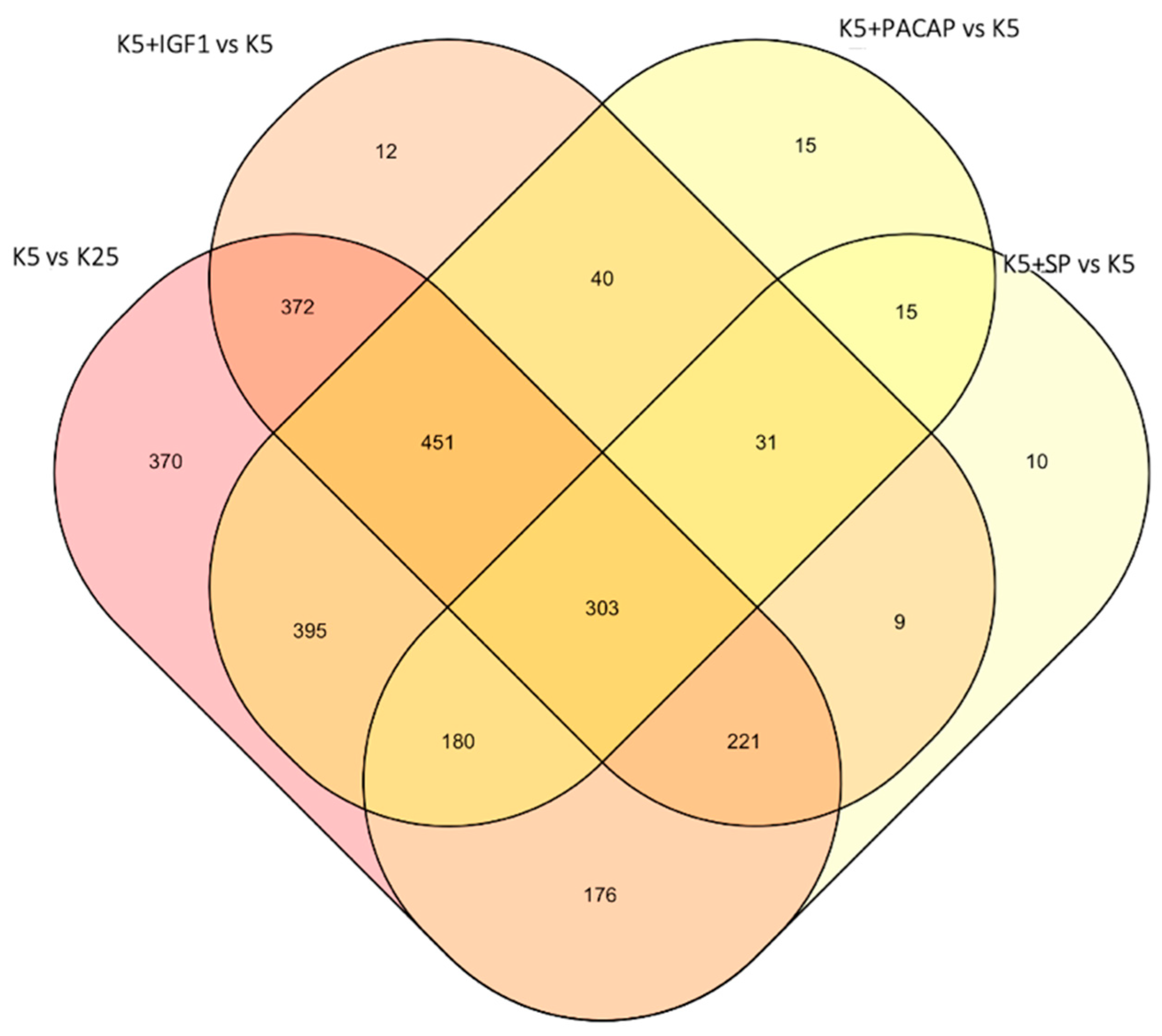
3.1. Identification of a Converging Set of Apoptosis and Survival-Related Genes
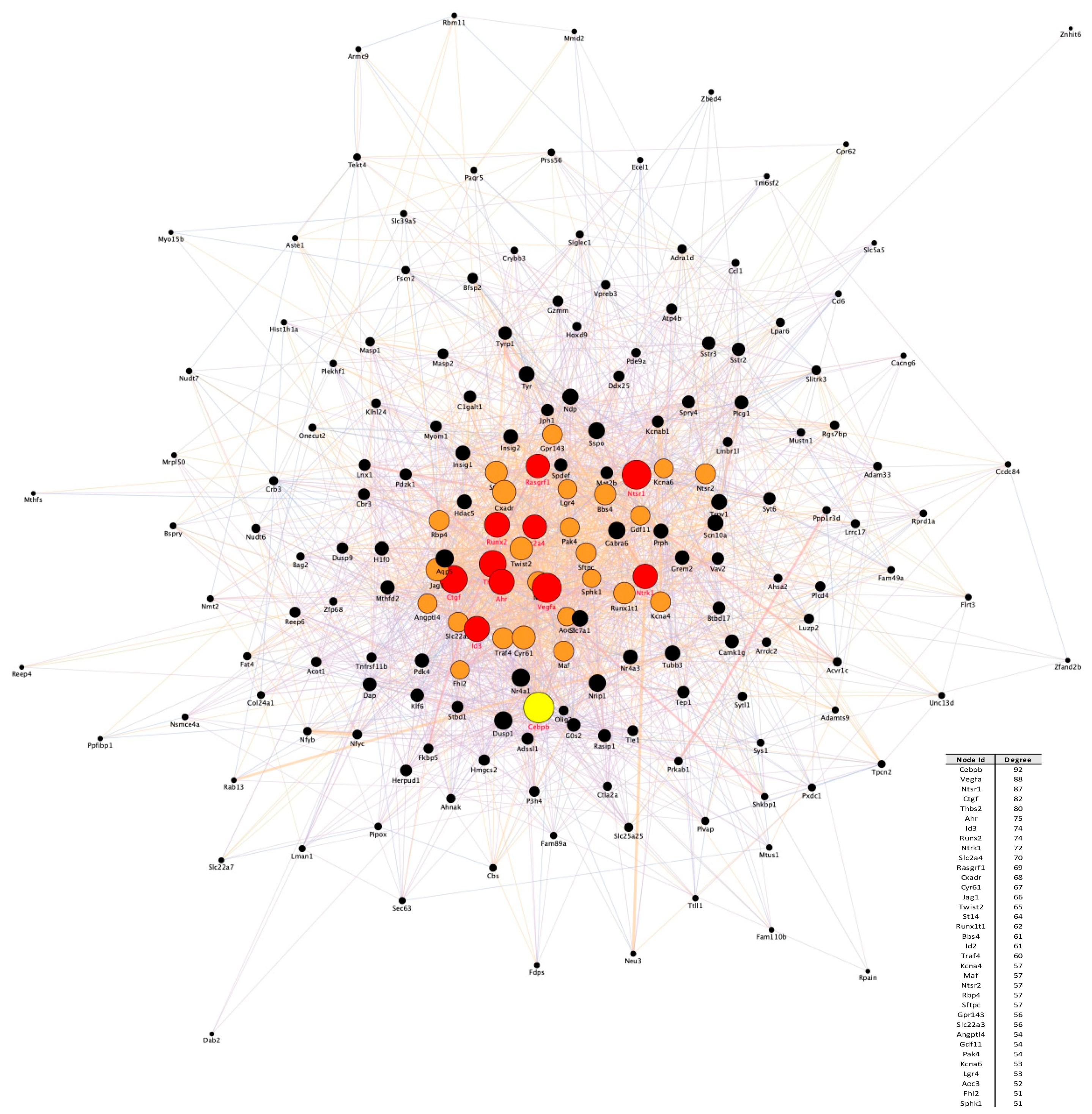
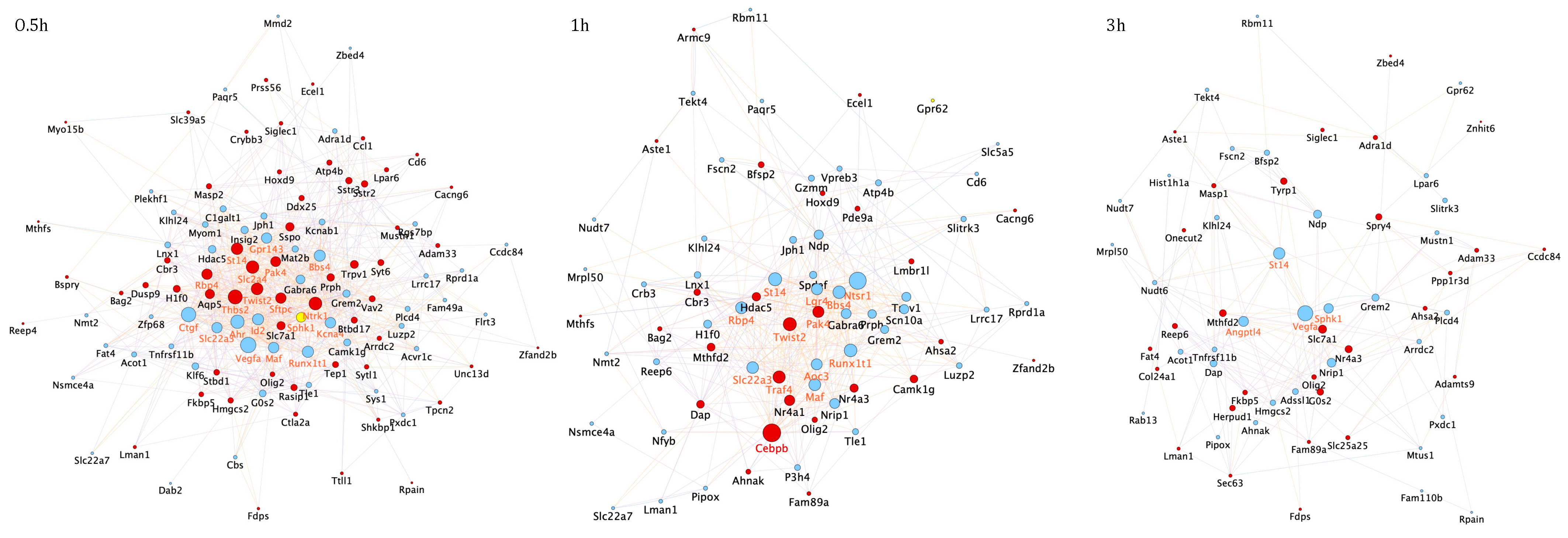
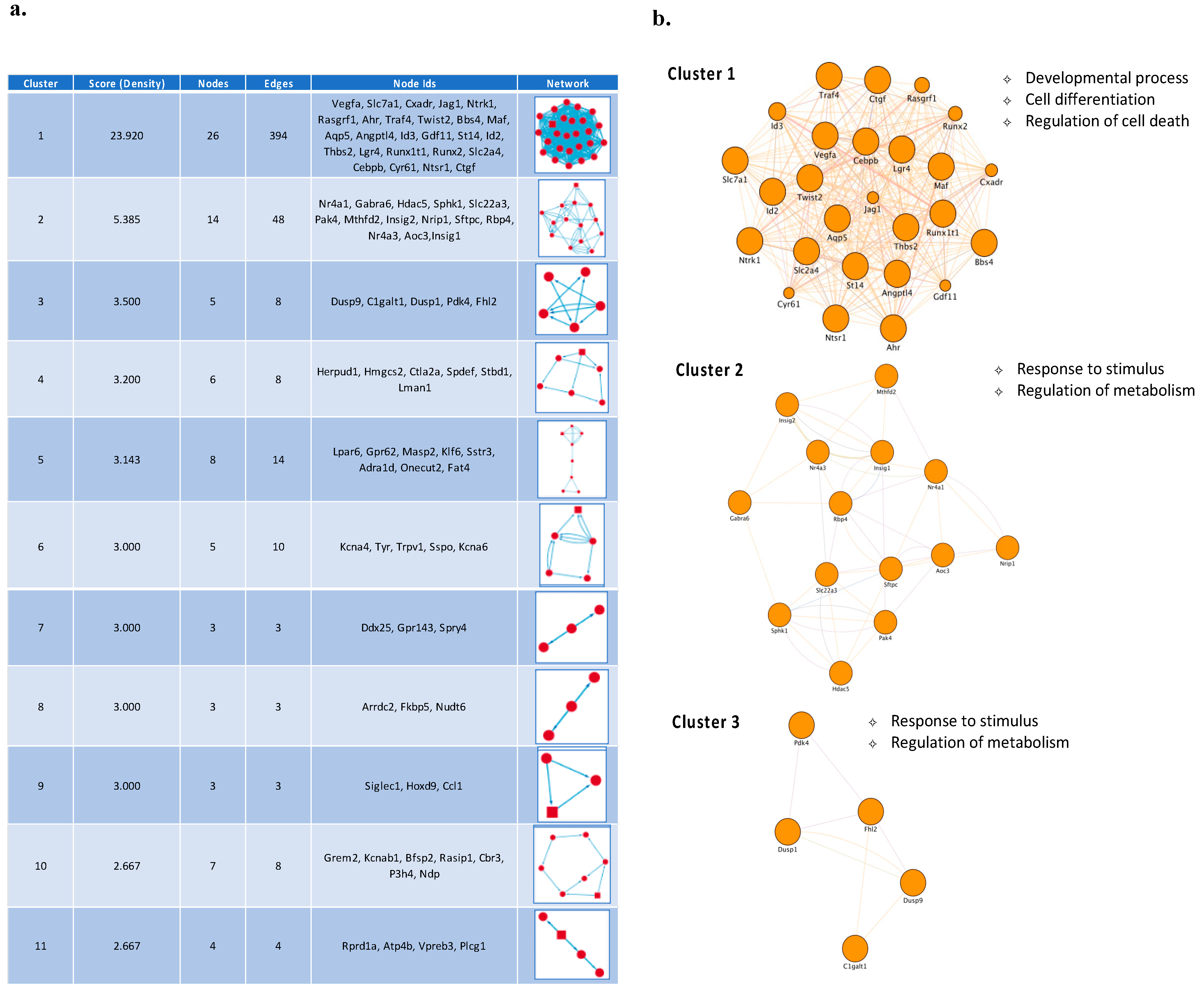
3.2. Temporal PPI Network Analysis Identified Distinct Functional Clusters of Proteins Promoting Neuronal Survival
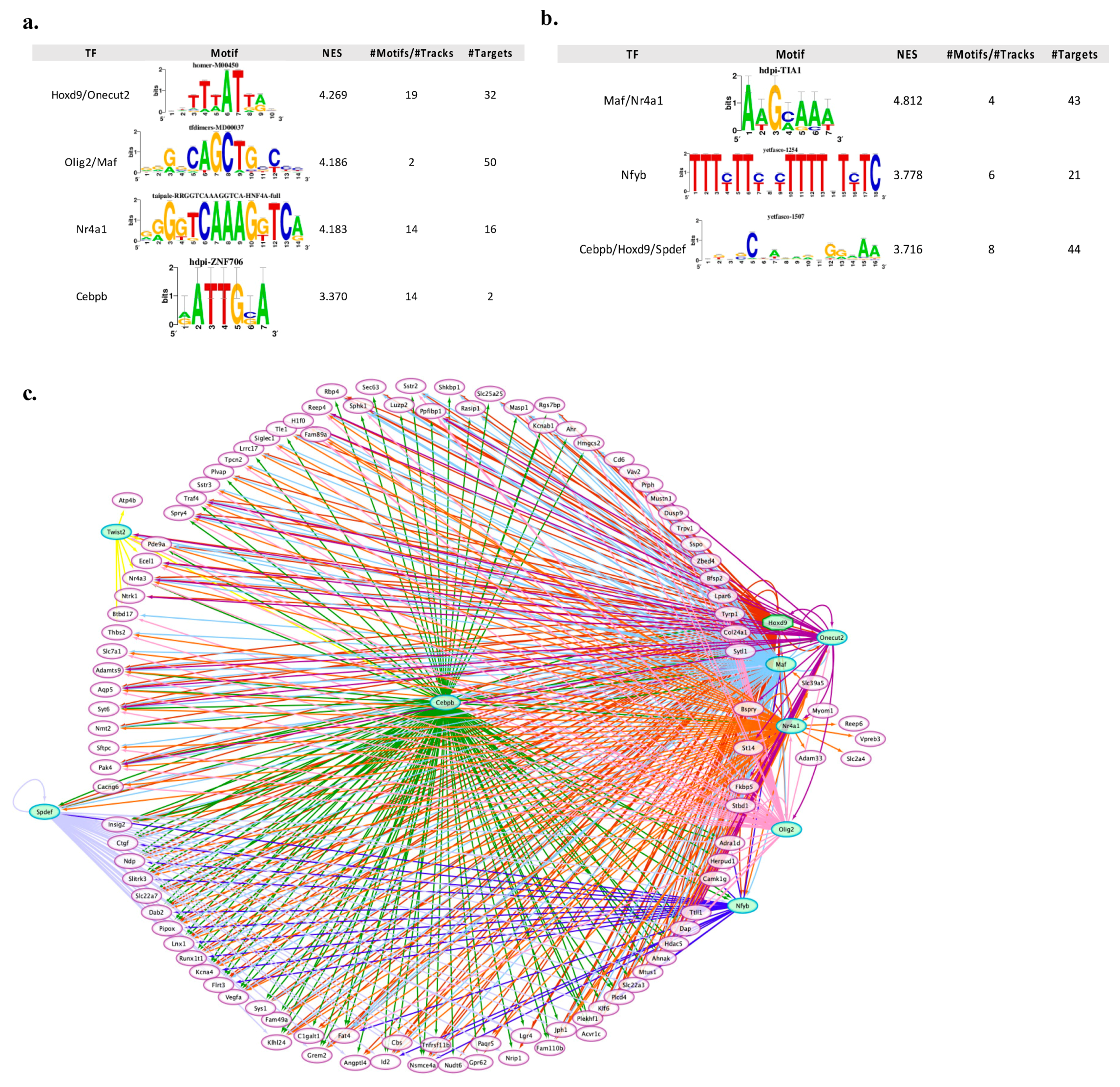
3.3. Transcription Regulatory Network Analysis Identified a Restricted Number of Master Regulators of Neuronal Apoptosis and Survival
3.4. Disease Enrichment Analysis Revealed a Strong Association of Core Set Genes with Neurological and Psychiatric Disorders
3.5. Identification of Repurposing Drugs That Could Revert the Transcriptional Regulation of the Core Set Genes during Neuronal Apoptosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mattson, M.P. Neuronal Life-and-Death Signaling, Apoptosis, and Neurodegenerative Disorders. Antioxid. Redox Signal. 2006, 8, 1997–2006. [Google Scholar] [CrossRef]
- Ceci, M.; Fazi, F.; Romano, N. The role of RNA-binding and ribosomal proteins as specific RNA translation regulators in cellular differentiation and carcinogenesis. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166046. [Google Scholar] [CrossRef] [PubMed]
- Loffreda, A.; Rigamonti, A.; Barabino, S.M.L.; Lenzken, S.C. RNA-Binding Proteins in the Regulation of miRNA Activity: A Focus on Neuronal Functions. Biomolecules 2015, 5, 2363–2387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, A.; Sarmah, D.; Mounica, L.; Kaur, H.; Kesharwani, R.; Verma, G.; Veeresh, P.; Kotian, V.; Kalia, K.; Borah, A.; et al. Cell Death Pathways in Ischemic Stroke and Targeted Pharmacotherapy. Transl. Stroke Res. 2020, 11, 1185–1202. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell Biol. 2000, 1, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, W.; Yang, H. Balancing apoptosis and autophagy for parkinson’s disease therapy: Targeting BCL-2. ACS Chem. Neurosci. 2019, 10, 792–802. [Google Scholar] [CrossRef] [PubMed]
- Tajbakhsh, A.; Read, M.; Barreto, G.E.; Ávila-Rodriguez, M.; Gheibi-Hayat, S.M.; Sahebkar, A. Apoptotic neurons and amyloid-beta clearance by phagocytosis in Alzheimer’s disease: Pathological mechanisms and therapeutic outlooks. Eur. J. Pharmacol. 2021, 895, 173873. [Google Scholar] [CrossRef] [PubMed]
- Ułamek-Kozioł, M.; Czuczwar, S.J.; Kocki, J.; Januszewski, S.; Bogucki, J.; Bogucka-Kocka, A.; Pluta, R. Dysregulation of Autophagy, Mitophagy, and Apoptosis Genes in the CA3 Region of the Hippocampus in the Ischemic Model of Alzheimer’s Disease in the Rat. J. Alzheimer’s Dis. 2019, 72, 1279–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetmańczyk-Sawicka, K.; Iwanicka-Nowicka, R.; Fogtman, A.; Cieśla, J.; Włodarski, P.; Żyżyńska-Granica, B.; Filocamo, M.; Dardis, A.; Peruzzo, P.; Bednarska-Makaruk, M.; et al. Changes in global gene expression indicate disordered autophagy, apoptosis and inflammatory processes and downregulation of cytoskeletal signalling and neuronal development in patients with Niemann–Pick C disease. Neurogenetics 2020, 21, 105–119. [Google Scholar] [CrossRef]
- Yalçinkaya, N.; Haytural, H.; Bilgiç, B.; Özdemir, Ö.; Hanağasi, H.; Küçükali, C.I.; Özbek, Z.; Akcan, U.; Idrisoğlu, H.A.; Gürvit, H.; et al. Expression changes of genes associated with apoptosis and survival processes in Parkinson’s disease. Neurosci. Lett. 2016, 615, 72–77. [Google Scholar] [CrossRef]
- Cavallaro, S. Cracking the code of neuronal apoptosis and survival. Cell Death Dis. 2015, 6, e1963. [Google Scholar] [CrossRef] [Green Version]
- Cavallaro, S. Neuronal apoptosis revealed by genomic analysis: Integrating gene expression profiles with functional information. Neuroinformatics 2007, 5, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Cavallaro, S.; Calissano, P. A Genomic Approach to Investigate Neuronal Apoptosis. Curr. Alzheimer Res. 2006, 3, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Paratore, S.; Parenti, R.; Torrisi, A.; Copani, A.; Cicirata, F.; Cavallaro, S. Genomic profiling of cortical neurons following exposure to β-amyloid. Genomics 2006, 88, 468–479. [Google Scholar] [CrossRef] [Green Version]
- Cavallaro, S.; D’Agata, V.; Alessi, E.; Coffa, S.; Alkon, D.L.; Manickam, P.; Ciotti, M.T.; Possenti, R.; Bonini, P.; Marlier, L.; et al. Gene expression profiles of apoptotic neurons. Genomics 2004, 84, 485–496. [Google Scholar] [CrossRef] [PubMed]
- D’Mello, S.R.; Galli, C.; Ciotti, T.; Calissano, P. Induction of apoptosis in cerebellar granule neurons by low potassium: Inhibition of death by insulin-like growth factor I and cAMP. Proc. Natl. Acad. Sci. USA 1993, 90, 10989–10993. [Google Scholar] [CrossRef] [Green Version]
- Contestabile, A. Cerebellar granule cells as a model to study mechanisms of neuronal apoptosis or survival in vivo and in vitro. Cerebellum 2002, 1, 41–55. [Google Scholar] [CrossRef]
- Amadoro, G.; Pieri, M.; Ciotti, M.T.; Carunchio, I.; Canu, N.; Calissano, P.; Zona, C.; Severini, C. Substance P provides neuroprotection in cerebellar granule cells through Akt and MAPK/Erk activation: Evidence for the involvement of the delayed rectifier potassium current. Neuropharmacology 2007, 52, 1366–1377. [Google Scholar] [CrossRef]
- Cavallaro, S.; Copani, A.; D’Agata, V.; Musco, S.; Petralia, S.; Ventra, C.; Stivala, F.; Travali, S.; Canonico, P.L. Pituitary adenylate cyclase activating polypeptide prevents apoptosis in cultured cerebellar granule neurons. Mol. Pharmacol. 1996, 50, 60–66. [Google Scholar]
- Paparone, S.; Severini, C.; Ciotti, M.T.; D’agata, V.; Calissano, P.; Cavallaro, S. Transcriptional landscapes at the intersection of neuronal apoptosis and substance P-induced survival: Exploring pathways and drug targets. Cell Death Discov. 2016, 2, 16050. [Google Scholar] [CrossRef] [Green Version]
- Maino, B.; D’Agata, V.; Severini, C.; Ciotti, M.T.; Calissano, P.; Copani, A.; Chang, Y.C.; Delisi, C.; Cavallaro, S. Igf1 and pacap rescue cerebellar granule neurons from apoptosis via a common transcriptional program. Cell Death Discov. 2015, 1, 15029. [Google Scholar] [CrossRef] [Green Version]
- Tendi, E.A.; Cunsolo, R.; Bellia, D.; Messina, R.L.; Paratore, S.; Calissano, P.; Cavallaro, S. Drug Target Identification for Neuronal Apoptosis through a Genome Scale Screening. Curr. Med. Chem. 2010, 17, 2906–2920. [Google Scholar] [CrossRef]
- Maino, B.; Paparone, S.; Severini, C.; Ciotti, M.T.; D’agata, V.; Calissano, P.; Cavallaro, S. Drug target identification at the crossroad of neuronal apoptosis and survival. Expert Opin. Drug Discov. 2017, 12, 249–259. [Google Scholar] [CrossRef] [Green Version]
- Maino, B.; Ciotti, M.T.; Calissano, P.; Cavallaro, S. Transcriptional analysis of apoptotic cerebellar granule neurons following rescue by gastric inhibitory polypeptide. Int. J. Mol. Sci. 2014, 15, 5596–5622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaudry, D.; Gonzalez, B.J.; Basille, M.; Pamantung, T.F.; Fontaine, M.; Fournier, A.; Vaudry, H. The neuroprotective effect of pituitary adenylate cyclase-activating polypeptide on cerebellar granule cells is mediated through inhibition of the CED3-related cysteine protease caspase-3/CPP32. Proc. Natl. Acad. Sci. USA 2000, 97, 13390–13395. [Google Scholar] [CrossRef] [Green Version]
- Conesa, A.; Nueda, M.J.; Ferrer, A.; Talón, M. maSigPro: A method to identify significantly differential expression profiles in time-course microarray experiments. Bioinformatics 2006, 22, 1096–1102. [Google Scholar] [CrossRef] [Green Version]
- von Mering, C.; Jensen, L.J.; Snel, B.; Hooper, S.D.; Krupp, M.; Foglierini, M.; Jouffre, N.; Huynen, M.A.; Bork, P. STRING: Known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 2005, 33, D433–D437. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Janky, R.; Verfaillie, A.; Imrichová, H.; van de Sande, B.; Standaert, L.; Christiaens, V.; Hulselmans, G.; Herten, K.; Naval Sanchez, M.; Potier, D.; et al. iRegulon: From a Gene List to a Gene Regulatory Network Using Large Motif and Track Collections. PLoS Comput. Biol. 2014, 10, e1003731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stathias, V.; Turner, J.; Koleti, A.; Vidovic, D.; Cooper, D.; Fazel-Najafabadi, M.; Pilarczyk, M.; Terryn, R.; Chung, C.; Umeano, A.; et al. LINCS Data Portal 2.0: Next generation access point for perturbation-response signatures. Nucleic Acids Res. 2020, 48, D431–D439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarskog, L.F.; Glantz, L.A.; Gilmore, J.H.; Lieberman, J.A. Apoptotic mechanisms in the pathophysiology of schizophrenia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2005, 29, 846–858. [Google Scholar] [CrossRef] [PubMed]
- Jarskog, L.F.; Selinger, E.S.; Lieberman, J.A.; Gilmore, J.H. Apoptotic Proteins in the Temporal Cortex in Schizophrenia: High Bax/Bcl-2 Ratio without Caspase-3 Activation. Am. J. Psychiatry 2004, 161, 109–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Role of Apoptosis in Neuronal Pathology in Schizophrenia…: Current Opinion in Psychiatry. Available online: https://journals.lww.com/co-psychiatry/Citation/2004/05000/The_role_of_apoptosis_in_neuronal_pathology_in.6.aspx (accessed on 6 July 2021).
- Uribe, E.; Wix, R. Neuronal migration, apoptosis and bipolar disorder. Rev. Psiquiatr. Salud Ment. 2012, 5, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Scaini, G.; Fries, G.R.; Valvassori, S.S.; Zeni, C.P.; Zunta-Soares, G.; Berk, M.; Soares, J.C.; Quevedo, J. Perturbations in the apoptotic pathway and mitochondrial network dynamics in peripheral blood mononuclear cells from bipolar disorder patients. Transl. Psychiatry 2017, 7, e1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlović, D. Apoptosis—The potential pathophysiological mechanism in mood disorders modifiable by lithium salts. Biochem. Med. 2008, 18, 291–310. [Google Scholar] [CrossRef]
- Semon, B.A. Dietary cyclic dipeptides, apoptosis and psychiatric disorders: A hypothesis. Med. Hypotheses 2014, 82, 740–743. [Google Scholar] [CrossRef]
- Beyazyüz, M.; Küfeciler, T.; Bulut, L.; Ünsal, C.; Albayrak, Y.; Akyol, E.S.; Baykal, S.; Kuloglu, M.; Hashimoto, K. Increased serum levels of apoptosis in deficit syndrome schizophrenia patients: A preliminary study. Neuropsychiatr. Dis. Treat. 2016, 12, 1261–1268. [Google Scholar] [CrossRef] [Green Version]
- Yin, C.; Huang, G.F.; Sun, X.C.; Guo, Z.; Zhang, J.H. Tozasertib attenuates neuronal apoptosis via DLK/JIP3/MA2K7/JNK pathway in early brain injury after SAH in rats. Neuropharmacology 2016, 108, 316–323. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.R.; Wang, J.; Zhou, S.K.; Yang, L.; Yin, J.L.; Cao, J.P.; Cheng, Y.B. Necrostatin-1 protection of dopaminergic neurons. Neural Regen. Res. 2015, 10, 1120–1124. [Google Scholar] [CrossRef]
- Wu, X.; Zhao, J.; Yu, S.; Chen, Y.; Wu, J.; Zhao, Y. Sulforaphane protects primary cultures of cortical neurons against injury induced by oxygen-glucose deprivation/reoxygenation via antiapoptosis. Neurosci. Bull. 2012, 28, 509–516. [Google Scholar] [CrossRef] [Green Version]
- Kolla, N.; Wei, Z.; Richardson, J.S.; Li, X.M. Amitriptyline and fluoxetine protect PC12 cells from cell death induced by hydrogen peroxide. J. Psychiatry Neurosci. 2005, 30, 196–201. [Google Scholar]
- Jantas, D.; Krawczyk, S.; Lason, W. The predominant protective effect of tianeptine over other antidepressants in models of neuronal apoptosis: The effect blocked by inhibitors of MAPK/ERK1/2 and PI3-K/Akt pathways. Neurotox. Res. 2014, 25, 208–225. [Google Scholar] [CrossRef] [Green Version]
- Lin, K.C.; Wang, C.C.; Wang, S.J. Bupropion attenuates kainic acid-induced seizures and neuronal cell death in rat hippocampus. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2013, 45, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.D.; Chen, X.L.; Ding, S.F.; Kou, D.; Hu, H.L.; Li, L. Promethazine inhibits neuronal apoptosis via PI3K/Akt signaling pathway in rats with cerebral infarction. Eur. Rev. Med. Pharmacol. Sci. 2021, 23, 126–134. [Google Scholar] [CrossRef]
- Lackie, R.E.; Maciejewski, A.; Ostapchenko, V.G.; Marques-Lopes, J.; Choy, W.-Y.; Duennwald, M.L.; Prado, V.F.; Prado, M.A.M. The Hsp70/Hsp90 Chaperone Machinery in Neurodegenerative Diseases. Front. Neurosci. 2017, 11, 254. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Bansal, A.; Hashimoto-Torii, K. HSP70 and HSP90 in neurodegenerative diseases. Neurosci. Lett. 2020, 716, 134678. [Google Scholar] [CrossRef] [PubMed]
- Bohush, A.; Bieganowski, P.; Filipek, A. Hsp90 and Its Co-Chaperones in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 4976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyon, M.S.; Milligan, C. Extracellular heat shock proteins in neurodegenerative diseases: New perspectives. Neurosci. Lett. 2019, 711, 134462. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.J.; Fort, P.E. Heat Shock Proteins Regulatory Role in Neurodevelopment. Front. Neurosci. 2018, 12, 821. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, D.; Jäger, R.; Mosser, D.D.; Samali, A. Regulation of apoptosis by heat shock proteins. IUBMB Life 2014, 66, 327–338. [Google Scholar] [CrossRef]
- Beere, H.M. Death versus survival: Functional interaction between the apoptotic and stress-inducible heat shock protein pathways. J. Clin. Investig. 2005, 115, 2633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, H.; Howarth, J.; Lee, Y.B.; Wong, L.F.; Bantounas, I.; Phylactou, L.; Verkade, P.; Uney, J.B. MiR-3120 Is a Mirror MicroRNA That Targets Heat Shock Cognate Protein 70 and Auxilin Messenger RNAs and Regulates Clathrin Vesicle Uncoating. J. Biol. Chem. 2012, 287, 14726–14733. [Google Scholar] [CrossRef] [Green Version]
- Pulido-Salgado, M.; Vidal-Taboada, J.M.; Saura, J. C/EBPβ and C/EBPδ transcription factors: Basic biology and roles in the CNS. Prog. Neurobiol. 2015, 132, 1–33. [Google Scholar] [CrossRef]
- Moore, F.; Santin, I.; Nogueira, T.C.; Gurzov, E.N.; Marselli, L.; Marchetti, P.; Eizirik, D.L. The transcription factor C/EBP delta has anti-apoptotic and anti-inflammatory roles in pancreatic beta cells. PLoS ONE 2012, 7, e31062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meir, O.; Dvash, E.; Werman, A.; Rubinstein, M. C/EBP-β regulates endoplasmic reticulum stress-triggered cell death in mouse and human models. PLoS ONE 2010, 5, e9516. [Google Scholar] [CrossRef]
- Cortes-Canteli, M.; Aguilar-Morante, D.; Sanz-SanCristobal, M.; Megias, D.; Santos, A.; Perez-Castillo, A. Role of C/EBPβ transcription factor in adult hippocampal neurogenesis. PLoS ONE 2011, 6, e24842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kfoury, N.; Kapatos, G. Identification of neuronal target genes for CCAAT/Enhancer Binding Proteins. Mol. Cell. Neurosci. 2009, 40, 313–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valente, T.; Dentesano, G.; Ezquerra, M.; Fernandez-Santiago, R.; Martinez-Martin, J.; Gallastegui, E.; Domuro, C.; Compta, Y.; Martí, M.J.; Bachs, O.; et al. CCAAT/enhancer binding protein δ is a transcriptional repressor of α-synuclein. Cell Death Differ. 2020, 27, 509–524. [Google Scholar] [CrossRef]
- Peña-Altamira, E.; Polazzi, E.; Moretto, E.; Lauriola, M.; Monti, B. The transcription factor CCAAT enhancer-binding protein β protects rat cerebellar granule neurons from apoptosis through its transcription-activating isoforms. Eur. J. Neurosci. 2014, 39, 176–185. [Google Scholar] [CrossRef]
- Calella, A.M.; Nerlov, C.; Lopez, R.G.; Sciarretta, C.; Von Bohlen Und Halbach, O.; Bereshchenko, O.; Minichiello, L. Neurotrophin/Trk receptor signaling mediates C/EBPα, -β and NeuroD recruitment to immediate-early gene promoters in neuronal cells and requires C/EBPs to induce immediate-early gene transcription. Neural Dev. 2007, 2, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Pan, H.C.; Yang, C.N.; Hung, Y.W.; Lee, W.J.; Tien, H.R.; Shen, C.C.; Sheehan, J.; Chou, C.T.; Sheu, M.L. Reciprocal modulation of C/EBP-α and C/EBP-β by IL-13 in activated microglia prevents neuronal death. Eur. J. Immunol. 2013, 43, 2854–2865. [Google Scholar] [CrossRef]
- Wang, Z.H.; Xiang, J.; Liu, X.; Yu, S.P.; Manfredsson, F.P.; Sandoval, I.M.; Wu, S.; Wang, J.Z.; Ye, K. Deficiency in BDNF/TrkB Neurotrophic Activity Stimulates δ-Secretase by Upregulating C/EBPβ in Alzheimer’s Disease. Cell Rep. 2019, 28, 655–669.e5. [Google Scholar] [CrossRef] [Green Version]
- Turovskaya, M.V.; Gaidin, S.G.; Vedunova, M.V.; Babaev, A.A.; Turovsky, E.A. BDNF Overexpression Enhances the Preconditioning Effect of Brief Episodes of Hypoxia, Promoting Survival of GABAergic Neurons. Neurosci. Bull. 2020, 36, 733–760. [Google Scholar] [CrossRef] [Green Version]
- Minichiello, L.; Klein, R. TrkB and TrkC neurotrophin receptors cooperate in promoting survival of hippocampal and cerebellar granule neurons. Genes Dev. 1996, 10, 2849–2858. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; He, F.; Zhong, Z.; Lv, R.; Xiao, S.; Liu, Z. Overexpression of NTRK1 Promotes Differentiation of Neural Stem Cells into Cholinergic Neurons. BioMed Res. Int. 2015, 2015, 857202. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.D.; Yang, J.L.; Lin, Y.C.; Chao, A.C.; Yang, D.I. Emerging Roles of Inhibitor of Differentiation-1 in Alzheimer’s Disease: Cell Cycle Reentry and Beyond. Cells 2020, 9, 1746. [Google Scholar] [CrossRef]
- Gleichmann, M.; Buchheim, G.; El-Bizri, H.; Yokota, Y.; Klockgether, T.; Kügler, S.; Bähr, M.; Weller, M.; Schulz, J.B. Identification of inhibitor-of-differentiation 2 (Id2) as a modulator of neuronal apoptosis. J. Neurochem. 2002, 80, 755–762. [Google Scholar] [CrossRef]
- Briscoe, J.; Wilkinson, D.G. Establishing neuronal circuitry: Hox genes make the connection. Genes Dev. 2004, 18, 1643–1648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatlekar, S.; Fields, J.Z.; Boman, B.M. Role of HOX genes in stem cell differentiation and cancer. Stem Cells Int. 2018, 2018, 3569493. [Google Scholar] [CrossRef] [Green Version]
- Kuert, P.A.; Hartenstein, V.; Bello, B.C.; Lovick, J.K.; Reichert, H. Neuroblast lineage identification and lineage-specific Hox gene action during postembryonic development of the subesophageal ganglion in the Drosophila central brain. Dev. Biol. 2014, 390, 102–115. [Google Scholar] [CrossRef] [Green Version]
- Seto, Y.; Ishiwata, S.; Hoshino, M. Characterization of Olig2 expression during cerebellar development. Gene Expr. Patterns 2014, 15, 1–7. [Google Scholar] [CrossRef]
- Liu, H.; Weng, W.; Guo, R.; Zhou, J.; Xue, J.; Zhong, S.; Cheng, J.; Zhu, M.X.; Pan, S.J.; Li, Y. Olig2 SUMOylation protects against genotoxic damage response by antagonizing p53 gene targeting. Cell Death Differ. 2020, 27, 3146–3161. [Google Scholar] [CrossRef]
- Gaber, Z.B.; Novitch, B.G. Previews All the Embryo’s a Stage, and Olig2 in Its Time Plays Many Parts. Neuron 2011, 69, 833–835. [Google Scholar] [CrossRef] [Green Version]
- Furusho, M.; Kaga, Y.; Ishii, A.; Hébert, J.M.; Bansal, R. Fibroblast growth factor signaling is required for the generation of oligodendrocyte progenitors from the embryonic forebrain. J. Neurosci. 2011, 31, 5055–5066. [Google Scholar] [CrossRef] [Green Version]
- Allahdadi, K.J.; De Santana, T.A.; Santos, G.C.; Azevedo, C.M.H.; Mota, R.A.; Nonaka, C.K.; Silva, D.N.; Valim, C.X.R.; Figueira, C.P.; Dos Santos, W.L.C.; et al. IGF-1 overexpression improves mesenchymal stem cell survival and promotes neurological recovery after spinal cord injury. Stem Cell Res. Ther. 2019, 10, 146. [Google Scholar] [CrossRef]
- Cui, Q.L.; Fragoso, G.; Miron, V.E.; Darlington, P.J.; Mushynski, W.E.; Antel, J.; Almazan, G. Response of human oligodendrocyte progenitors to growth factors and axon signals. J. Neuropathol. Exp. Neurol. 2010, 69, 930–944. [Google Scholar] [CrossRef] [Green Version]
- Cortés, D.; Carballo-Molina, O.A.; Castellanos-Montiel, M.J.; Velasco, I. The non-survival effects of Glial cell line-derived neurotrophic factor on neural cells. Front. Mol. Neurosci. 2017, 10, 258. [Google Scholar] [CrossRef]
- Stewart, S.E.; Platko, J.; Fagerness, J.; Birns, J.; Jenike, E.; Smoller, J.W.; Perlis, R.; Leboyer, M.; Delorme, R.; Chabane, N.; et al. A genetic family-based association study of OLIG2 in obsessive-compulsive disorder. Arch. Gen. Psychiatry 2007, 64, 209–215. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Wang, F.; Gan, J.; Zhang, Z.; Liang, X.; Li, T.; Huang, N.; Zhao, X.; Mei, F.; Xiao, L. Myelin Deficits Caused by Olig2 Deficiency Lead to Cognitive Dysfunction and Increase Vulnerability to Social Withdrawal in Adult Mice. Neurosci. Bull. 2020, 36, 419–426. [Google Scholar] [CrossRef]
- Komatsu, H.; Takeuchi, H.; Kikuchi, Y.; Ono, C.; Yu, Z.; Iizuka, K.; Takano, Y.; Kakuto, Y.; Funakoshi, S.; Ono, T.; et al. Ethnicity-Dependent Effects of Schizophrenia Risk Variants of the OLIG2 Gene on OLIG2 Transcription and White Matter Integrity. Schizophr. Bull. 2020, 46, 1619–1628. [Google Scholar] [CrossRef]
- Sims, R.; Hollingworth, P.; Moskvina, V.; Dowzell, K.; O’Donovan, M.C.; Powell, J.; Lovestone, S.; Brayne, C.; Rubinsztein, D.; Owen, M.J.; et al. Evidence that variation in the oligodendrocyte lineage transcription factor 2 (OLIG2) gene is associated with psychosis in Alzheimer’s disease. Neurosci. Lett. 2009, 461, 54–59. [Google Scholar] [CrossRef]
- Tan, B.T.; Yu, J.; Yin, Y.; Jia, G.W.; Jiang, W.; Yu, L.H. The Olig family affects central nervous system development and disease. Neural Regen. Res. 2014, 9, 329–336. [Google Scholar]
- Satoh, J.-I.; Asahina, N.; Kitano, S.; Kino, Y. A Comprehensive Profile of ChIP-Seq-Based Olig2 Target Genes in Motor Neuron Progenitor Cells Suggests the Possible Involvement of Olig2 in the Pathogenesis of Amyotrophic Lateral Sclerosis. J. Cent. Nerv. Syst. Dis. 2015, 7, JCNSD-S23210. [Google Scholar] [CrossRef]
- Benatti, P.; Basile, V.; Merico, D.; Fantoni, L.I.; Tagliafico, E.; Imbriano, C. A balance between NF-Y and p53 governs the pro- and anti-apoptotic transcriptional response. Nucleic Acids Res. 2008, 36, 1415–1428. [Google Scholar] [CrossRef] [Green Version]
- Ly, L.L.; Yoshida, H.; Yamaguchi, M. Nuclear transcription factor Y and its roles in cellular processes related to human disease. Am. J. Cancer Res. 2013, 3, 339–346. [Google Scholar]
- Hale, T.K.; Myers, C.; Maitra, R.; Kolzau, T.; Nishizawa, M.; Braithwaite, A.W. Maf transcriptionally activates the mouse p53 promoter and causes a p53-dependent cell death. J. Biol. Chem. 2000, 275, 17991–17999. [Google Scholar] [CrossRef] [Green Version]
- Herring, J.A.; Elison, W.S.; Tessem, J.S. Function of Nr4a Orphan Nuclear Receptors in Proliferation, Apoptosis and Fuel Utilization across Tissues. Cells 2019, 8, 1373. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Wang, Y.; Ertürk, A.; Kallop, D.; Jiang, Z.; Weimer, R.M.; Kaminker, J.; Sheng, M. Activity-induced Nr4a1 regulates spine density and distribution pattern of excitatory synapses in pyramidal neurons. Neuron 2014, 83, 431–443. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, M.A.; Muscat, G.E.O. The NR4A Subgroup: Immediate Early Response Genes with Pleiotropic Physiological Roles. Nucl. Recept. Signal. 2006, 4, nrs-04002. [Google Scholar] [CrossRef] [Green Version]
- Volakakis, N.; Kadkhodaei, B.; Joodmardi, E.; Wallis, K.; Panman, L.; Silvaggi, J.; Spiegelman, B.M.; Perlmann, T. NR4A orphan nuclear receptors as mediators of CREB-dependent neuroprotection. Proc. Natl. Acad. Sci. USA 2010, 107, 12317–12322. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Walsh, E.N.; Yan, A.L.; Giese, K.P.; Safe, S.; Abel, T. Pharmacological activation of Nr4a rescues age-associated memory decline. Neurobiol. Aging 2020, 85, 140–144. [Google Scholar] [CrossRef]
- Munoz-Tello, P.; Lin, H.; Khan, P.; de Vera, I.M.; Kamenecka, T.; Kojetin, D. Assessment of NR4A Ligands that Directly Bind and Modulate the Orphan Nuclear Receptor Nurr1. J. Med. Chem. 2020, 63, 15639–15654. [Google Scholar] [CrossRef]
- Bridi, M.S.; Hawk, J.D.; Chatterjee, S.; Safe, S.; Abel, T. Pharmacological Activators of the NR4A Nuclear Receptors Enhance LTP in a CREB/CBP-Dependent Manner. Neuropsychopharmacology 2016, 42, 1243–1253. [Google Scholar] [CrossRef] [Green Version]
- Jakaria, M.; Haque, M.E.; Cho, D.-Y.; Azam, S.; Kim, I.-S.; Choi, D.-K. Molecular Insights into NR4A2(Nurr1): An Emerging Target for Neuroprotective Therapy against Neuroinflammation and Neuronal Cell Death. Mol. Neurobiol. 2019, 56, 5799–5814. [Google Scholar] [CrossRef]
- Corley, S.M.; Tsai, S.Y.; Wilkins, M.R.; Weickert, C.S. Transcriptomic analysis shows decreased cortical expression of nr4a1, nr4a2 and rxrb in schizophrenia and provides evidence for nuclear receptor dysregulation. PLoS ONE 2016, 11, e0166944. [Google Scholar] [CrossRef] [Green Version]
- Tsai, S.-Y.; Catts, V.S.; Fullerton, J.M.; Corley, S.M.; Fillman, S.G.; Weickert, C.S. Nuclear Receptors and Neuroinflammation in Schizophrenia. Mol. Neuropsychiatry 2017, 3, 181–191. [Google Scholar] [CrossRef]
- Jeanneteau, F.; Barrè, C.; Vos, M.; De Vries, C.J.M.; Rouillard, X.; Levesque, D.; Dromard, Y.; Moisan, M.-P.; Duric, V.; Franklin, T.C.; et al. The Stress-Induced Transcription Factor NR4A1 Adjusts Mitochondrial Function and Synapse Number in Prefrontal Cortex. J. Neurosci. 2018, 38, 1335–1350. [Google Scholar] [CrossRef] [Green Version]
- Rouillard, C.; Baillargeon, J.; Paquet, B.; St-Hilaire, M.; Maheux, J.; Lévesque, C.; Darlix, N.; Majeur, S.; Lévesque, D. Genetic disruption of the nuclear receptor Nur77 (Nr4a1) in rat reduces dopamine cell loss and L-Dopa-induced dyskinesia in experimental Parkinson’s disease. Exp. Neurol. 2018, 304, 143–153. [Google Scholar] [CrossRef]
- Zhao, L.G.; Tang, Y.; Tan, J.Z.; Wang, J.W.; Chen, G.J.; Zhu, B.L. The effect of NR4A1 on APP metabolism and tau phosphorylation. Genes Dis. 2018, 5, 342–348. [Google Scholar] [CrossRef]
- Bao, X.J.; Wang, G.C.; Zuo, F.X.; Li, X.Y.; Wu, J.; Chen, G.; Dou, W.C.; Guo, Y.; Shen, Q.; Wang, R.Z. Transcriptome profiling of the subventricular zone and dentate gyrus in an animal model of Parkinson’s disease. Int. J. Mol. Med. 2017, 40, 771–783. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.-F.; Lu, Y.-J.; Wang, Z.-G. MicroRNAs and apoptosis: Implications in the molecular therapy of human disease. Clin. Exp. Pharmacol. Physiol. 2009, 36, 951–960. [Google Scholar] [CrossRef]
- Marson, A.; Levine, S.S.; Cole, M.F.; Frampton, G.M.; Brambrink, T.; Johnstone, S.; Guenther, M.G.; Johnston, W.K.; Wernig, M.; Newman, J.; et al. Connecting microRNA Genes to the Core Transcriptional Regulatory Circuitry of Embryonic Stem Cells. Cell 2008, 134, 521–533. [Google Scholar] [CrossRef] [Green Version]
- Zolboot, N.; Du, J.X.; Zampa, F.; Lippi, G. MicroRNAs Instruct and Maintain Cell Type Diversity in the Nervous System. Front. Mol. Neurosci. 2021, 14, 69. [Google Scholar] [CrossRef]
- Gagliardi, D.; Comi, G.P.; Bresolin, N.; Corti, S. MicroRNAs as regulators of cell death mechanisms in amyotrophic lateral sclerosis. J. Cell. Mol. Med. 2019, 23, 1647. [Google Scholar] [CrossRef]
- Lang, M.-F.; Shi, Y. Dynamic Roles of microRNAs in Neurogenesis. Front. Neurosci. 2012, 6, 71. [Google Scholar] [CrossRef] [Green Version]
- Jung, H.J.; Suh, Y. Regulation of IGF -1 signaling by microRNAs. Front. Genet. 2015, 5, 472. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Ni, S.; Luo, Z.; Lang, Y.; Hu, J.; Lu, H. The protective effect of microRNA-21 in neurons after spinal cord injury. Spinal Cord 2018, 57, 141–149. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease Class | p Value | Genes Number |
|---|---|---|
| Vision (Glaucoma, Macular retinal edema, …) | 4.9 × 10−3 | 15 |
| Psychiatric disorders | 1.4 × 10−2 | 29 |
| Hematological disease | 4.4 × 10−2 | 22 |
| Cardiovascular disease | 4.7 × 10−2 | 50 |
| Immune disease | 5.0 × 10−2 | 37 |
| Gene Symbol | Gene Name | Disease |
|---|---|---|
| ATP4B | ATPase H+/K+ transporting beta subunit | Bipolar Disorder |
| FAT4 | FAT atypical cadherin 4 | Bipolar Disorder |
| FKBP5 | FK506 binding protein 5 | Depression, affective psychoses, post-traumatic stress disorder, bipolar disorders |
| RASIP1 | Ras interacting protein 1 | Bipolar Disorder |
| ADRA1D | Adrenoceptor alpha 1D | Several psychiatric disorders |
| AHR | Aryl hydrocarbon receptor | Dementia |
| C1GALT1 | Core 1 synthase, glycoprotein-N-acetylgalactosamine 3-beta-galactosyltransferase 1 | Bipolar Disorder |
| CBS | Cystathionine-beta-synthase | Dementia (AD), migraine disorders, schizophrenia |
| DAP | Death-associated protein | Schizophrenia |
| GABRA6 | Gamma-aminobutyric acid type A receptor alpha-6-subunit | Schizophrenia, anxiety disorder |
| ID2 | Inhibitor of DNA binding 2, HLH protein | Attention-deficit hyperactivity disorder |
| INSIG2 | Insulin-induced gene 2 | Schizophrenia |
| MASP2 | Mannan binding lectin serine peptidase 2 | Dementia |
| NTSR1 | Neurotensin receptor 1 | Schizophrenia, several psychiatric disorders |
| NTRK1 | Neurotrophic receptor tyrosine kinase 1 | Several psychiatric disorders, autism, dementia |
| NR4A1 | Nuclear receptor subfamily 4 group A member 1 | Schizophrenia, bipolar disorder |
| NR4A3 | Nuclear receptor subfamily 4 group A member 3 | Schizophrenia, bipolar disorder |
| NUDT6 | Nudix hydrolase 6 | Schizophrenia, bipolar disorder |
| OLIG2 | Oligodendrocyte lineage transcription factor 2 | Schizophrenia, obsessive compulsive disorder, Tourette syndrome, dementia |
| PDE9A | Phosphodiesterase 9A | Depression |
| PLCD4 | Phospholipase C delta 4 | Several psychiatric disorders |
| PAQR5 | Progestin and adipoQ receptor family member 5 | Mental Disorders |
| SSTR2 | Somatostatin receptor 2 | Several psychiatric disorders |
| SSTR3 | Somatostatin receptor 3 | Several psychiatric disorders |
| SPRY4 | Sprouty RTK signaling antagonist 4 | Schizophrenia |
| SYT6 | Synaptotagmin 6 | Mental Disorders |
| TRPV1 | Transient receptor potential cation channel subfamily V member 1 | Autism |
| VEGFA | Vascular endothelial growth factor A | Major depressive disorder, autism, dementia |
| ZBED4 | Zinc finger BED-type containing 4 | Schizophrenia, bipolar disorder |
| Disease State | Concordance | p Value | No. of Genes |
|---|---|---|---|
| Lean | 0.64 | 1.50 × 10−3 | 21 |
| Adenocarcinoma | 0.58 | 1.17 × 10−6 | 159 |
| Hypernephroma | 0.50 | 1.23 × 10−2 | 159 |
| Carcinosarcoma | 0.49 | 3.01 × 10−2 | 159 |
| Renal_cell_carcinoma | 0.49 | 4.11 × 10−2 | 159 |
| Amyotrophic_lateral_sclerosis_ | 0.48 | 4.95 × 10−5 | 65 |
| No_atrial_fibrillation | 0.47 | 5.53 × 10−5 | 65 |
| Duchenne_muscular_dystrophy | 0.46 | 5.58 × 10−5 | 87 |
| Carcinoma | 0.42 | 1.22 × 10−4 | 80 |
| B-cell acute lymphoblastic leukemia | 0.42 | 3.19 × 10−2 | 26 |
| Rank | Perturbation | p-Value | Correlation Score | Mechanism of Action | Pharmacological Class (Current Indication) |
|---|---|---|---|---|---|
| 1 | Tozasertib | 2.08 × 10−5 | −0.98 | Aurora A/B/C kinases inhibitor | Chemotherapeutic |
| 2 | Necrostatin | 5.29 × 10−5 | −0.97 | RIP1 kinase inhibitor | Inhibitor of necroptosis |
| 3 | Tianeptine | 8.71 × 10−5 | −0.97 | Mu-type opioid receptor agonist | Tricyclic antidepressant |
| 4 | L-Sulforaphane | 1.00 × 10−4 | −0.97 | N/A | Anticancer |
| 5 | Pentoxifylline | 1.77 × 10−4 | −0.96 | Phosphodiesterase inhibitor | Hemorheological agent |
| 6 | Purmorphamine | 1.80 × 10−4 | −0.96 | Sonic Hedgehog agonist | - |
| 7 | Nicergoline | 1.91 × 10−4 | −0.96 | Alpha-1A adrenergic receptor antagonist | Vasodilator Agent |
| 8 | Pifithrin | 2.95 × 10−30 | −0.95 | p53 inhibitor | - |
| 9 | 5-Nonyloxytryptamine | 2.72 × 10−4 | −0.95 | Serotonin Receptor Agonist | - |
| 10 | Nifedipine | 2.79 × 10−4 | −0.95 | Specific blocker of L-type calcium channels | Antihypertensive, Antianginal |
| 11 | Tyrphostin | 1.54 × 10−15 | −0.92 | EGFR inhibitor | Antineoplastic |
| 12 | Parthenolide | 4.11 × 10−15 | −0.92 | NFKB inhibitor | - |
| 13 | Atorvastatin | 1.03 × 10−6 | −0.90 | HMG-CoA inhibitor | Statin (used to lower lipid levels and reduce the risk of cardiovascular disease) |
| 14 | Tanespimycin | 7.69 × 10−5 | −0.86 | HSP inhibitor | Anticancer |
| 15 | Monorden/Radicicol | 6.94 × 10−3 | −0.85 | HSP inhibitor | - |
| 16 | Azacyclonol | 1.55 × 10−2 | −0.81 | N/A | Antipsychotic |
| 17 | Rapamycin | 1.66 × 10−6 | −0.54 | mTOR inhibitor | Immunosuppressive |
| 18 | Amitriptyline | 7.97 × 10−6 | −0.50 | Norepinephrine and serotonin reuptake inhibitor | Tricyclic antidepressant |
| 19 | Allopurinol | 8.01 × 10−6 | −0.50 | Xanthine dehydrogenase/oxidase inhibitor | Xanthine Oxidase Inhibitors; Antigout Agents |
| 20 | Nortriptyline | 8.62 × 10−6 | −0.50 | Multiple | Tricyclic antidepressant |
| 21 | Bupropion | 1.57 × 10−5 | −0.49 | Norepinephrine/dopamine-reuptake inhibitor | Antidepressant |
| 22 | Roflumilast | 1.84 × 10−5 | −0.48 | Phosphodiesterase-4 inhibitor | Tricyclic antidepressant |
| 23 | Tranilast | 3.41 × 10−5 | −0.47 | Hematopoietic prostaglandin D synthase inhibitor | Antiallergic |
| 24 | Indomethacin | 3.10 × 10−5 | −0.47 | COX inhibitor | Non-steroidal anti-inflammatory drug |
| 25 | Nystatin | 3.24 × 10−5 | −0.47 | Channel-forming ionophore | Antifungal |
| 26 | Theophylline | 3.80 × 10−5 | −0.47 | Adenosine receptor antagonist | Bronchodilator |
| 27 | Citalopram | 3.85 × 10−5 | −0.47 | Reuptake of serotonin inhibitor | Antidepressant |
| 28 | Piracetam | 4.11 × 10−5 | −0.47 | Acetylcholine receptor agonist | Antipsychotic |
| 29 | Tacrolimus | 5.22 × 10−5 | −0.46 | Peptidyl-prolyl cis-trans isomerase FKBP1A, inhibitor | Immunosuppressive |
| 30 | Diazepam | 8.10 × 10−5 | −0.45 | GABA(A) Receptor positive allosteric modulator | Anxiolytic, sedative |
| 31 | Iproniazid | 7.33 × 10−5 | −0.45 | MAO inhibitor | Antidepressant |
| 32 | Promazine hydrochloride | 5.32 × 10−4 | −0.45 | Dopamine receptor antagonist | Antipsychotic |
| 33 | Cyproheptadine | 1.62 × 10−2 | −0.45 | Histamine receptor antagonist | Antiallergic |
| 34 | Dipyrone | 1.07 × 10−4 | −0.44 | N/A | Non-steroidal anti-inflammatory drug |
| 35 | Ethosuximide | 1.0 × 10−4 | −0.44 | T-type voltage sensitive calcium channels inhibitor | Anticonvulsants |
| 36 | Phenotiazine | 1.09 × 10−4 | −0.44 | N/A | Antipsychotic |
| 37 | Sulfanilamide | 1.12 × 10−4 | −0.44 | Dihydropteroate synthase inhibitor | Antibiotic |
| 38 | Clozapine | 1.35 × 10−4 | −0.44 | Dopamine/Serotonin antagonist | Antipsychotic |
| 39 | Lamotrigine | 1.68 × 10−4 | −0.43 | Multiple | Antiepileptic |
| 40 | Doxepin | 1.94 × 10−4 | −0.43 | Selective histamine H1 receptor blocker | Tricyclic antidepressant |
| 41 | Moclobemide | 2.22 × 10−4 | −0.43 | MAO inhibitor | Antidepressant |
| 42 | Rifabutin | 2.34 × 10−4 | −0.43 | DNA-dependent RNA polymerase inhibitor | Antibiotic |
| 43 | Rolipram | 2.15 × 10−4 | −0.43 | N/A | Antidepressant |
| 44 | Enaplapril | 2.75 × 10−4 | −0.42 | ACE inhibitor | Antihypertensive |
| 45 | Geldanamycin | 1.62 × 10−6 | −0.42 | N/A | Anticancer |
| 46 | Sibutramine | 3.77 × 10−4 | −0.41 | Dopamine, norepinephrine, serotonin transporter inhibitor | Antiobesity |
| 47 | Phenelzine | 9.79 × 10−4 | −0.38 | MAO inhibitor | Antidepressant |
| 48 | Thioridazine | 4.51 × 10−5 | −0.36 | Dopamine receptor antagonist | Antipsychotic |
| 49 | Artemisinin | 1.09 × 10−5 | −0.36 | N/A | Antimalarials |
| 50 | Withaferin A | 9.48 × 10−5 | −0.35 | N/A | Anticancer |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morello, G.; Villari, A.; Spampinato, A.G.; La Cognata, V.; Guarnaccia, M.; Gentile, G.; Ciotti, M.T.; Calissano, P.; D’Agata, V.; Severini, C.; et al. Transcriptional Profiles of Cell Fate Transitions Reveal Early Drivers of Neuronal Apoptosis and Survival. Cells 2021, 10, 3238. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10113238
Morello G, Villari A, Spampinato AG, La Cognata V, Guarnaccia M, Gentile G, Ciotti MT, Calissano P, D’Agata V, Severini C, et al. Transcriptional Profiles of Cell Fate Transitions Reveal Early Drivers of Neuronal Apoptosis and Survival. Cells. 2021; 10(11):3238. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10113238
Chicago/Turabian StyleMorello, Giovanna, Ambra Villari, Antonio Gianmaria Spampinato, Valentina La Cognata, Maria Guarnaccia, Giulia Gentile, Maria Teresa Ciotti, Pietro Calissano, Velia D’Agata, Cinzia Severini, and et al. 2021. "Transcriptional Profiles of Cell Fate Transitions Reveal Early Drivers of Neuronal Apoptosis and Survival" Cells 10, no. 11: 3238. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10113238