
Metabolomics of Acute vs. Chronic Spinach Intake in an Apc–Mutant Genetic Background: Linoleate and Butanoate Metabolites Targeting HDAC Activity and IFN–γ Signaling

 ,
,  , ,
, , 

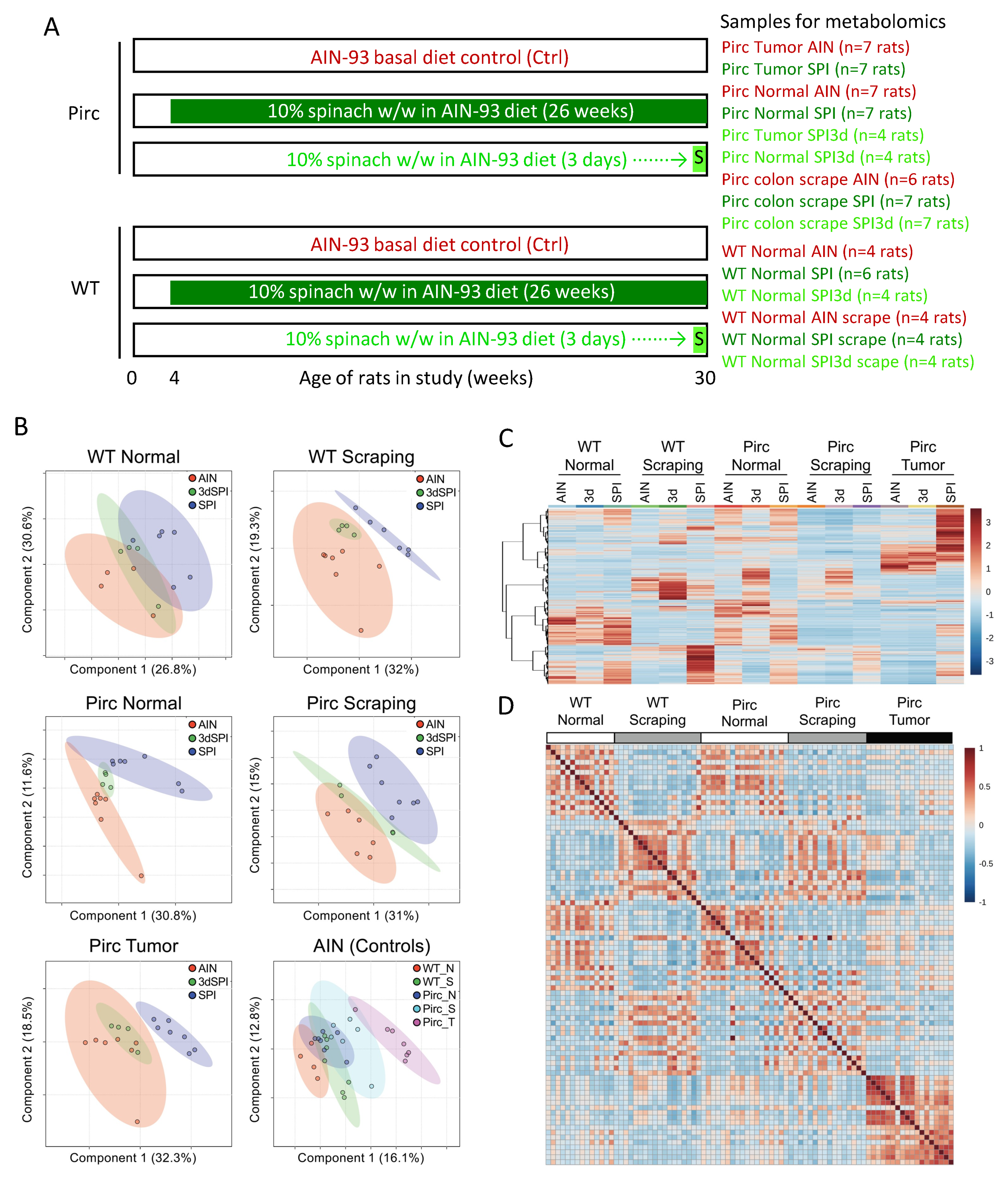{kind=link}
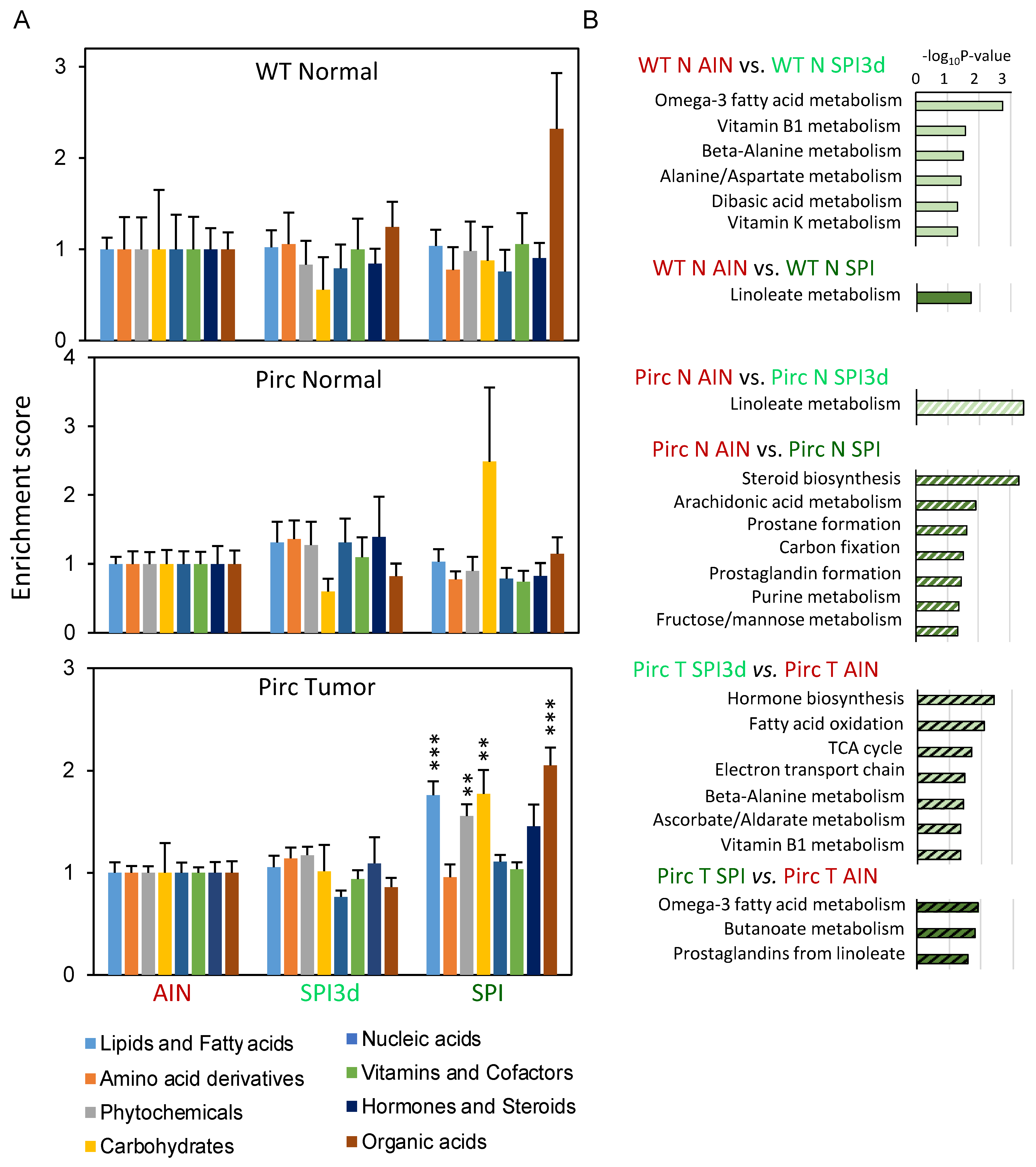{kind=link}
{kind=link}
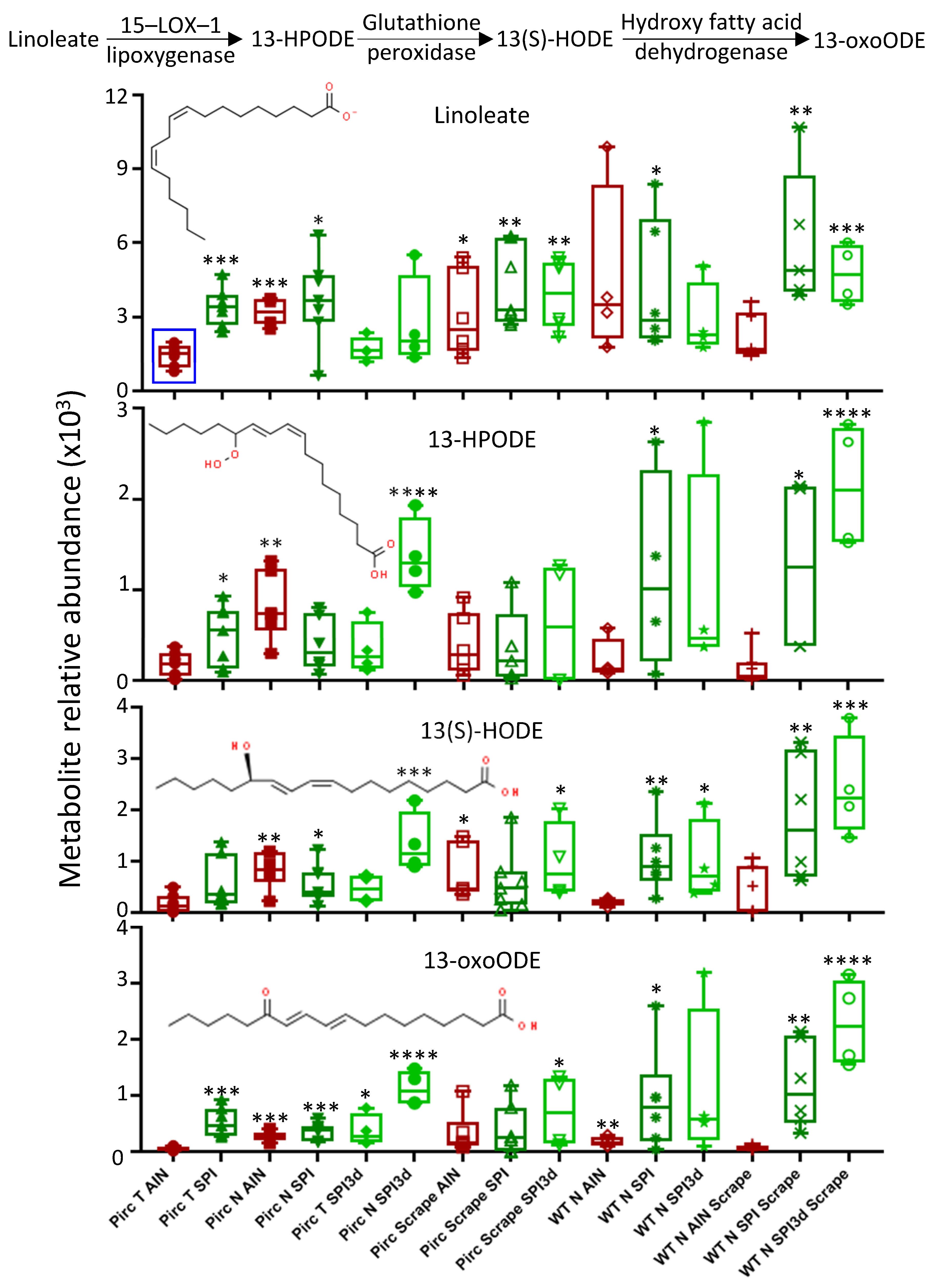{kind=link}
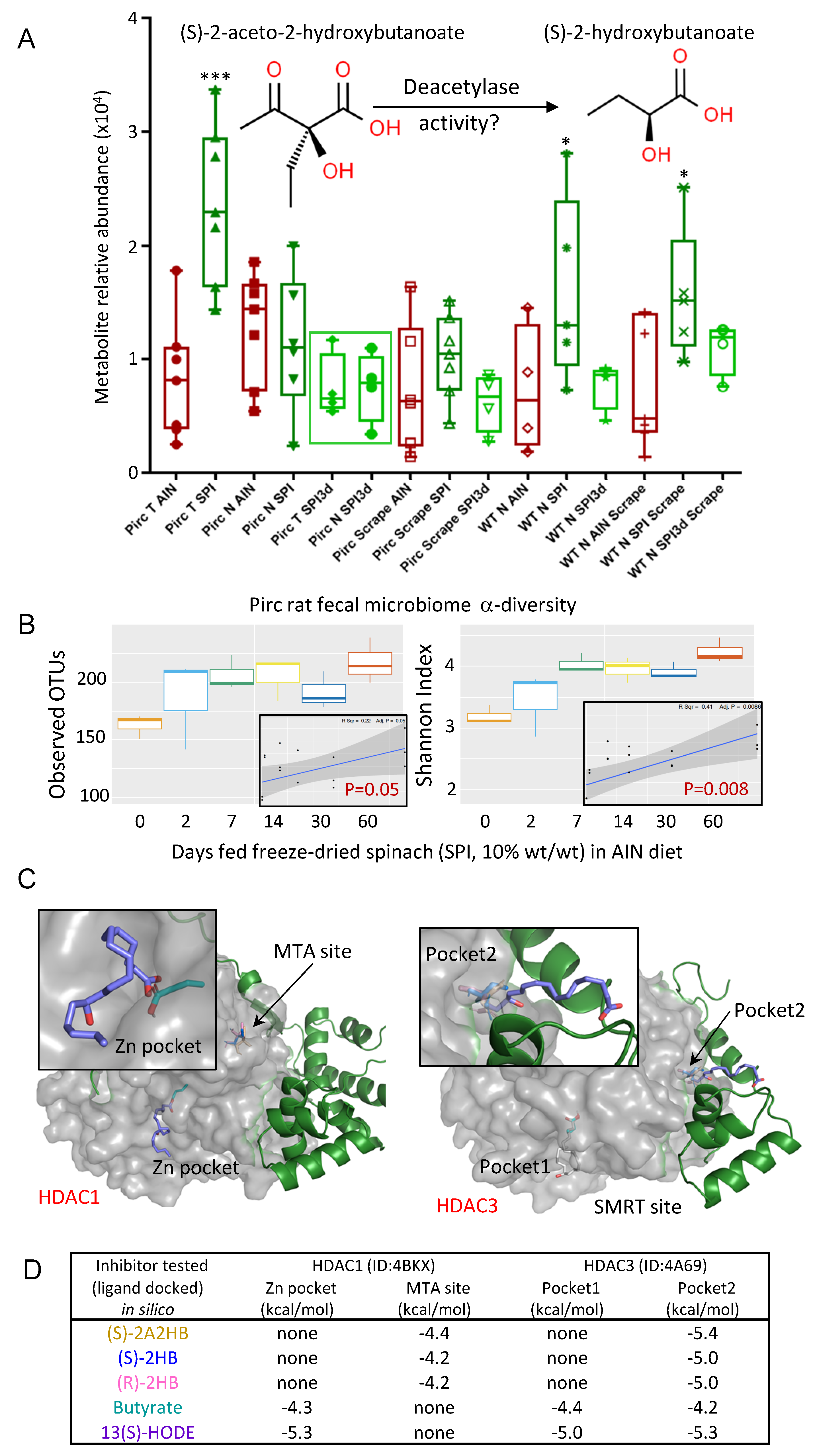{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Metabolomics Segregated Pirc and WT Rats According to Acute vs. Chronic SPI Intake
3.2. Fatty Acids and Other Compound Categories Were Altered by SPI Intake
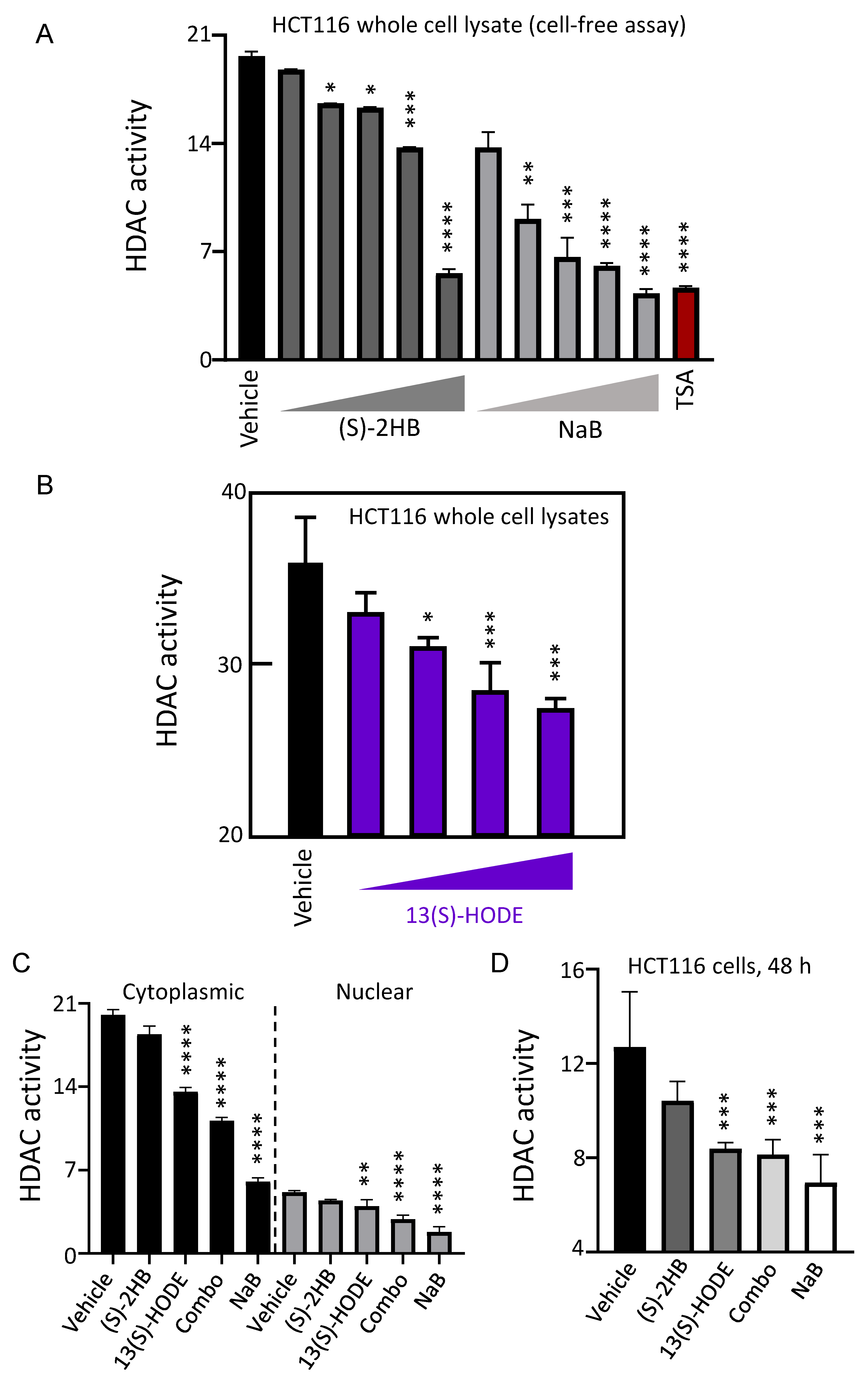
3.3. Butanoate Metabolites and 13(S)–HODE Inhibited HDAC Activity
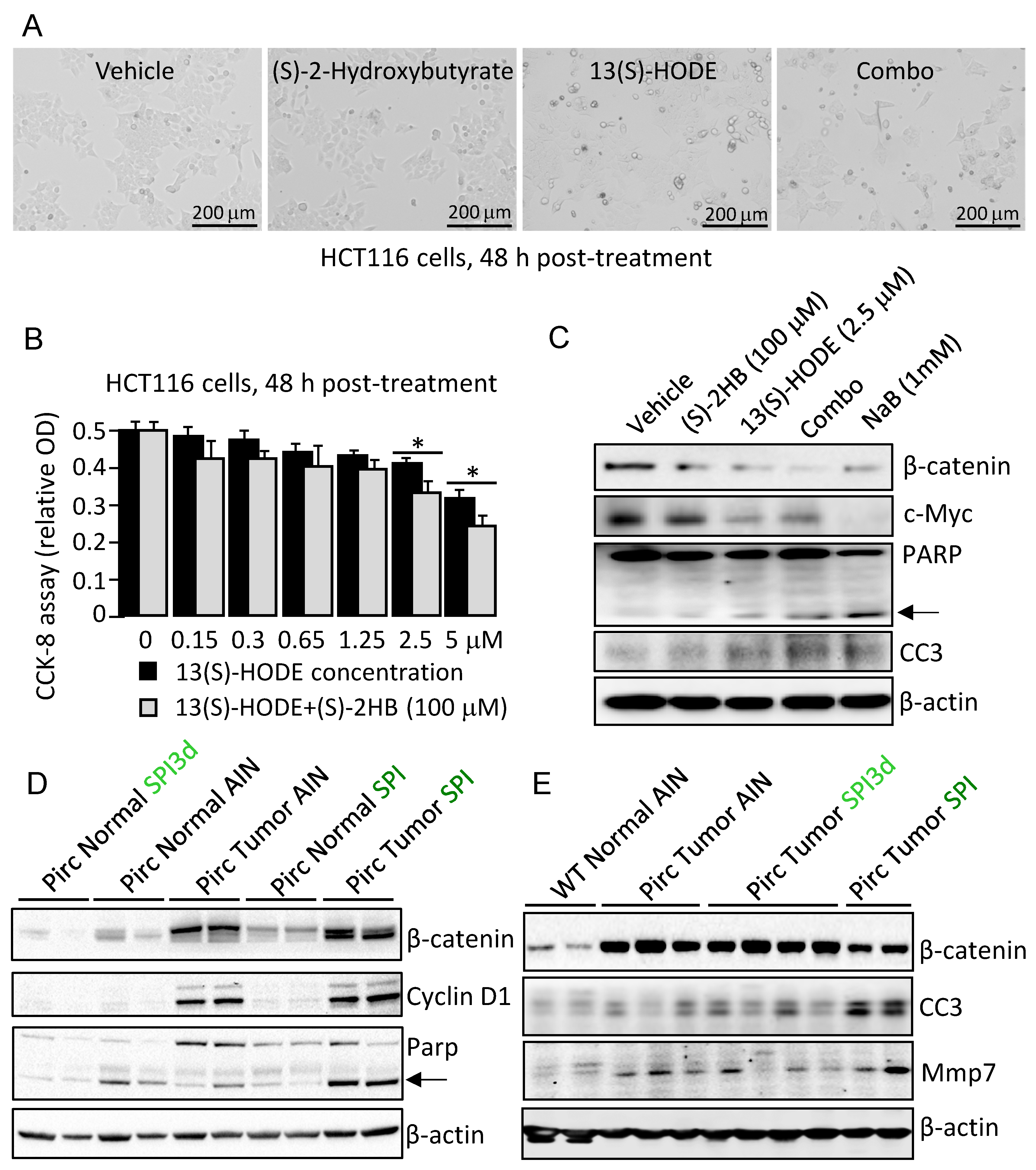
3.4. Apoptosis Induction Was Observed by 13(S)–HODE ± (S)–2HB
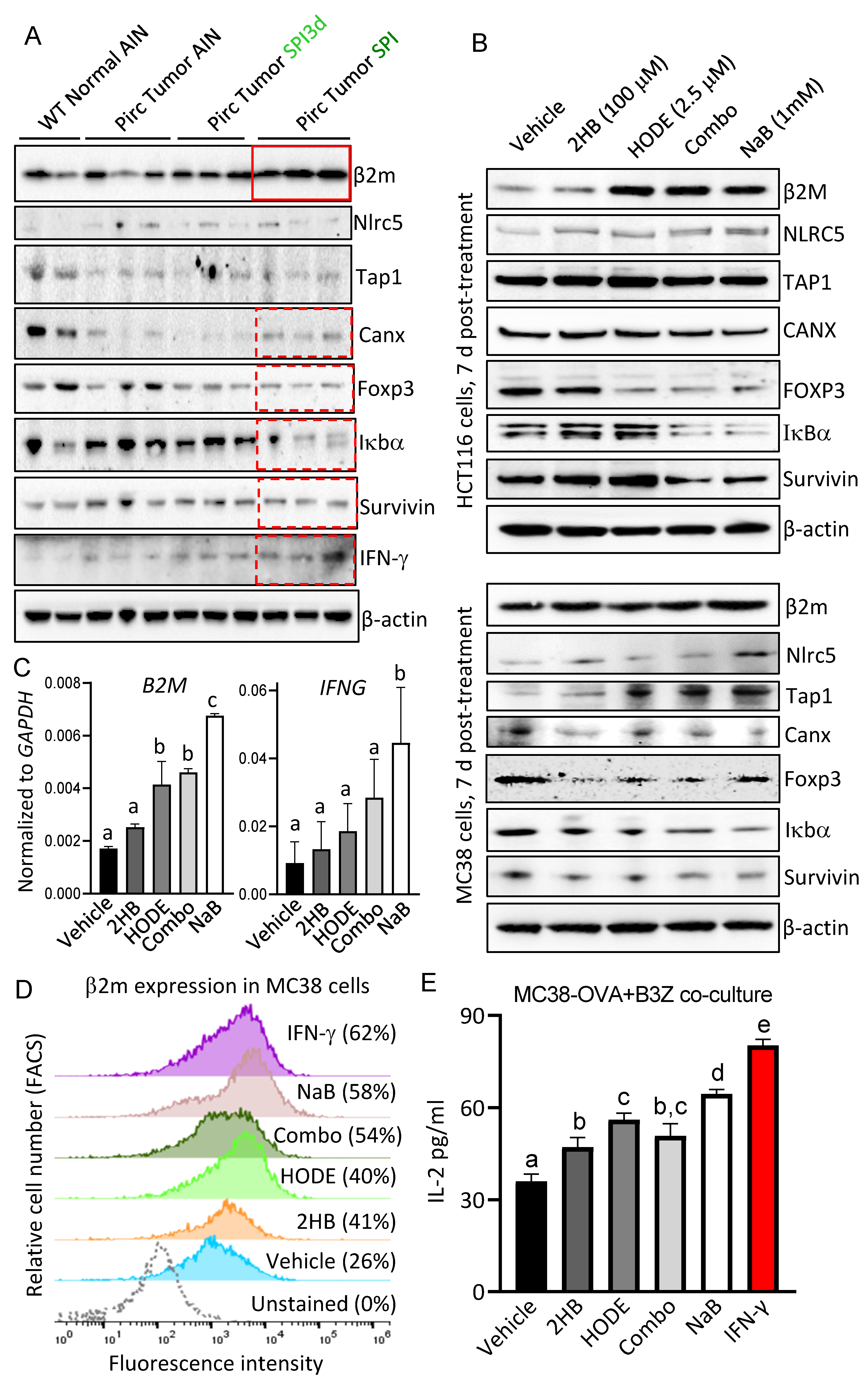
3.5. 13(S)–HODE and (S)–2HB Targeted the IFN-γ Signaling Axis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fan, Y.; Pedersen, O. Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol. 2021, 19, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Mazier, W.; Le Corf, K.; Martinez, C.; Tudela, H.; Kissi, D.; Kropp, C.; Coubard, C.; Soto, M.; Elustondo, F.; Rawadi, G.; et al. A new strain of Christensenella minuta as a potential biotherapy for obesity and associated metabolic diseases. Cells 2021, 10, 823. [Google Scholar] [CrossRef] [PubMed]
- Rivas–Dominguez, A.; Pastor, N.; Martínez–López, L.; Colon–Perez, J.; Bermudez, B.; Orta, M.L. The role of DNA damage response in dysbiosis–induced colorectal cancer. Cells 2021, 10, 1934. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, B.; Iribarren, C.; Magnusson, M.K.; Sundin, J.; Clevers, E.; Savolainen, O.; Ross, A.B.; Tornblom, H.; Simren, M.; Öhman, L. A distinct faecal microbiota and metabolite profile linked to bowel habits in patients with irritable bowel syndrome. Cells 2021, 10, 1459. [Google Scholar] [CrossRef]
- Xi, M.; La Barbera, G.; Eriksen, J.N.; Prahm, A.P.; Jeppesen, P.B.; Dragsted, L.O. Discovery of urinary biomarkers of spinach consumption using untargeted LC–MS metabolomics in a human intervention trial. Mol. Nutr. Food Res. 2022, 66, 202100260. [Google Scholar] [CrossRef]
- Chen, Y.-S.; Li, J.; Menon, R.; Jayaraman, A.; Lee, K.; Huang, Y.; Dashwood, W.D.; Zhang, K.; Sun, D.; Dashwood, R.H. Dietary spinach reshapes the gut microbiome in an Apc–mutant genetic background: Mechanistic insights from integrated multi–omics. Gut Microbes 2021, 13, 1972756. [Google Scholar] [CrossRef] [PubMed]
- Amos–Landgraf, J.A.; Kwong, L.N.; Kendziorski, C.M.; Reicheldefer, M.; Torrealba, J.; Weichert, J.; Haag, J.D.; Chen, K.S.; Waller, J.L.; Gould, M.N.; et al. A target–selected Apc–mutant rat kindred enhances the modeling of familial human colon cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 4036–4041. [Google Scholar] [CrossRef] [Green Version]
- Myzak, M.C.; Karplus, P.A.; Chung, F.L.; Dashwood, R.H. A novel mechanism of chemoprotection by sulforaphane: Inhibition of histone deacetylase. Cancer Res. 2004, 64, 5767–5774. [Google Scholar] [CrossRef] [Green Version]
- Rajendran, P.; Delage, B.; Dashwood, W.D.; Yu, T.W.; Wuth, B.; Williams, D.E.; Ho, E.; Dashwood, R.H. Histone deacetylase turnover and recovery in sulforaphane–treated colon cancer cells: Competing actions of 14–3–3 and Pin1 in HDAC/SMRT corepressor complex dissociation/reassembly. Mol. Cancer 2011, 10, 68. [Google Scholar] [CrossRef] [Green Version]
- Rajendran, P.; Kidane, A.I.; Yu, T.W.; Dashwood, W.M.; Bisson, W.H.; Löhr, C.V.; Ho, E.; Williams, D.E.; Dashwood, R.H. HDAC turnover, CtIP acetylation and dysregulated DNA damage signaling in colon cancer cells treated with sulforaphane and related dietary isothiocyanates. Epigenetics 2013, 8, 612–623. [Google Scholar] [CrossRef]
- Kang, Y.; Nian, H.; Rajendran, P.; Kim, E.; Dashwood, W.M.; Pinto, J.T.; Boardman, L.A.; Thibodeau, S.N.; Limburg, P.J.; Löhr, C.V.; et al. HDAC8 and STAT3 repress BMF gene activity in colon cancer cells. Cell Death Dis. 2014, 5, e1476. [Google Scholar] [CrossRef] [Green Version]
- Rajendran, P.; Dashwood, W.M.; Li, L.; Kang, Y.; Kim, E.; Johnson, G.; Fischer, K.A.; Löhr, C.V.; Williams, D.E.; Ho, E.; et al. Nrf2 status affects tumor growth, HDAC3 promoter associations, and the response to sulforaphane in the colon. Clin. Epigenet. 2015, 7, 102. [Google Scholar] [CrossRef] [Green Version]
- Okonkwo, A.; Mitra, J.; Johnson, G.S.; Li, L.; Dashwood, W.M.; Hedge, M.L.; Yue, C.; Dashwood, R.H.; Rajendran, P. Heterocyclic analogs of sulforaphane trigger DNA damage and impede DNA repair in colon cancer cells: Interplay of HATs and HDACs. Mol. Nutr. Food Res. 2018, 62, e1800228. [Google Scholar] [CrossRef]
- Rajendran, P.; Johnson, G.; Li, L.; Chen, Y.S.; Dashwood, M.; Nguyen, N.; Ulusan, A.; Ertem, F.; Zhang, M.; Li, J.; et al. Acetylation of CCAR2 establishes a BET/BRD9 acetyl switch in response to combined deacetylase and bromodomain inhibition. Cancer Res. 2019, 79, 918–927. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, S.; Gustafson, T.; Zhang, M.; Chen, Y.S.; Li, J.; Nguyen, N.; Perez, J.E.T.; Dashwood, W.M.; Rajendran, P.; Dashwood, R.H. Deacetylase plus bromodomain inhibition downregulates ERCC2 and suppresses the growth of metastatic colon cancer cells. Cancers 2021, 13, 1438. [Google Scholar] [CrossRef] [PubMed]
- LaFleur, M.W.; Nguyen, T.H.; Coxe, M.A.; Miller, B.C.; Yates, K.B.; Gillis, J.E.; Sen, D.R.; Gaudiano, E.F.; Al Abosy, R.; Freeman, G.J.; et al. PTPN2 regulates the generation of exhausted CD8+ T cell subpopulations and restrains tumor immunity. Nat. Immunol. 2019, 20, 1335–1347. [Google Scholar] [CrossRef]
- Sasaki, K.; Himeno, A.; Nakagawa, T.; Sasaki, Y.; Kiyonari, H.; Iwai, K. Modulation of autoimmune pathogenesis by T cell-triggered inflammatory cell death. Nat. Commun. 2019, 10, 3878. [Google Scholar] [CrossRef]
- Lee, S.I.; Zuo, X.; Shureiqi, I. 15–lipoxygenase–1 as a tumor suppressor gene in colon cancer: Is the verdict in? Cancer Metastasis Rev. 2011, 30, 481–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shureiqi, I.; Wonjo, K.J.; Poore, J.A.; Reddy, R.G.; Moussalli, M.J.; Spindler, S.A.; Greenson, J.K.; Normolle, D.; Hasan, A.A.; Lawrence, T.S.; et al. Decreased 13–S–hydroxyoctadecadienoic acid levels and 15–lipoxygenase–1 expression in human colon cancers. Carcinogenesis 1999, 20, 1985–1995. [Google Scholar] [CrossRef] [Green Version]
- Shureiqi, I.; Chen, D.; Lee, J.J.; Yang, P.; Newman, R.A.; Brenner, D.E.; Lotan, R.; Fischer, S.M.; Lippman, S.M. 15–LOX–1: A novel molecular target of nonsteroidal anti–inflammatory drug–induced apoptosis in colorectal cancer cells. J. Natl. Cancer Inst. 2000, 92, 1136–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shureiqi, I.; Chen, D.; Lotan, R.; Yang, P.; Newman, R.A.; Fischer, S.M.; Lippman, S.M. 15–lipoxygenase–1 mediates nonsteroidal anti–inflammatory drug–induced apoptosis independently of cyclooxygenase–2 in colon cancer cells. Cancer Res. 2000, 60, 6846–6850. [Google Scholar] [PubMed]
- Shureiqi, I.; Jiang, W.; Zuo, X.; Wu, Y.; Stimmel, J.B.; Leesnitzer, L.M.; Morris, J.S.; Fan, H.-Z.; Fischer, S.M.; Lippman, S.M. The 15–lipoxygenase–1 product 13–S–hydroxyoctadecadienoic acid down–regulates PPAR–delta to induce apoptosis in colorectal cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 9968–9973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, X.; Wu, Y.; Morris, J.S.; Stimmel, J.B.; Leesnitzer, L.M.; Fischer, S.M.; Lippman, S.M.; Shureiqi, I. Oxidative metabolism of linoleic acid modulates PPAR–beta/delta suppression of PPAR–gamma activity. Oncogene 2006, 25, 1225–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Zuo, X.; Liu, Y.; Deguchi, Y.; Moussalli, M.J.; Chen, W.; Yang, P.; Wei, B.; Tan, L.; Lorenzi, P.L.; et al. Suppression of membranous LPR5 recycling, Wnt/β–catenin signaling, and colon carcinogenesis by 15–LOX–1 peroxidation of linoleic acid in PI3P. Cell Rep. 2020, 32, 108049. [Google Scholar] [CrossRef] [PubMed]
- Thangaraju, M.; Carswell, K.N.; Prasad, P.D.; Ganapathy, V. Colon cancer cells maintain low levels of pyruvate to avoid cell death caused by inhibition of HDAC1/HDAC3. Biochem. J. 2009, 417, 379–389. [Google Scholar] [CrossRef]
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 2016, 7, 189–200. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.X.; Henen, M.A.; Lee, J.S.; Vögeli, B.; Colgan, S.P. Microbiota–derived butyrate is an endogenous HIF prolyl hydroxylase inhibitor. Gut Microbes 2021, 12, 1938380. [Google Scholar] [CrossRef]
- Thangaraju, M.; Cresci, G.A.; Liu, K.; Anath, S.; Gnanaprakasam, J.P.; Browning, D.D.; Mellinger, J.D.; Smith, S.B.; Digby, G.J.; Lambert, N.A.; et al. GPR109A is a G–protein–coupled receptor for the bacterial fermentation product butyrate and functions as a tumor suppressor in colon. Cancer Res. 2009, 69, 2826–2832. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, K.S.; van den Elsen, P.J. NLRC5: A key regulator of MHC class I–dependent immune responses. Nat. Rev. Immunol. 2012, 12, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Yoshihama, S.; Roszik, J.; Downs, I.; Meissner, T.B.; Vijayan, S.; Chapuy, B.; Sidiq, T.; Shipp, M.A.; Lizee, G.A.; Kobayashi, K.S. NLRC5/MHC class I transactivator is a target for immune evasion in cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 5999–6004. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.X.; Vijayan, S.; Yoo, J.-S.; Watanabe, T.; Ouda, R.; An, N.; Kobayashi, K.S. MHC class I transactivator NLRC5 in host immunity, cancer and beyond. Immunology 2020, 163, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Cornel, A.M.; Mimpen, I.L.; Nierkens, S. MHC class I downregulation in cancer: Underlying mechanisms and potential targets for cancer immunotherapy. Cancers 2020, 12, 1760. [Google Scholar] [CrossRef]
- Zheng, J.; Yang, T.; Gao, S.; Cheng, M.; Shao, Y.; Xi, Y.; Guo, L.; Zhang, D.; Gao, W.; Zhang, G.; et al. miR–148–3p silences the CANX/MHC–I pathway and impairs CD8+ T cell–mediated immune attack in colorectal cancer. FASEB J. 2021, 35, e21776. [Google Scholar] [CrossRef]
- Pérez–Piñero, S.; Ávila–Gandía, V.; Rubio Arias, J.A.; Muñoz–Carrillo, J.C.; Losada–Zafrilla, P.; López–Román, F.J. A 12-week randomized double-blind placebo-controlled clinical trial evaluating the effect of supplementation with a spinach extract on skeletal muscle fitness in adults older than 50 years of age. Nutrients 2021, 13, 4373. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, F.P.F.; Farhangi, M.A.; Vaezi, M.; Hemmati, S. The effects of spinach–derived thylakoid supplementation in combination with calorie restriction on anthropometric parameters and metabolic profiles in obese women with polycystic ovary syndrome: A randomized, double–blind, placebo–controlled clinical trial. Nutr. J. 2020, 19, 82. [Google Scholar] [PubMed]
- Jovanovski, E.; Bosco, L.; Khan, K.; Au–Yeung, F.; Ho, H.; Zurbau, A.; Jenkins, A.L.; Vuksan, V. Effect of spinach, a high dietary nitrate source, on arterial stiffness and related hemodynamic measures: A randomized, controlled trial in healthy adults. Clin. Nutr. Res. 2015, 4, 160–167. [Google Scholar] [CrossRef] [Green Version]
- Roberts, J.L.; Moreau, R. Functional properties of spinach (Spinacia oleracea L.) phytochemicals and bioactives. Food Funct. 2016, 7, 3337–3353. [Google Scholar] [CrossRef]
- Gutierrez, R.M.P.; Velazquez, E.G.; Carrera, S.P.P. Spinacia oleracea Linn considered as one of the most perfect foods: A pharmacological and phytochemical review. Mini Rev. Med. Chem. 2019, 19, 1666–1680. [Google Scholar] [CrossRef]
- Zheng, Y.; Xiang, S.; Zhang, H.; Ye, H.; Zhang, Y.; Ge, Y.; Feng, X.; Bao, X.; Chen, J.; Zhu, X. Vitamin B12 enriched in spinach and its effects on gut microbiome. J. Agric. Food Chem. 2021, 69, 2204–2212. [Google Scholar] [CrossRef]
- Parasramka, M.A.; Dashwood, W.M.; Wang, R.; Abdelli, A.; Bailey, G.S.; Williams, D.E.; Ho, E.; Dashwood, R.H. MicroRNA profiling of carcinogen–induced rat colon tumors and the influence of dietary spinach. Mol. Nutr. Food Res. 2012, 56, 1259–1269. [Google Scholar] [CrossRef] [Green Version]
- Parasramka, M.A.; Ho, E.; Williams, D.E.; Dashwood, R.H. MicroRNAs, diet, and cancer: New mechanistic insights on the epigenetic actions of phytochemicals. Mol. Carcinog. 2012, 51, 213–230. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.; Taggart, M.W.; Reyes–Uribe, L.; Borras, E.; Riquelme, E.; Barnett, R.M.; Leoni, G.; San Lucas, F.A.; Catanese, M.T.; Mori, F.; et al. Immune profiling of premalignant lesions in patients with Lynch Syndrome. JAMA Oncol. 2018, 4, 1085–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S.; et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter–inhibitory checkpoints. Cancer Discov. 2015, 5, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Samadder, N.J.; Neklason, D.W.; Boucher, K.M.; Byrne, K.R.; Kanth, P.; Samowitz, W.; Jones, D.; Tavtigian, S.W.; Done, M.W.; Berry, T.; et al. Effect of sulindac and erlotinib vs placebo on duodenal neoplasia in Familial Adenomatous Polyposis: A randomized clinical trial. JAMA 2016, 315, 1266–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samadder, N.J.; Kuwada, S.K.; Boucher, K.M.; Byrne, K.; Samowitz, W.; Jones, D.; Tavtigian, S.V.; Westover, M.; Berry, T.; Jasperson, K.; et al. Association of sulindac and erlotinib vs placebo with colorectal neoplasia in Familial Adenomatous Polyposis: Secondary analysis of a randomized clinical trial. JAMA Oncol. 2018, 4, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Ulusan, A.M.; Rajendran, P.; Dashwood, W.M.; Yavuz, O.F.; Kapoor, S.; Gustafson, T.A.; Savage, M.I.; Brown, P.H.; Sei, S.; Mohammed, A.; et al. Optimization of erlotinib plus sulindac dosing regimens for intestinal cancer prevention in an Apc–mutant model of Familial Adenomatous Polyposis (FAP). Cancer Prev. Res. 2021, 14, 325–336. [Google Scholar] [CrossRef]
- Ricciardiello, L.; Ahnen, D.J.; Lynch, P.M. Chemoprevention of hereditary colon cancers: Time for new strategies. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 352–361. [Google Scholar] [CrossRef]
- Shahbaz, S.K.; Koushki, K.; Ayati, S.H.; Bland, A.R.; Bezsonov, E.E.; Sahebkar, E. Inflammasomes and colorectal cancer. Cells 2021, 10, 2172. [Google Scholar] [CrossRef]
- Del Corno, M.; Vari, R.; Scazzocchio, B.; Varano, B.; Masella, R.; Conti, L. Dietary fatty acids at the crossroad between obesity and colorectal cancer: Fine regulators of adipose tissue homoeostasis and immune response. Cells 2021, 10, 1738. [Google Scholar] [CrossRef]
- Alvarez, M.D.; Quintana, I.; Terradas, M.; Mur, P.; Balaguer, F.; Valle, L. The inherited and familial component of early-onset colorectal cancer. Cells 2021, 10, 710. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-S.; Li, J.; Neja, S.; Kapoor, S.; Tovar Perez, J.E.; Tripathi, C.; Menon, R.; Jayaraman, A.; Lee, K.; Dashwood, W.M.; et al. Metabolomics of Acute vs. Chronic Spinach Intake in an Apc–Mutant Genetic Background: Linoleate and Butanoate Metabolites Targeting HDAC Activity and IFN–γ Signaling. Cells 2022, 11, 573. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11030573
Chen Y-S, Li J, Neja S, Kapoor S, Tovar Perez JE, Tripathi C, Menon R, Jayaraman A, Lee K, Dashwood WM, et al. Metabolomics of Acute vs. Chronic Spinach Intake in an Apc–Mutant Genetic Background: Linoleate and Butanoate Metabolites Targeting HDAC Activity and IFN–γ Signaling. Cells. 2022; 11(3):573. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11030573
Chicago/Turabian StyleChen, Ying-Shiuan, Jia Li, Sultan Neja, Sabeeta Kapoor, Jorge Enrique Tovar Perez, Chakrapani Tripathi, Rani Menon, Arul Jayaraman, Kyongbum Lee, Wan Mohaiza Dashwood, and et al. 2022. "Metabolomics of Acute vs. Chronic Spinach Intake in an Apc–Mutant Genetic Background: Linoleate and Butanoate Metabolites Targeting HDAC Activity and IFN–γ Signaling" Cells 11, no. 3: 573. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11030573