
Multiomics Approach Reveals an Important Role of BNIP3 in Myocardial Remodeling and the Pathogenesis of Heart Failure with Reduced Ejection Fraction

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Production of Recombinant Adenoviruses and Adeno-Associated Virus
2.2. Isolation and Culture of Adult Rat Cardiomyocytes and Design of In Vitro Experiments
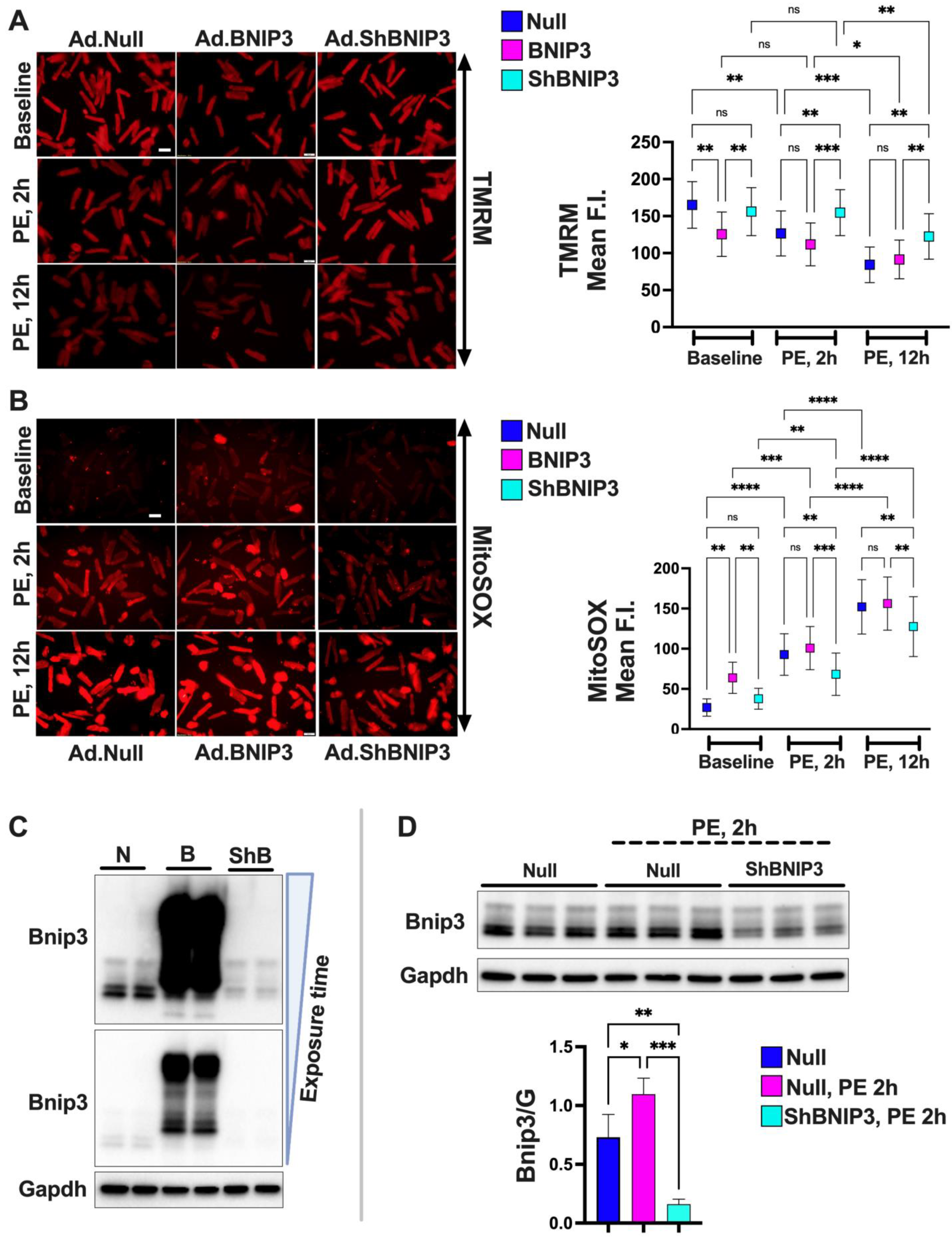
2.2.1. Assessment of Mt-Reactive Oxygen Species (ROS) and Mt-Membrane Potential
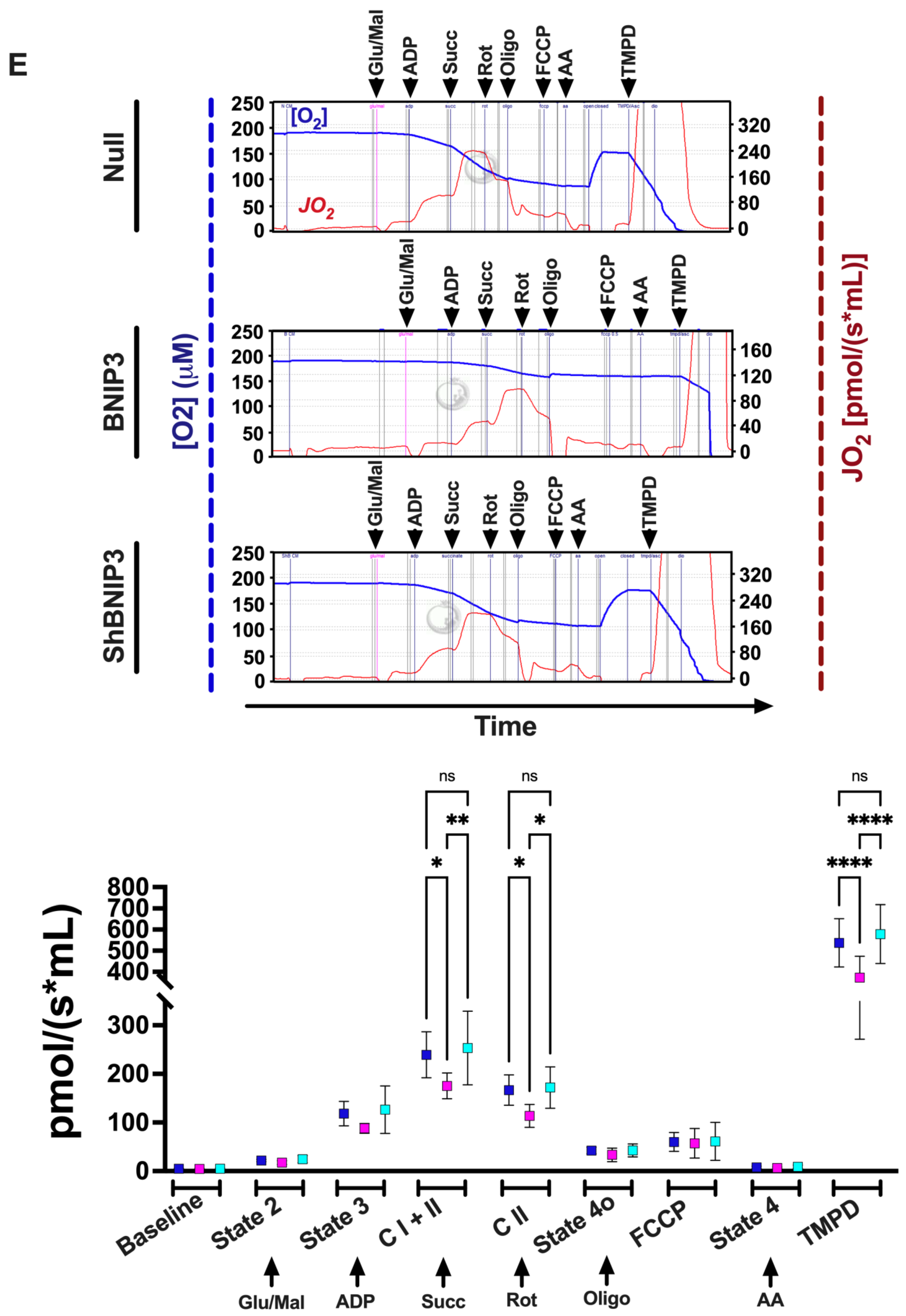
2.2.2. Measurement of Mt-Oxygen (O2) Flux (JO2) and Oxidation Phosphorylation (OXPHOS)
2.3. Animal Model of Ascending Aortic Banding and Gene Delivery
2.4. Discovery-Based Proteomic and Phosphoproteomic Quantitative Analysis Using Isobaric Labeling and Liquid Chromatography–Mass Spectrometry
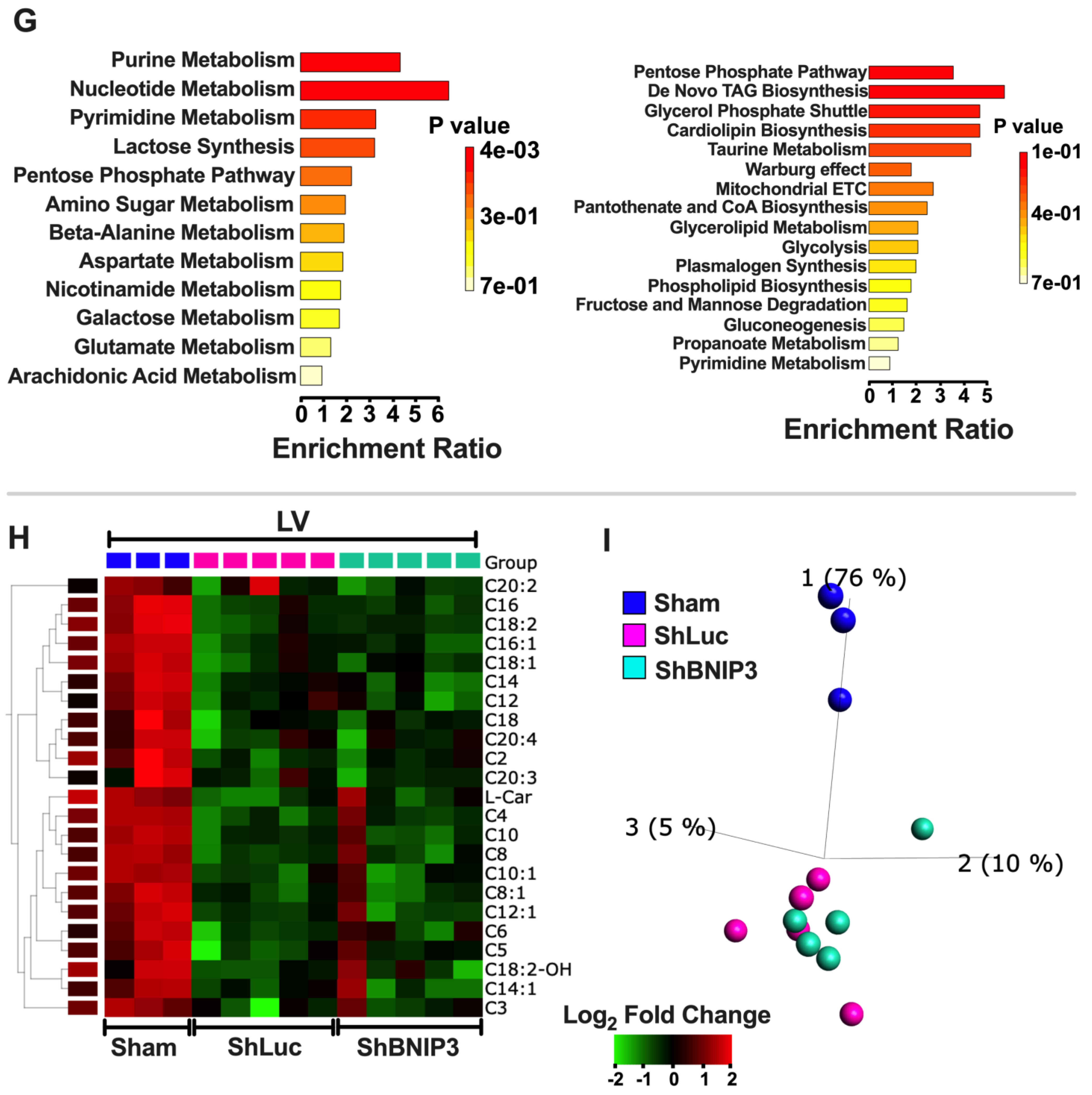
2.5. Discovery-Based Non-Targeted Central Carbon Metabolism and Acyl-Carnitine Analysis via LC–MS
2.6. Co-Immunoprecipitation and MS-Based Protein Identification
2.7. Validation by Western Blot
2.8. Validation by Co-IP and Western Blot
2.9. Statistical Analysis
3. Results
3.1. Echocardiographic Phenotyping of the Studied Animals in Proteomic Studies
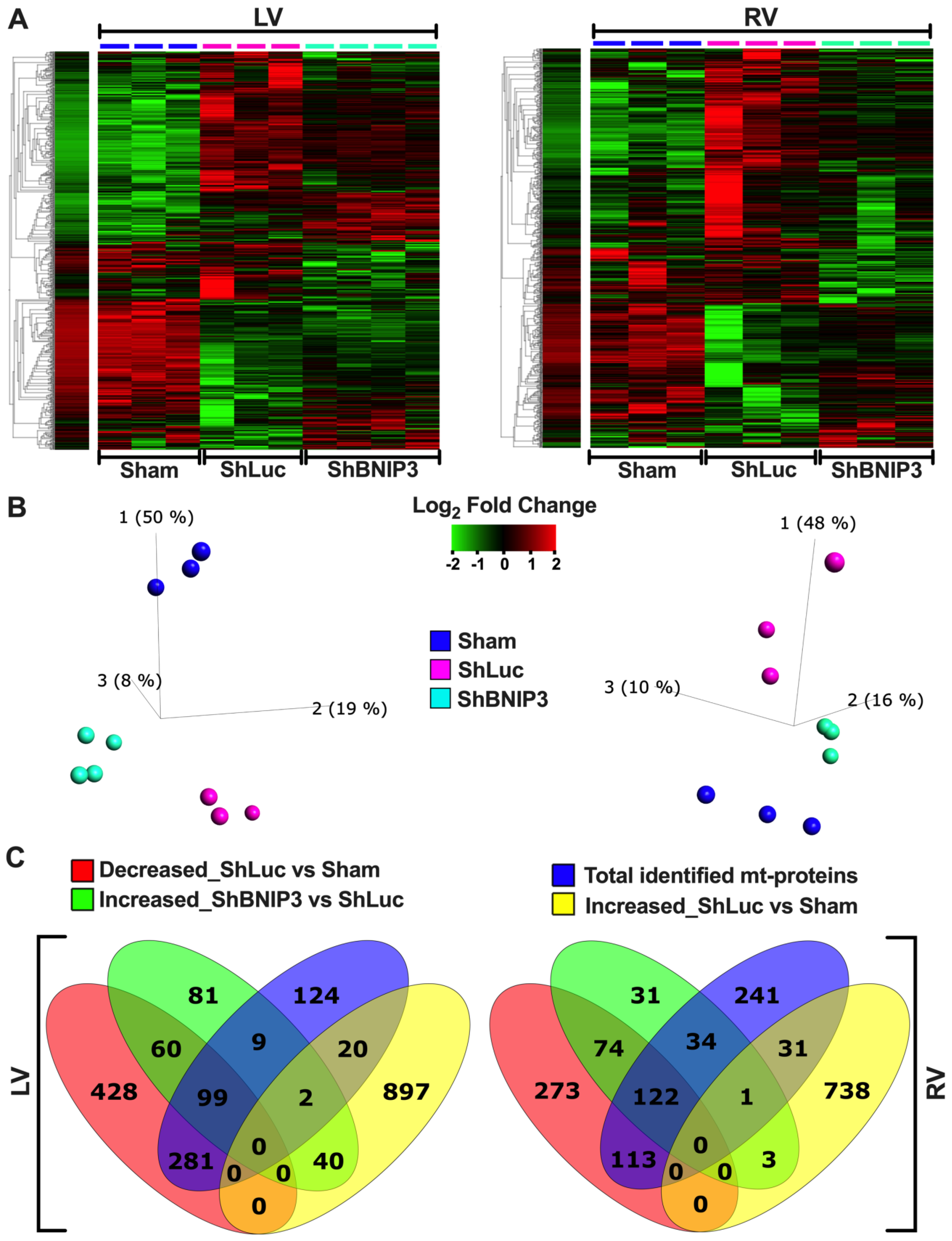
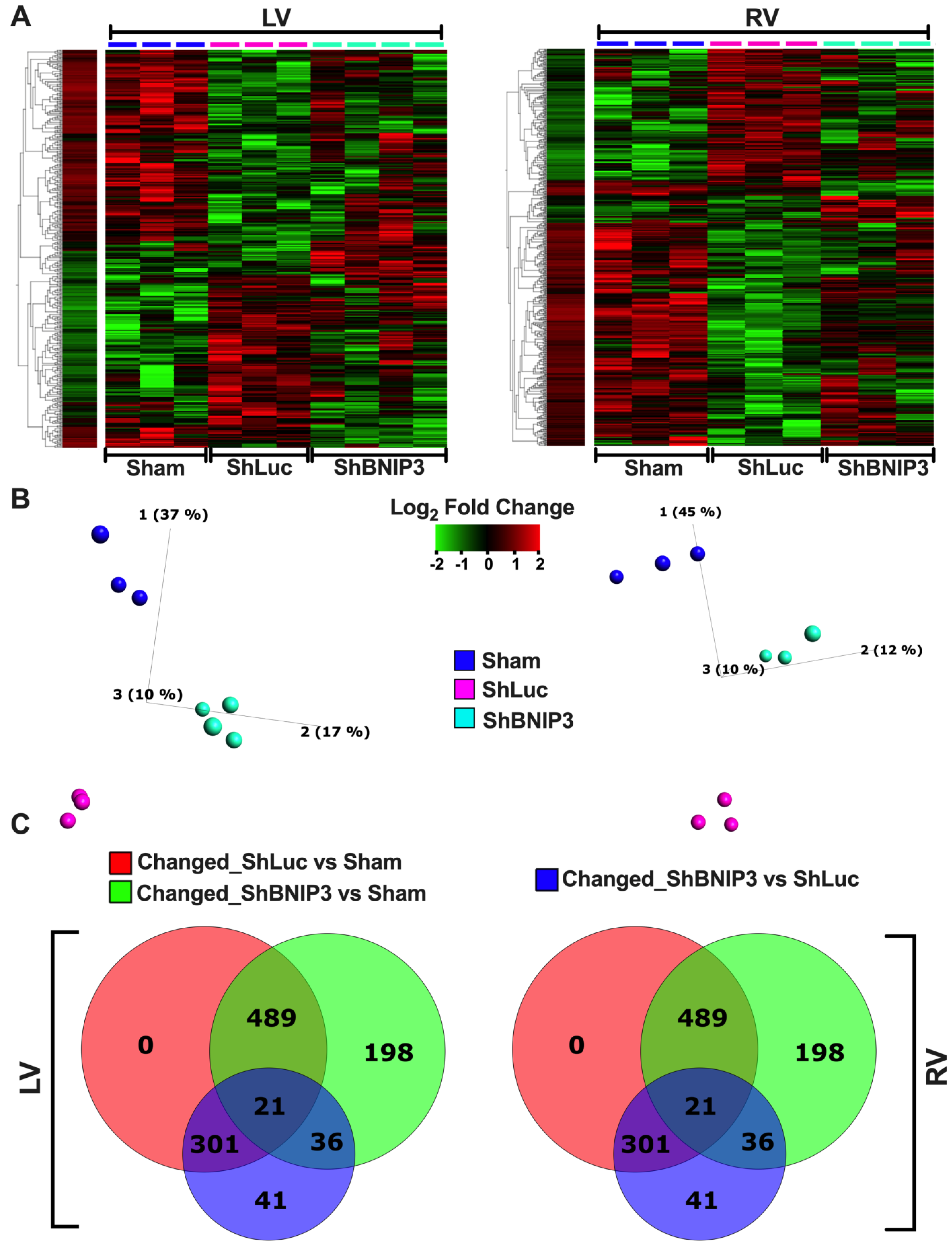
3.2. Visualization of the LV and RV Proteomic Datasets
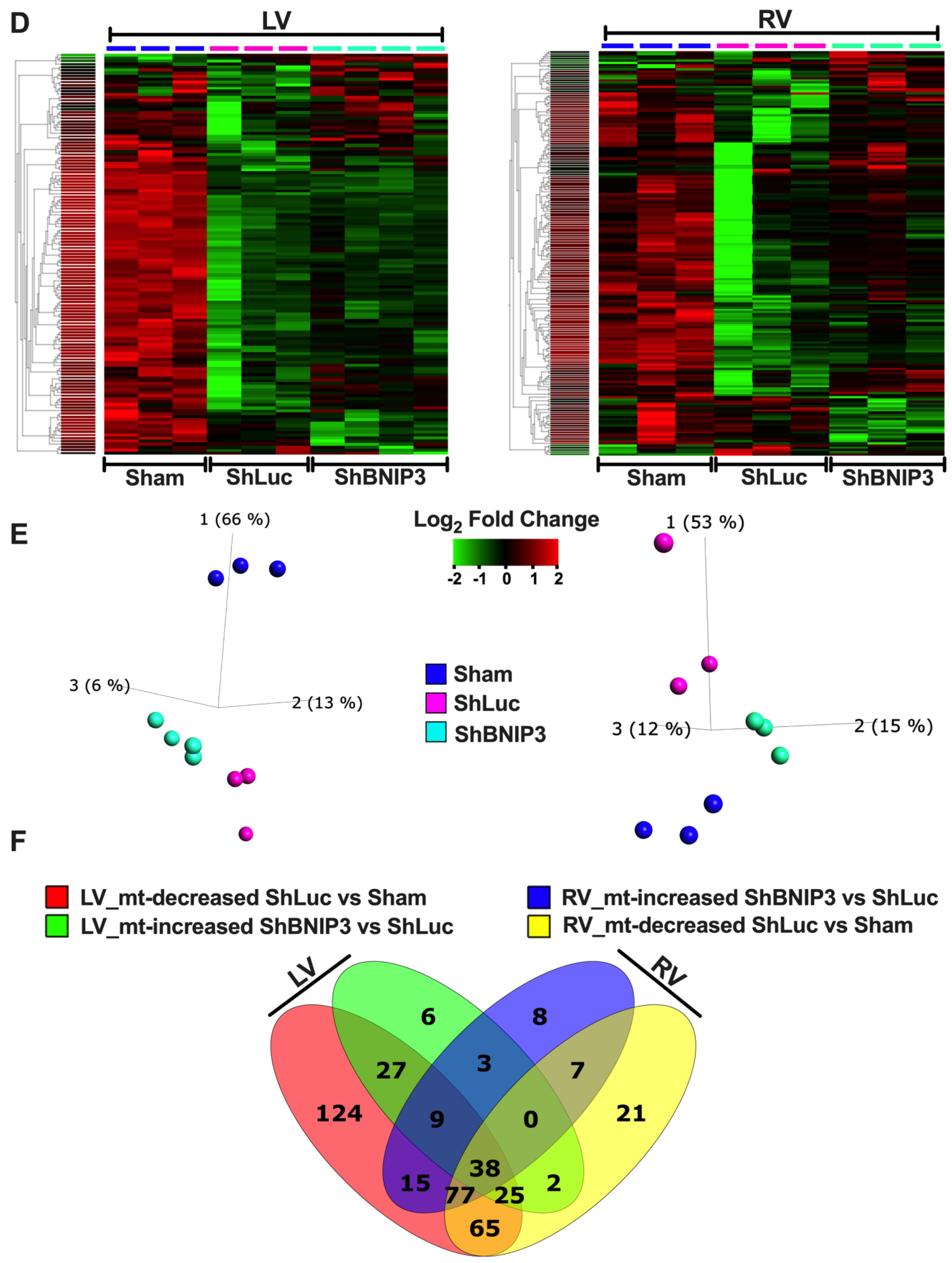
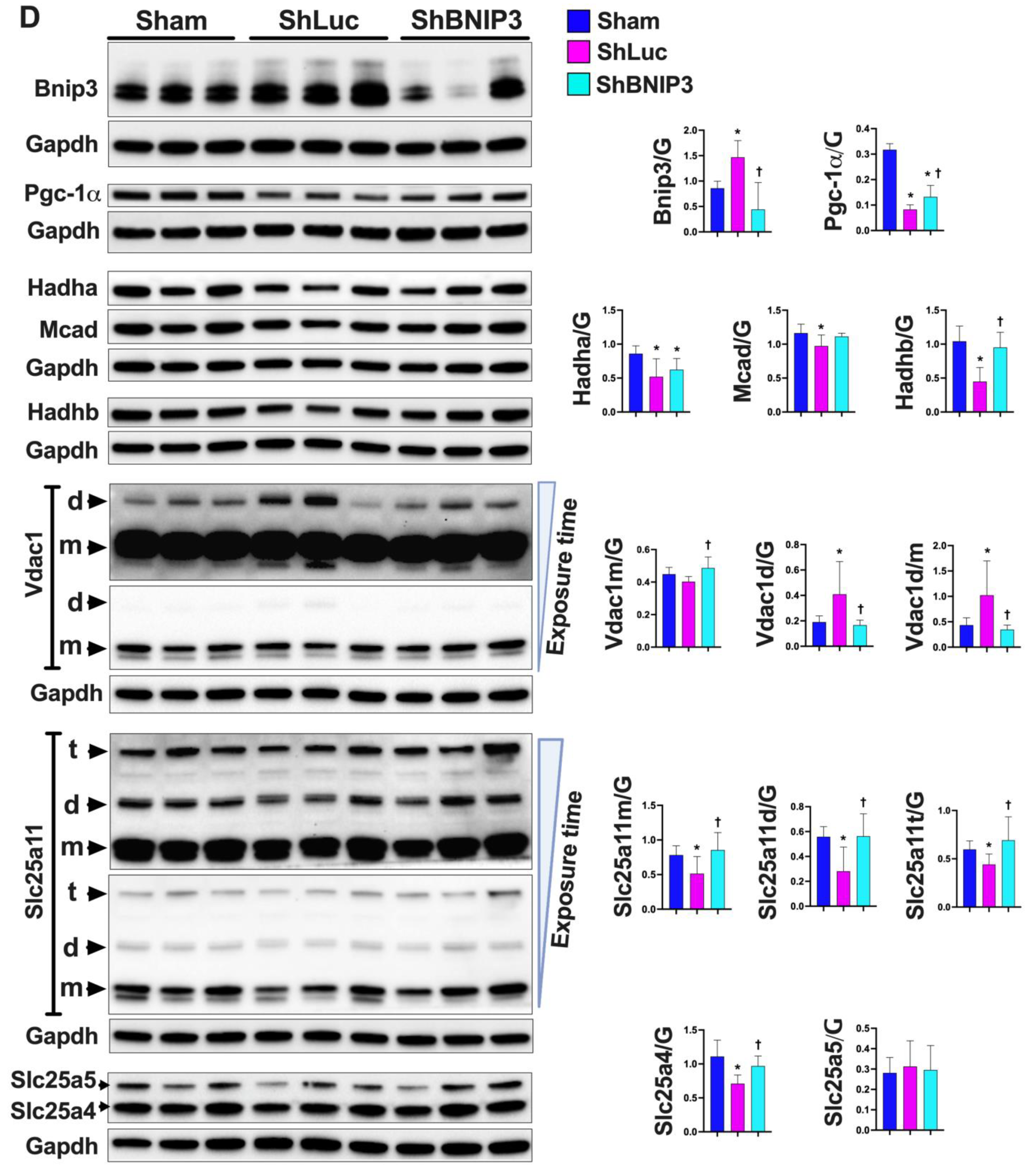
3.3. Validation of the LV and RV Proteomic Findings
3.4. Effect of BNIP3 on Mitochondrial Function and Respiration in Cardiac Myocytes
3.5. Visualization and Validation of the Untargeted Metabolomic Dataset in LV Myocardia
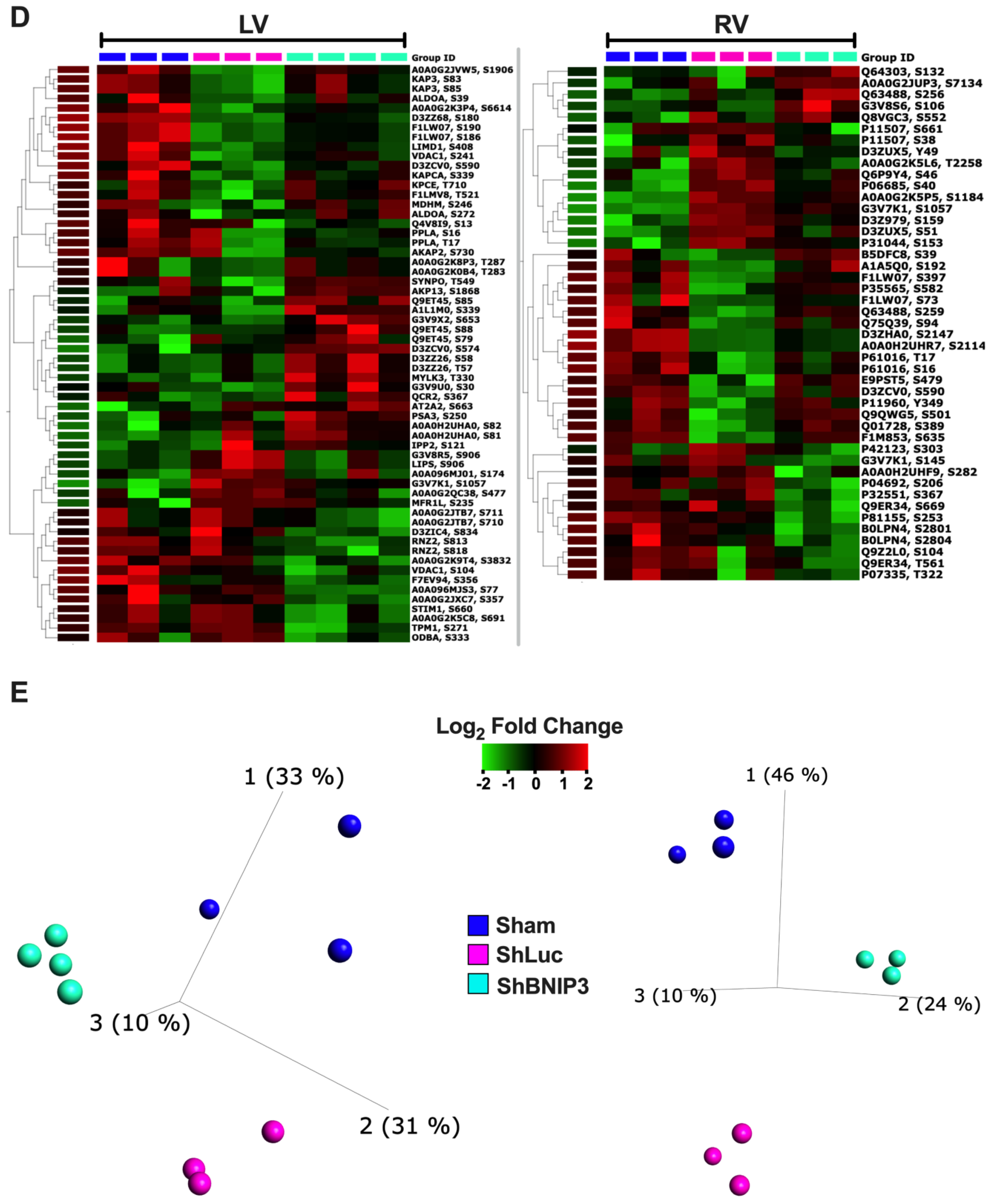
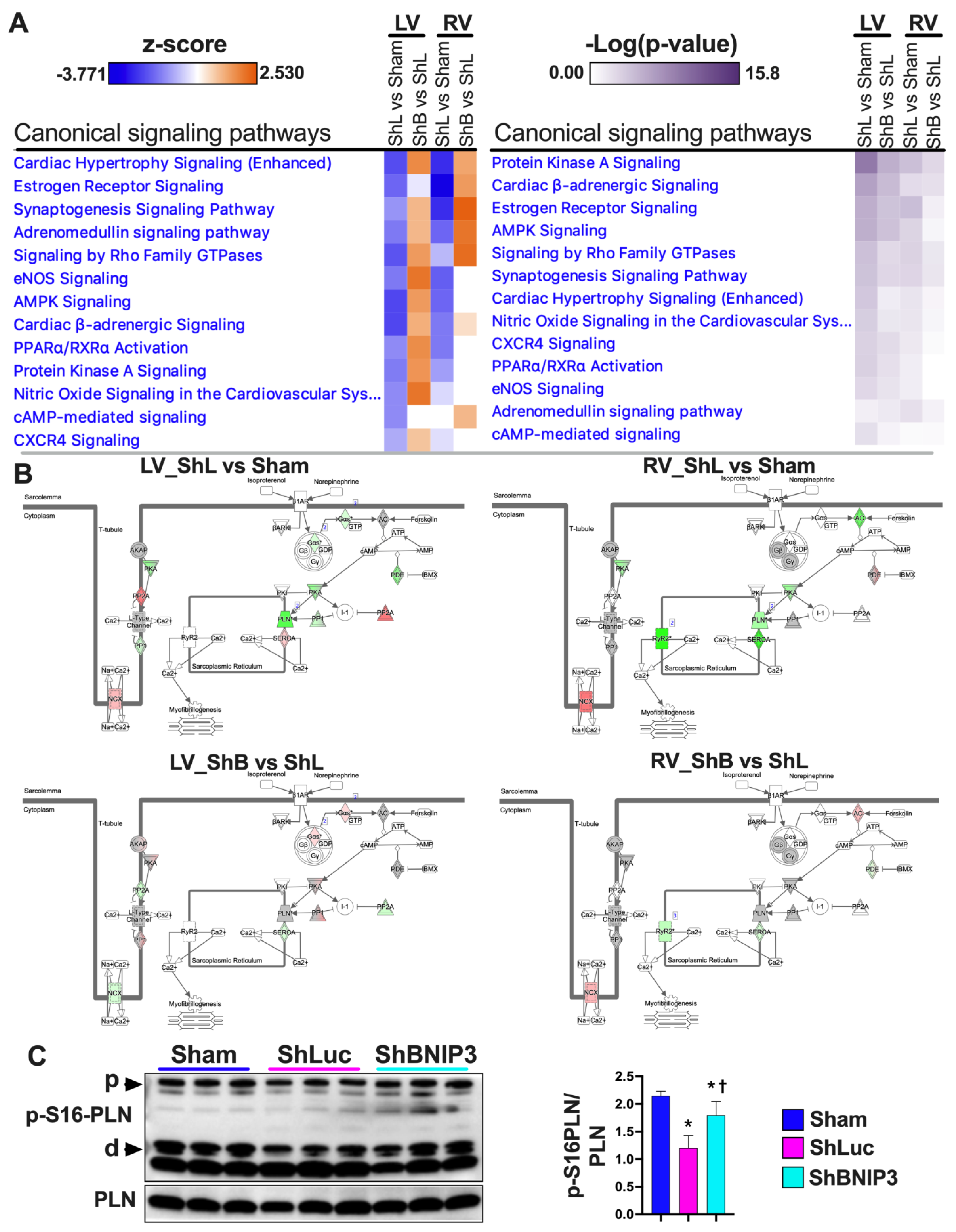
3.6. Visualization and Validation of the LV and RV Phosphoproteomic Datasets
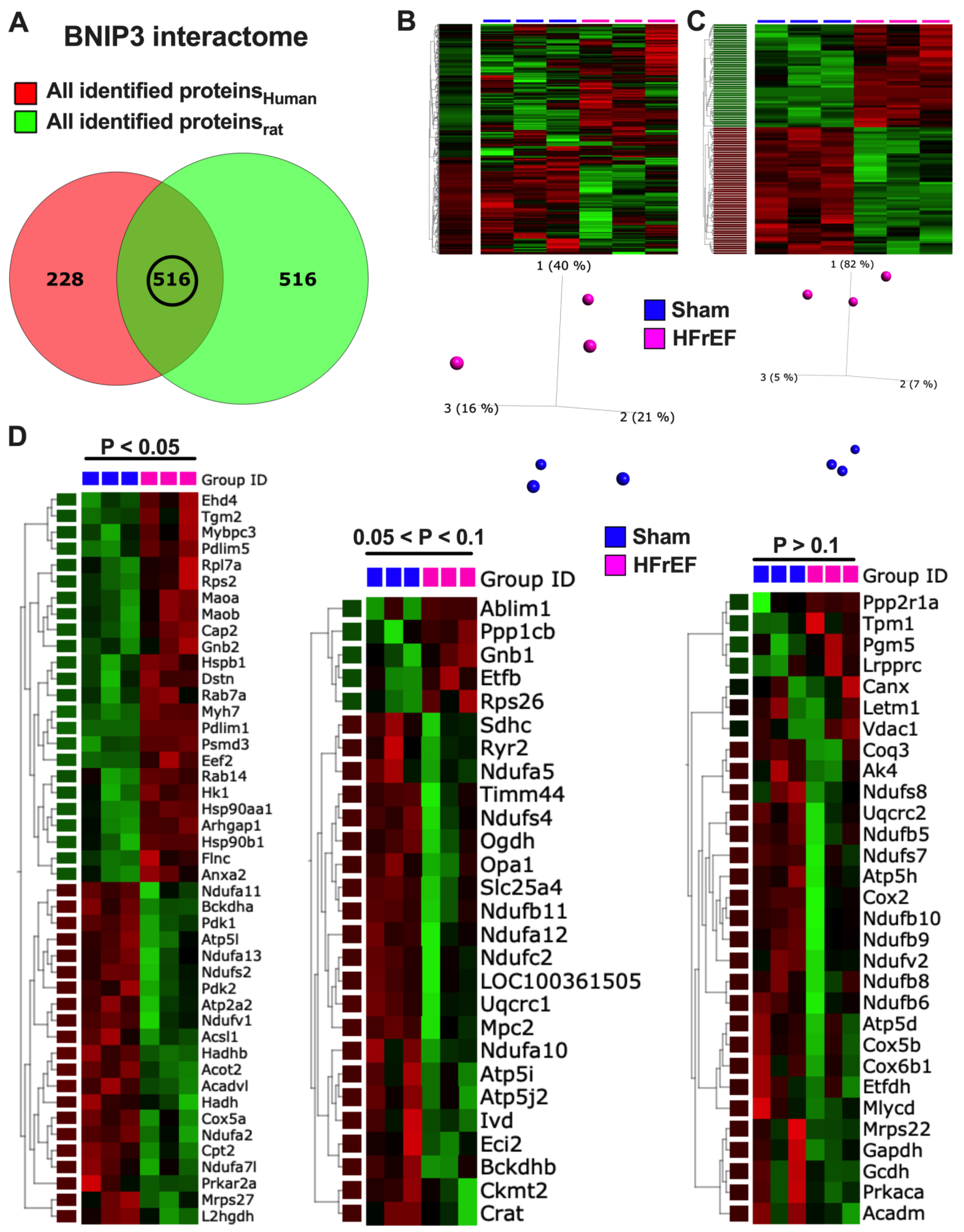
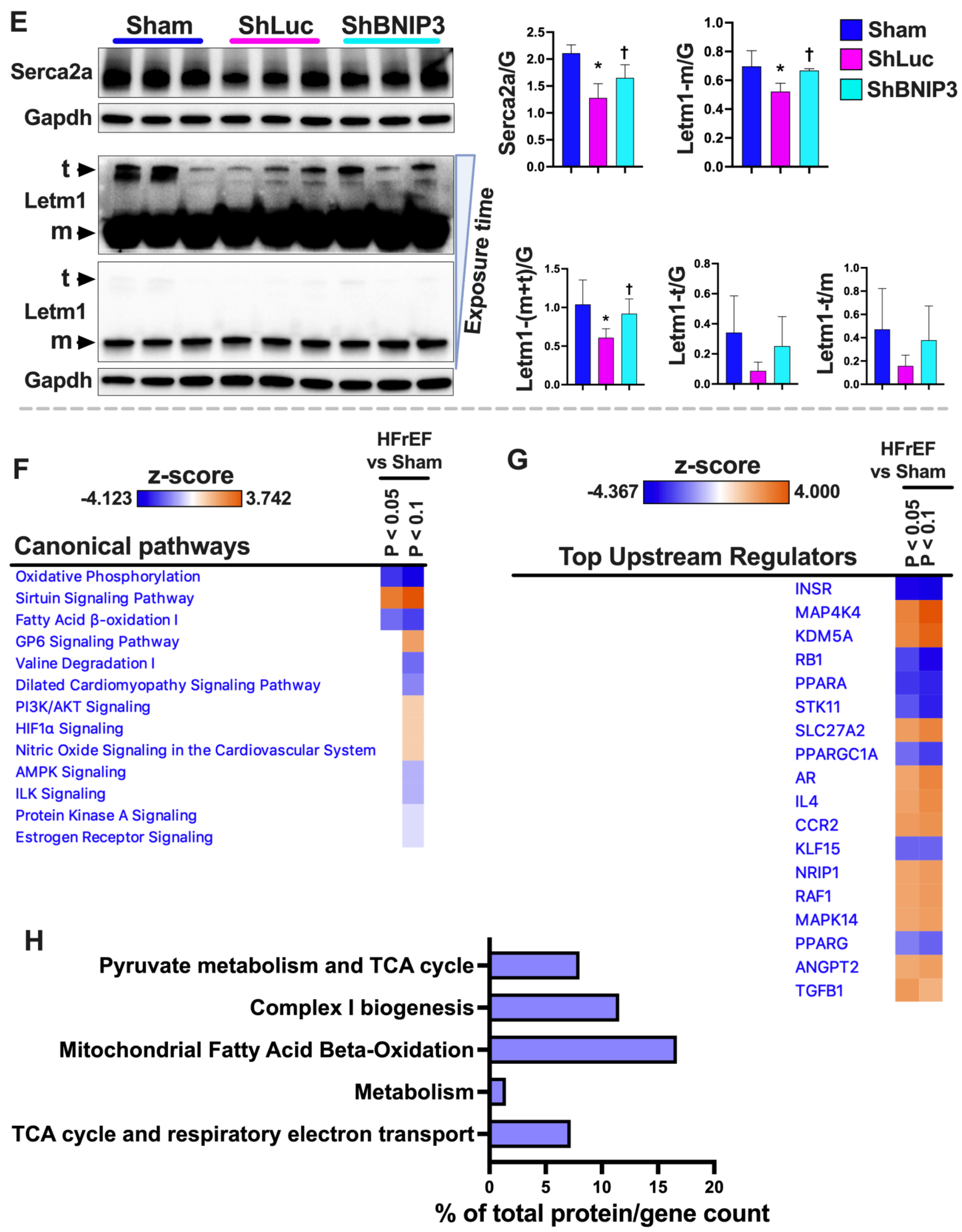
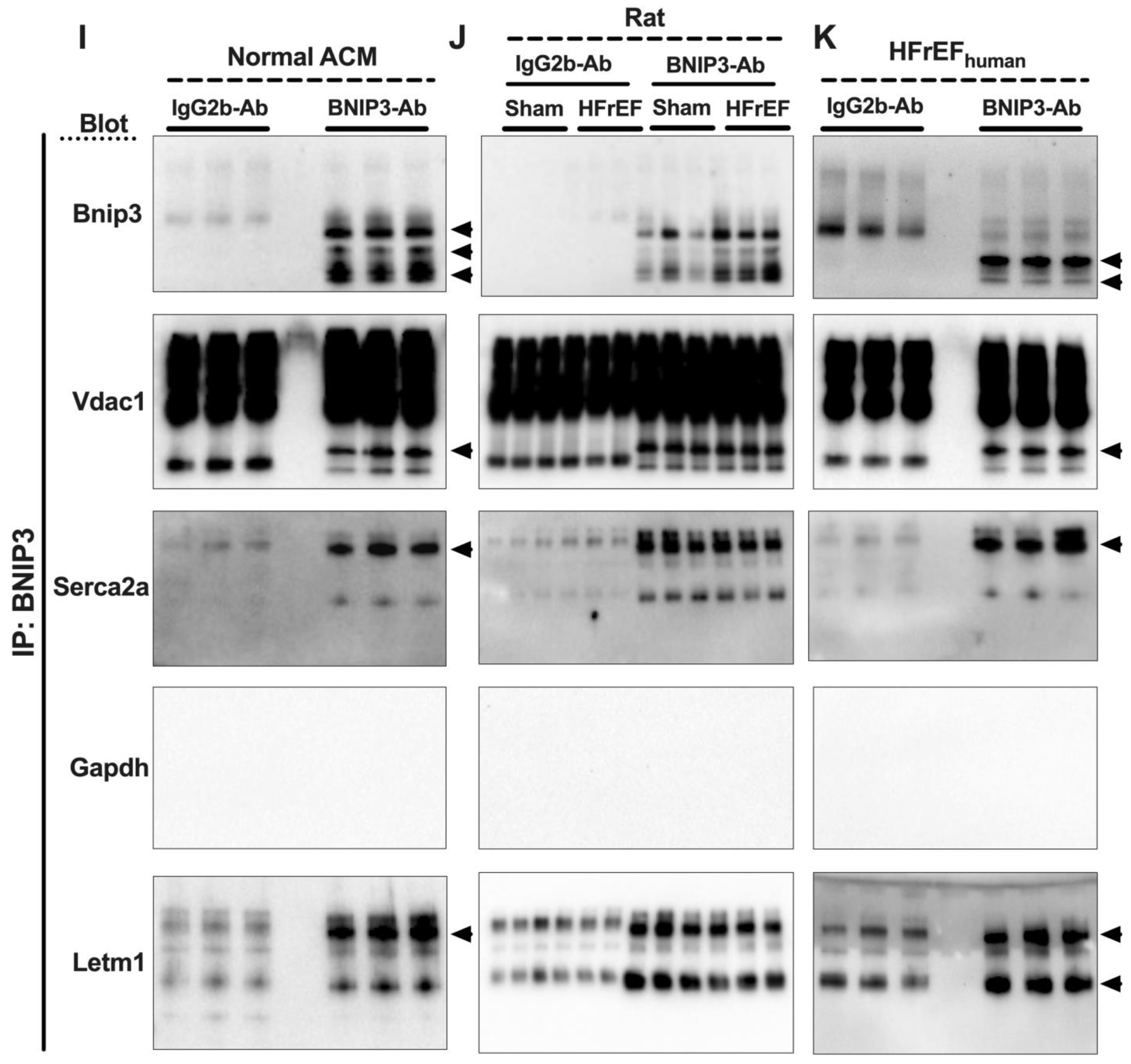
3.7. Visualization and Validation of BNIP3 Interactome in Rat and Human LV Myocardia
4. Discussion
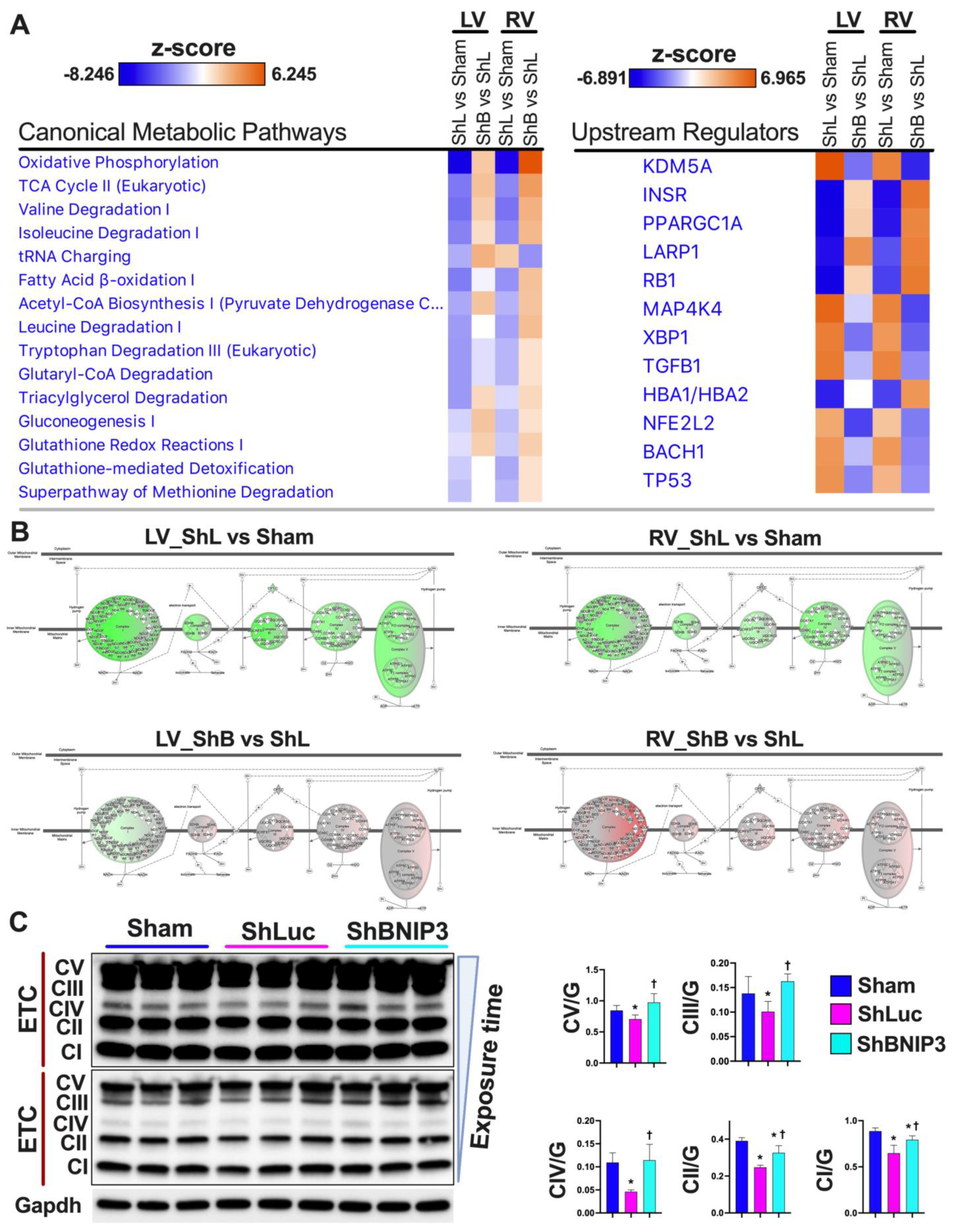
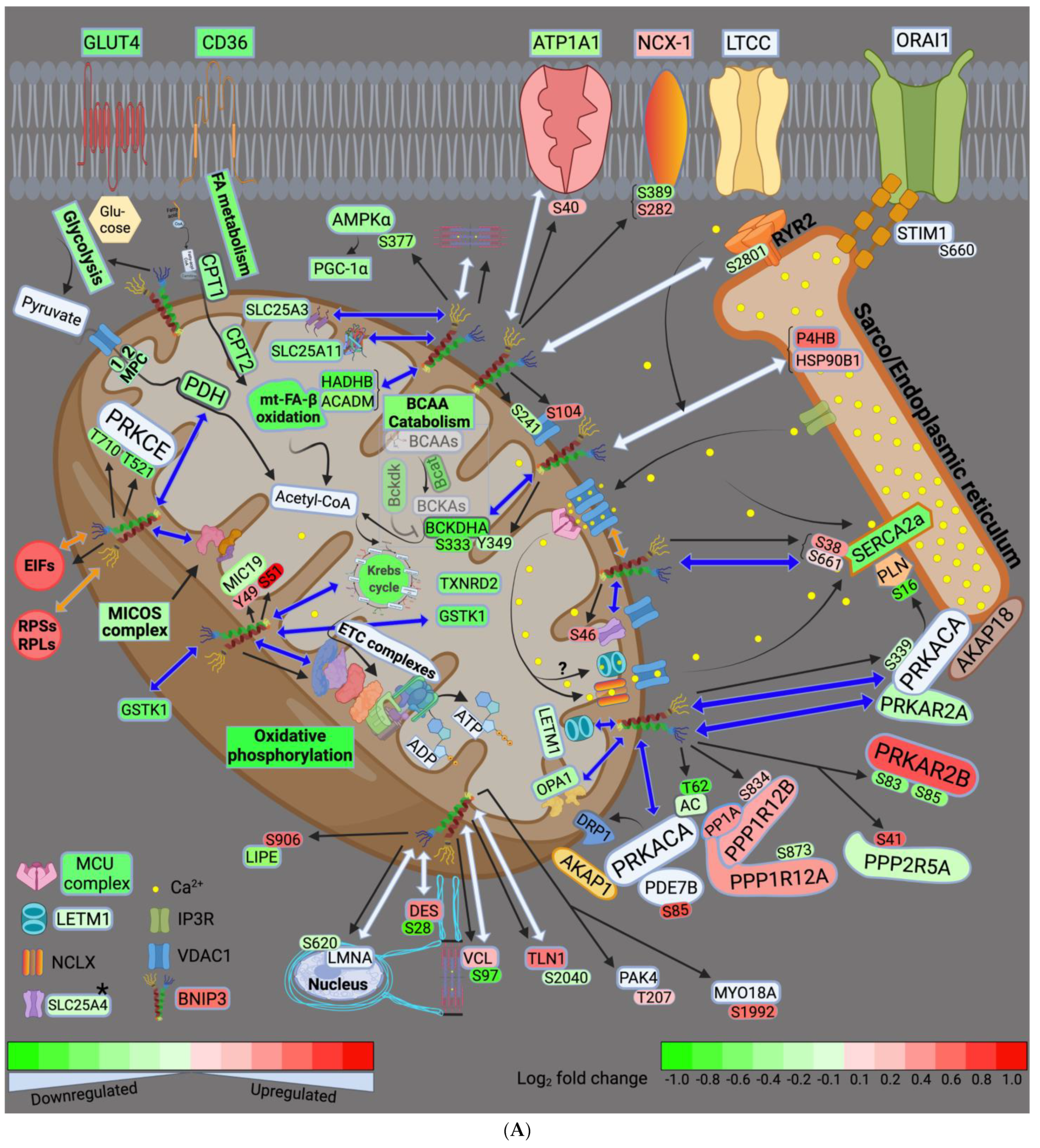
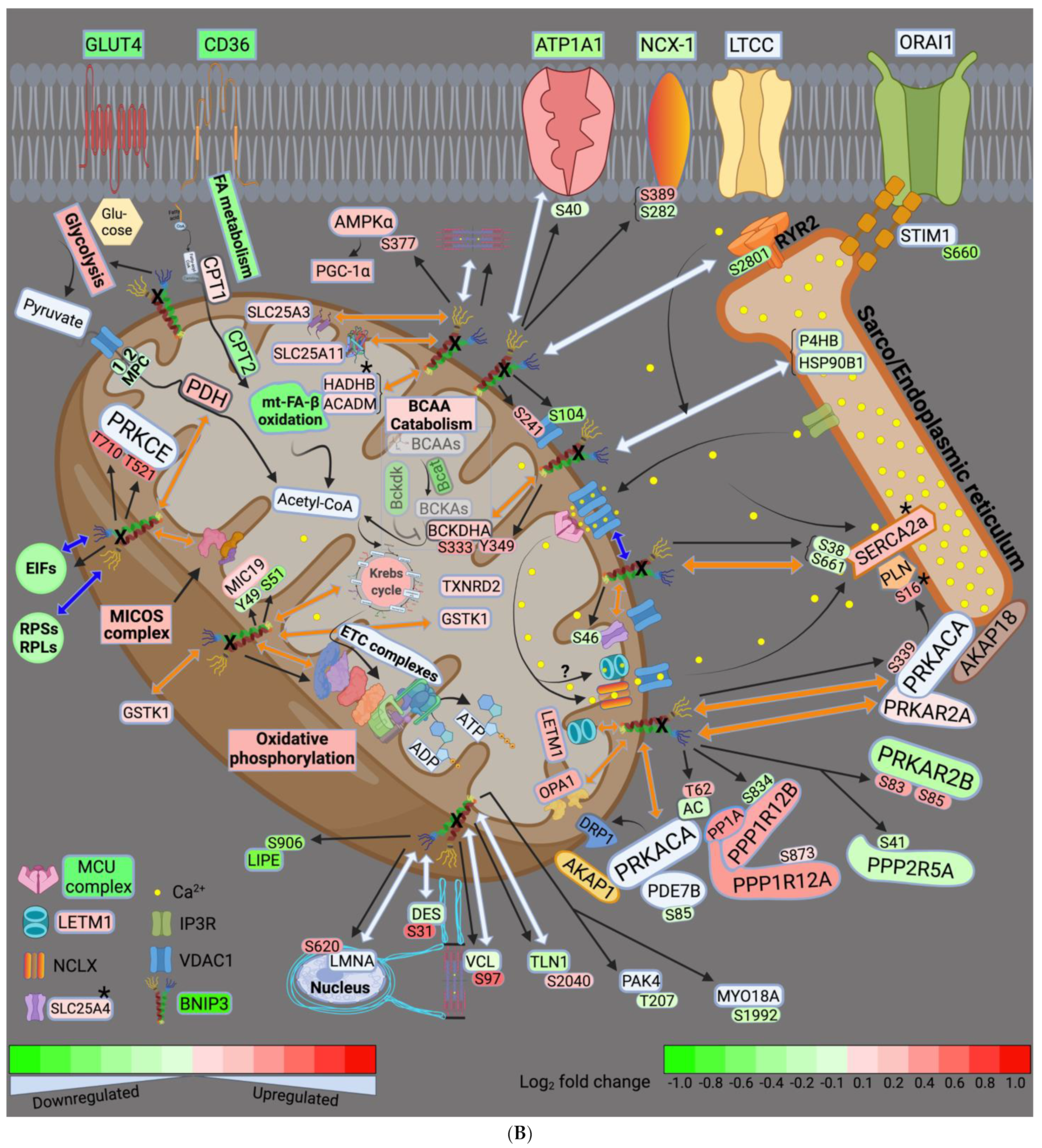
4.1. BNIP3 and Remodeling of the Mt-Proteome
4.2. BNIP3 and the Regulation of ER–Mt-Calcium and Ion Homeostasis
4.3. BNIP3 and PKA, PRKCE, and AMPK Signaling
4.4. Other Effects of BNIP3
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mann, D.L.; Felker, G.M. Mechanisms and Models in Heart Failure: A Translational Approach. Circ. Res. 2021, 128, 1435–1450. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.A.; Olson, E.N. Cardiac plasticity. N. Engl. J. Med. 2008, 358, 1370–1380. [Google Scholar] [CrossRef] [PubMed]
- Chaanine, A.H. Metabolic Remodeling and Implicated Calcium and Signal Transduction Pathways in the Pathogenesis of Heart Failure. Int. J. Mol. Sci. 2021, 22, 10579. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. The Extracellular Matrix in Ischemic and Nonischemic Heart Failure. Circ. Res. 2019, 125, 117–146. [Google Scholar] [CrossRef]
- Quinsay, M.N.; Lee, Y.; Rikka, S.; Sayen, M.R.; Molkentin, J.D.; Gottlieb, R.A.; Gustafsson, A.B. Bnip3 mediates permeabilization of mitochondria and release of cytochrome c via a novel mechanism. J. Mol. Cell Cardiol. 2010, 48, 1146–1156. [Google Scholar] [CrossRef] [Green Version]
- Quinsay, M.N.; Thomas, R.L.; Lee, Y.; Gustafsson, A.B. Bnip3-mediated mitochondrial autophagy is independent of the mitochondrial permeability transition pore. Autophagy 2010, 6, 855–862. [Google Scholar] [CrossRef] [Green Version]
- Ray, R.; Chen, G.; Vande Velde, C.; Cizeau, J.; Park, J.H.; Reed, J.C.; Gietz, R.D.; Greenberg, A.H. BNIP3 heterodimerizes with Bcl-2/Bcl-X(L) and induces cell death independent of a Bcl-2 homology 3 (BH3) domain at both mitochondrial and nonmitochondrial sites. J. Biol. Chem. 2000, 275, 1439–1448. [Google Scholar] [CrossRef] [Green Version]
- Regula, K.M.; Ens, K.; Kirshenbaum, L.A. Inducible expression of BNIP3 provokes mitochondrial defects and hypoxia-mediated cell death of ventricular myocytes. Circ. Res. 2002, 91, 226–231. [Google Scholar] [CrossRef] [Green Version]
- Chaanine, A.H.; Jeong, D.; Liang, L.; Chemaly, E.R.; Fish, K.; Gordon, R.E.; Hajjar, R.J. JNK modulates FOXO3a for the expression of the mitochondrial death and mitophagy marker BNIP3 in pathological hypertrophy and in heart failure. Cell Death Dis. 2012, 3, 265. [Google Scholar] [CrossRef] [Green Version]
- Chaanine, A.H.; Joyce, L.D.; Stulak, J.M.; Maltais, S.; Joyce, D.L.; Dearani, J.A.; Klaus, K.; Nair, K.S.; Hajjar, R.J.; Redfield, M.M. Mitochondrial Morphology, Dynamics, and Function in Human Pressure Overload or Ischemic Heart Disease With Preserved or Reduced Ejection Fraction. Circ. Heart Fail. 2019, 12, e005131. [Google Scholar] [CrossRef]
- Diwan, A.; Krenz, M.; Syed, F.M.; Wansapura, J.; Ren, X.; Koesters, A.G.; Li, H.; Kirshenbaum, L.A.; Hahn, H.S.; Robbins, J.; et al. Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. J. Clin. Investig. 2007, 117, 2825–2833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaanine, A.H.; Gordon, R.E.; Kohlbrenner, E.; Benard, L.; Jeong, D.; Hajjar, R.J. Potential role of BNIP3 in cardiac remodeling, myocardial stiffness, and endoplasmic reticulum: Mitochondrial calcium homeostasis in diastolic and systolic heart failure. Circ. Heart Fail. 2013, 6, 572–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaanine, A.H.; Kohlbrenner, E.; Gamb, S.I.; Guenzel, A.J.; Klaus, K.; Fayyaz, A.U.; Nair, K.S.; Hajjar, R.J.; Redfield, M.M. FOXO3a regulates BNIP3 and modulates mitochondrial calcium, dynamics, and function in cardiac stress. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H1540–H1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapti, K.; Louis-Jeune, V.; Kohlbrenner, E.; Ishikawa, K.; Ladage, D.; Zolotukhin, S.; Hajjar, R.J.; Weber, T. Neutralizing antibodies against AAV serotypes 1, 2, 6, and 9 in sera of commonly used animal models. Mol. Ther. J. Am. Soc. Gene Ther. 2012, 20, 73–83. [Google Scholar] [CrossRef] [Green Version]
- Lanza, I.R.; Nair, K.S. Functional assessment of isolated mitochondria in vitro. Methods Enzym. 2009, 457, 349–372. [Google Scholar] [CrossRef] [Green Version]
- Picard, M.; Wright, K.J.; Ritchie, D.; Thomas, M.M.; Hepple, R.T. Mitochondrial function in permeabilized cardiomyocytes is largely preserved in the senescent rat myocardium. PLoS ONE 2012, 7, e43003. [Google Scholar] [CrossRef] [Green Version]
- Chaanine, A.H.; Navar, L.G.; Delafontaine, P. A Rat Model of Pressure Overload Induced Moderate Remodeling and Systolic Dysfunction as Opposed to Overt Systolic Heart Failure. J. Vis. Exp. JoVE 2020, 158, e60954. [Google Scholar] [CrossRef]
- Chaanine, A.H.; Higgins, L.; Markowski, T.; Harman, J.; Kachman, M.; Burant, C.; Navar, L.G.; Busija, D.; Delafontaine, P. Multi-Omics Approach Profiling Metabolic Remodeling in Early Systolic Dysfunction and in Overt Systolic Heart Failure. Int. J. Mol. Sci. 2021, 23, 235. [Google Scholar] [CrossRef]
- Shadforth, I.P.; Dunkley, T.P.; Lilley, K.S.; Bessant, C. i-Tracker: For quantitative proteomics using iTRAQ. BMC Genom. 2005, 6, 145. [Google Scholar] [CrossRef] [Green Version]
- Oberg, A.L.; Mahoney, D.W.; Eckel-Passow, J.E.; Malone, C.J.; Wolfinger, R.D.; Hill, E.G.; Cooper, L.T.; Onuma, O.K.; Spiro, C.; Therneau, T.M.; et al. Statistical analysis of relative labeled mass spectrometry data from complex samples using ANOVA. J. Proteome Res. 2008, 7, 225–233. [Google Scholar] [CrossRef]
- Glickman, M.E.; Rao, S.R.; Schultz, M.R. False discovery rate control is a recommended alternative to Bonferroni-type adjustments in health studies. J. Clin. Epidemiol. 2014, 67, 850–857. [Google Scholar] [CrossRef] [PubMed]
- Thonusin, C.; IglayReger, H.B.; Soni, T.; Rothberg, A.E.; Burant, C.F.; Evans, C.R. Evaluation of intensity drift correction strategies using MetaboDrift, a normalization tool for multi-batch metabolomics data. J. Chromatogr. A 2017, 1523, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Kachman, M.; Habra, H.; Duren, W.; Wigginton, J.; Sajjakulnukit, P.; Michailidis, G.; Burant, C.; Karnovsky, A. Deep annotation of untargeted LC-MS metabolomics data with Binner. Bioinformatics 2020, 36, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Takahashi, Y. Analysis of Protein-Protein Interaction by Co-IP in Human Cells. Methods Mol. Biol. 2018, 1794, 289–296. [Google Scholar] [CrossRef]
- Dai, D.F.; Hsieh, E.J.; Chen, T.; Menendez, L.G.; Basisty, N.B.; Tsai, L.; Beyer, R.P.; Crispin, D.A.; Shulman, N.J.; Szeto, H.H.; et al. Global proteomics and pathway analysis of pressure-overload-induced heart failure and its attenuation by mitochondrial-targeted peptides. Circ. Heart Fail. 2013, 6, 1067–1076. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Zhang, Z.; Kolwicz, S.C., Jr.; Abell, L.; Roe, N.D.; Kim, M.; Zhou, B.; Cao, Y.; Ritterhoff, J.; Gu, H.; et al. Defective Branched-Chain Amino Acid Catabolism Disrupts Glucose Metabolism and Sensitizes the Heart to Ischemia-Reperfusion Injury. Cell Metab. 2017, 25, 374–385. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Olson, K.C.; Gao, C.; Prosdocimo, D.A.; Zhou, M.; Wang, Z.; Jeyaraj, D.; Youn, J.Y.; Ren, S.; Liu, Y.; et al. Catabolic Defect of Branched-Chain Amino Acids Promotes Heart Failure. Circulation 2016, 133, 2038–2049. [Google Scholar] [CrossRef]
- Peoples, J.N.; Ghazal, N.; Duong, D.M.; Hardin, K.R.; Manning, J.R.; Seyfried, N.T.; Faundez, V.; Kwong, J.Q. Loss of the mitochondrial phosphate carrier SLC25A3 induces remodeling of the cardiac mitochondrial protein acylome. Am. J. Physiol. Cell Physiol. 2021, 321, C519–C534. [Google Scholar] [CrossRef]
- Gallo, M.; Park, D.; Luciani, D.S.; Kida, K.; Palmieri, F.; Blacque, O.E.; Johnson, J.D.; Riddle, D.L. MISC-1/OGC links mitochondrial metabolism, apoptosis and insulin secretion. PLoS ONE 2011, 6, e17827. [Google Scholar] [CrossRef]
- Tosserams, A.; Papadopoulos, C.; Jardel, C.; Lemière, I.; Romero, N.B.; De Lonlay, P.; Wahbi, K.; Voermans, N.; Hogrel, J.Y.; Laforêt, P. Two new cases of mitochondrial myopathy with exercise intolerance, hyperlactatemia and cardiomyopathy, caused by recessive SLC25A4 mutations. Mitochondrion 2018, 39, 26–29. [Google Scholar] [CrossRef]
- McManus, M.J.; Picard, M.; Chen, H.W.; De Haas, H.J.; Potluri, P.; Leipzig, J.; Towheed, A.; Angelin, A.; Sengupta, P.; Morrow, R.M.; et al. Mitochondrial DNA Variation Dictates Expressivity and Progression of Nuclear DNA Mutations Causing Cardiomyopathy. Cell Metab. 2019, 29, 78–90.e75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landes, T.; Emorine, L.J.; Courilleau, D.; Rojo, M.; Belenguer, P.; Arnaune-Pelloquin, L. The BH3-only Bnip3 binds to the dynamin Opa1 to promote mitochondrial fragmentation and apoptosis by distinct mechanisms. EMBO Rep. 2010, 11, 459–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callegari, S.; Müller, T.; Schulz, C.; Lenz, C.; Jans, D.C.; Wissel, M.; Opazo, F.; Rizzoli, S.O.; Jakobs, S.; Urlaub, H.; et al. A MICOS-TIM22 Association Promotes Carrier Import into Human Mitochondria. J. Mol. Biol. 2019, 431, 2835–2851. [Google Scholar] [CrossRef] [PubMed]
- Ott, C.; Ross, K.; Straub, S.; Thiede, B.; Götz, M.; Goosmann, C.; Krischke, M.; Mueller, M.J.; Krohne, G.; Rudel, T.; et al. Sam50 functions in mitochondrial intermembrane space bridging and biogenesis of respiratory complexes. Mol. Cell. Biol. 2012, 32, 1173–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, C.; Dorsch, E.; Fraunholz, M.; Straub, S.; Kozjak-Pavlovic, V. Detailed analysis of the human mitochondrial contact site complex indicate a hierarchy of subunits. PLoS ONE 2015, 10, e0120213. [Google Scholar] [CrossRef]
- Rikka, S.; Quinsay, M.N.; Thomas, R.L.; Kubli, D.A.; Zhang, X.; Murphy, A.N.; Gustafsson, Å.B. Bnip3 impairs mitochondrial bioenergetics and stimulates mitochondrial turnover. Cell Death Differ. 2011, 18, 721–731. [Google Scholar] [CrossRef] [Green Version]
- Gang, H.; Hai, Y.; Dhingra, R.; Gordon, J.W.; Yurkova, N.; Aviv, Y.; Li, H.; Aguilar, F.; Marshall, A.; Leygue, E.; et al. A novel hypoxia-inducible spliced variant of mitochondrial death gene Bnip3 promotes survival of ventricular myocytes. Circ. Res. 2011, 108, 1084–1092. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.E.; Frazier, W.A. Phosphorylation of the BNIP3 C-Terminus Inhibits Mitochondrial Damage and Cell Death without Blocking Autophagy. PLoS ONE 2015, 10, e0129667. [Google Scholar] [CrossRef] [Green Version]
- Shi, R.Y.; Zhu, S.H.; Li, V.; Gibson, S.B.; Xu, X.S.; Kong, J.M. BNIP3 interacting with LC3 triggers excessive mitophagy in delayed neuronal death in stroke. CNS Neurosci. Ther. 2014, 20, 1045–1055. [Google Scholar] [CrossRef]
- Yuan, S.; Fu, Y.; Wang, X.; Shi, H.; Huang, Y.; Song, X.; Li, L.; Song, N.; Luo, Y. Voltage-dependent anion channel 1 is involved in endostatin-induced endothelial cell apoptosis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2008, 22, 2809–2820. [Google Scholar] [CrossRef]
- De Stefani, D.; Bononi, A.; Romagnoli, A.; Messina, A.; De Pinto, V.; Pinton, P.; Rizzuto, R. VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death Differ. 2012, 19, 267–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finkel, T.; Menazza, S.; Holmstrom, K.M.; Parks, R.J.; Liu, J.; Sun, J.; Liu, J.; Pan, X.; Murphy, E. The ins and outs of mitochondrial calcium. Circ. Res. 2015, 116, 1810–1819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joiner, M.L.; Koval, O.M.; Li, J.; He, B.J.; Allamargot, C.; Gao, Z.; Luczak, E.D.; Hall, D.D.; Fink, B.D.; Chen, B.; et al. CaMKII determines mitochondrial stress responses in heart. Nature 2012, 491, 269–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luongo, T.S.; Lambert, J.P.; Gross, P.; Nwokedi, M.; Lombardi, A.A.; Shanmughapriya, S.; Carpenter, A.C.; Kolmetzky, D.; Gao, E.; van Berlo, J.H.; et al. The mitochondrial Na(+)/Ca(2+) exchanger is essential for Ca(2+) homeostasis and viability. Nature 2017, 545, 93–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA 2010, 107, 436–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, S.; Matsui, A.; Akabane, S.; Tamura, Y.; Hatano, A.; Miyano, Y.; Omote, H.; Kajikawa, M.; Maenaka, K.; Moriyama, Y.; et al. The mitochondrial inner membrane protein LETM1 modulates cristae organization through its LETM domain. Commun. Biol. 2020, 3, 99. [Google Scholar] [CrossRef]
- Doonan, P.J.; Chandramoorthy, H.C.; Hoffman, N.E.; Zhang, X.; Cárdenas, C.; Shanmughapriya, S.; Rajan, S.; Vallem, S.; Chen, X.; Foskett, J.K.; et al. LETM1-dependent mitochondrial Ca2+ flux modulates cellular bioenergetics and proliferation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2014, 28, 4936–4949. [Google Scholar] [CrossRef] [Green Version]
- Shao, J.; Fu, Z.; Ji, Y.; Guan, X.; Guo, S.; Ding, Z.; Yang, X.; Cong, Y.; Shen, Y. Leucine zipper-EF-hand containing transmembrane protein 1 (LETM1) forms a Ca(2+)/H(+) antiporter. Sci. Rep. 2016, 6, 34174. [Google Scholar] [CrossRef] [Green Version]
- Dimmer, K.S.; Navoni, F.; Casarin, A.; Trevisson, E.; Endele, S.; Winterpacht, A.; Salviati, L.; Scorrano, L. LETM1, deleted in Wolf-Hirschhorn syndrome is required for normal mitochondrial morphology and cellular viability. Hum. Mol. Genet. 2008, 17, 201–214. [Google Scholar] [CrossRef]
- Tamai, S.; Iida, H.; Yokota, S.; Sayano, T.; Kiguchiya, S.; Ishihara, N.; Hayashi, J.; Mihara, K.; Oka, T. Characterization of the mitochondrial protein LETM1, which maintains the mitochondrial tubular shapes and interacts with the AAA-ATPase BCS1L. J. Cell Sci. 2008, 121, 2588–2600. [Google Scholar] [CrossRef] [Green Version]
- Keshwani, M.M.; Klammt, C.; von Daake, S.; Ma, Y.; Kornev, A.P.; Choe, S.; Insel, P.A.; Taylor, S.S. Cotranslational cis-phosphorylation of the COOH-terminal tail is a key priming step in the maturation of cAMP-dependent protein kinase. Proc. Natl. Acad. Sci. USA 2012, 109, E1221–E1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbin, J.D.; Sugden, P.H.; Lincoln, T.M.; Keely, S.L. Compartmentalization of adenosine 3’:5’-monophosphate and adenosine 3’:5’-monophosphate-dependent protein kinase in heart tissue. J. Biol. Chem. 1977, 252, 3854–3861. [Google Scholar] [CrossRef]
- Hayes, J.S.; Brunton, L.L.; Mayer, S.E. Selective activation of particulate cAMP-dependent protein kinase by isoproterenol and prostaglandin E1. J. Biol. Chem. 1980, 255, 5113–5119. [Google Scholar] [CrossRef]
- Wong, W.; Scott, J.D. AKAP signalling complexes: Focal points in space and time. Nat. Reviews. Mol. Cell Biol. 2004, 5, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Lin, R.Y.; Rubin, C.S. Organelle-specific targeting of protein kinase AII (PKAII). Molecular and in situ characterization of murine A kinase anchor proteins that recruit regulatory subunits of PKAII to the cytoplasmic surface of mitochondria. J. Biol. Chem. 1997, 272, 15247–15257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lygren, B.; Carlson, C.R.; Santamaria, K.; Lissandron, V.; McSorley, T.; Litzenberg, J.; Lorenz, D.; Wiesner, B.; Rosenthal, W.; Zaccolo, M.; et al. AKAP complex regulates Ca2+ re-uptake into heart sarcoplasmic reticulum. EMBO Rep. 2007, 8, 1061–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Ma, Y.; Taylor, S.S.; Tsien, R.Y. Genetically encoded reporters of protein kinase A activity reveal impact of substrate tethering. Proc. Natl. Acad. Sci. USA 2001, 98, 14997–15002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beene, D.L.; Scott, J.D. A-kinase anchoring proteins take shape. Curr. Opin. Cell Biol. 2007, 19, 192–198. [Google Scholar] [CrossRef] [Green Version]
- Gustavsson, M.; Verardi, R.; Mullen, D.G.; Mote, K.R.; Traaseth, N.J.; Gopinath, T.; Veglia, G. Allosteric regulation of SERCA by phosphorylation-mediated conformational shift of phospholamban. Proc. Natl. Acad. Sci. USA 2013, 110, 17338–17343. [Google Scholar] [CrossRef] [Green Version]
- Merrill, R.A.; Dagda, R.K.; Dickey, A.S.; Cribbs, J.T.; Green, S.H.; Usachev, Y.M.; Strack, S. Mechanism of neuroprotective mitochondrial remodeling by PKA/AKAP1. PLoS Biol. 2011, 9, e1000612. [Google Scholar] [CrossRef] [Green Version]
- Carlucci, A.; Lignitto, L.; Feliciello, A. Control of mitochondria dynamics and oxidative metabolism by cAMP, AKAPs and the proteasome. Trends Cell Biol. 2008, 18, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Bender, E.; Kadenbach, B. The allosteric ATP-inhibition of cytochrome c oxidase activity is reversibly switched on by cAMP-dependent phosphorylation. FEBS Lett. 2000, 466, 130–134. [Google Scholar] [CrossRef] [Green Version]
- De Rasmo, D.; Micelli, L.; Santeramo, A.; Signorile, A.; Lattanzio, P.; Papa, S. cAMP regulates the functional activity, coupling efficiency and structural organization of mammalian FOF1 ATP synthase. Biochim. Et Biophys. Acta 2016, 1857, 350–358. [Google Scholar] [CrossRef] [PubMed]
- De Rasmo, D.; Panelli, D.; Sardanelli, A.M.; Papa, S. cAMP-dependent protein kinase regulates the mitochondrial import of the nuclear encoded NDUFS4 subunit of complex I. Cell. Signal. 2008, 20, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Robin, M.A.; Prabu, S.K.; Raza, H.; Anandatheerthavarada, H.K.; Avadhani, N.G. Phosphorylation enhances mitochondrial targeting of GSTA4-4 through increased affinity for binding to cytoplasmic Hsp70. J. Biol. Chem. 2003, 278, 18960–18970. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.R.; Blackstone, C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J. Biol. Chem. 2007, 282, 21583–21587. [Google Scholar] [CrossRef] [Green Version]
- Horman, S.; Vertommen, D.; Heath, R.; Neumann, D.; Mouton, V.; Woods, A.; Schlattner, U.; Wallimann, T.; Carling, D.; Hue, L.; et al. Insulin antagonizes ischemia-induced Thr172 phosphorylation of AMP-activated protein kinase alpha-subunits in heart via hierarchical phosphorylation of Ser485/491. J. Biol. Chem. 2006, 281, 5335–5340. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Nie, J.; Wu, L.; Hu, Y.; Wen, Z.; Dong, L.; Zou, M.H.; Chen, C.; Wang, D.W. AMPKα2 Protects Against the Development of Heart Failure by Enhancing Mitophagy via PINK1 Phosphorylation. Circ. Res. 2018, 122, 712–729. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, N.J.; Parker, B.L.; Chaudhuri, R.; Fisher-Wellman, K.H.; Kleinert, M.; Humphrey, S.J.; Yang, P.; Holliday, M.; Trefely, S.; Fazakerley, D.J.; et al. Global Phosphoproteomic Analysis of Human Skeletal Muscle Reveals a Network of Exercise-Regulated Kinases and AMPK Substrates. Cell Metab. 2015, 22, 922–935. [Google Scholar] [CrossRef] [Green Version]
- Bogoyevitch, M.A.; Parker, P.J.; Sugden, P.H. Characterization of protein kinase C isotype expression in adult rat heart. Protein kinase C-epsilon is a major isotype present, and it is activated by phorbol esters, epinephrine, and endothelin. Circ. Res. 1993, 72, 757–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baines, C.P.; Song, C.X.; Zheng, Y.T.; Wang, G.W.; Zhang, J.; Wang, O.L.; Guo, Y.; Bolli, R.; Cardwell, E.M.; Ping, P. Protein kinase Cepsilon interacts with and inhibits the permeability transition pore in cardiac mitochondria. Circ. Res. 2003, 92, 873–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, M.O.; Karliner, J.S.; Mochly-Rosen, D. A selective epsilon-protein kinase C antagonist inhibits protection of cardiac myocytes from hypoxia-induced cell death. J. Biol. Chem. 1997, 272, 30945–30951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edmondson, R.D.; Vondriska, T.M.; Biederman, K.J.; Zhang, J.; Jones, R.C.; Zheng, Y.; Allen, D.L.; Xiu, J.X.; Cardwell, E.M.; Pisano, M.R.; et al. Protein kinase C epsilon signaling complexes include metabolism- and transcription/translation-related proteins: Complimentary separation techniques with LC/MS/MS. Mol. Cell. Proteom. MCP 2002, 1, 421–433. [Google Scholar] [CrossRef] [Green Version]
- Ping, P.; Zhang, J.; Pierce, W.M., Jr.; Bolli, R. Functional proteomic analysis of protein kinase C epsilon signaling complexes in the normal heart and during cardioprotection. Circ. Res. 2001, 88, 59–62. [Google Scholar] [CrossRef] [Green Version]
- Robles-Flores, M.; Meléndez, L.; García, W.; Mendoza-Hernández, G.; Lam, T.T.; Castañeda-Patlán, C.; González-Aguilar, H. Posttranslational modifications on protein kinase c isozymes. Effects of epinephrine and phorbol esters. Biochim. Et Biophys. Acta 2008, 1783, 695–712. [Google Scholar] [CrossRef] [Green Version]
- Hucho, T.B.; Dina, O.A.; Levine, J.D. Epac mediates a cAMP-to-PKC signaling in inflammatory pain: An isolectin B4(+) neuron-specific mechanism. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 6119–6126. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Cann, M.J.; Litvin, T.N.; Iourgenko, V.; Sinclair, M.L.; Levin, L.R.; Buck, J. Soluble adenylyl cyclase as an evolutionarily conserved bicarbonate sensor. Science 2000, 289, 625–628. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, B.S.; Conti, M. Calcium regulation of the soluble adenylyl cyclase expressed in mammalian spermatozoa. Proc. Natl. Acad. Sci. USA 2003, 100, 10676–10681. [Google Scholar] [CrossRef] [Green Version]
- Zippin, J.H.; Chen, Y.; Straub, S.G.; Hess, K.C.; Diaz, A.; Lee, D.; Tso, P.; Holz, G.G.; Sharp, G.W.; Levin, L.R.; et al. CO2/HCO3(-)- and calcium-regulated soluble adenylyl cyclase as a physiological ATP sensor. J. Biol. Chem. 2013, 288, 33283–33291. [Google Scholar] [CrossRef] [Green Version]
- Krebs, J.; Agellon, L.B.; Michalak, M. Ca(2+) homeostasis and endoplasmic reticulum (ER) stress: An integrated view of calcium signaling. Biochem. Biophys. Res. Commun. 2015, 460, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Giovarelli, M.; Zecchini, S.; Martini, E.; Garrè, M.; Barozzi, S.; Ripolone, M.; Napoli, L.; Coazzoli, M.; Vantaggiato, C.; Roux-Biejat, P.; et al. Drp1 overexpression induces desmin disassembling and drives kinesin-1 activation promoting mitochondrial trafficking in skeletal muscle. Cell Death Differ. 2020, 27, 2383–2401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Captur, G.; Arbustini, E.; Bonne, G.; Syrris, P.; Mills, K.; Wahbi, K.; Mohiddin, S.A.; McKenna, W.J.; Pettit, S.; Ho, C.Y.; et al. Lamin and the heart. Heart 2018, 104, 468–479. [Google Scholar] [CrossRef] [PubMed]
- Zemljic-Harpf, A.E.; Miller, J.C.; Henderson, S.A.; Wright, A.T.; Manso, A.M.; Elsherif, L.; Dalton, N.D.; Thor, A.K.; Perkins, G.A.; McCulloch, A.D.; et al. Cardiac-myocyte-specific excision of the vinculin gene disrupts cellular junctions, causing sudden death or dilated cardiomyopathy. Mol. Cell. Biol. 2007, 27, 7522–7537. [Google Scholar] [CrossRef] [Green Version]
- Zemljic-Harpf, A.; Manso, A.M.; Ross, R.S. Vinculin and talin: Focus on the myocardium. J. Investig. Med. Off. Publ. Am. Fed. Clin. Res. 2009, 57, 849–855. [Google Scholar] [CrossRef]
- Taylor, M.R.; Slavov, D.; Ku, L.; Di Lenarda, A.; Sinagra, G.; Carniel, E.; Haubold, K.; Boucek, M.M.; Ferguson, D.; Graw, S.L.; et al. Prevalence of desmin mutations in dilated cardiomyopathy. Circulation 2007, 115, 1244–1251. [Google Scholar] [CrossRef] [Green Version]
- Dalakas, M.C.; Park, K.Y.; Semino-Mora, C.; Lee, H.S.; Sivakumar, K.; Goldfarb, L.G. Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. N. Engl. J. Med. 2000, 342, 770–780. [Google Scholar] [CrossRef]

















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaanine, A.H.; Higgins, L.; Lauterboeck, L.; Markowski, T.; Yang, Q.; Delafontaine, P. Multiomics Approach Reveals an Important Role of BNIP3 in Myocardial Remodeling and the Pathogenesis of Heart Failure with Reduced Ejection Fraction. Cells 2022, 11, 1572. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11091572
Chaanine AH, Higgins L, Lauterboeck L, Markowski T, Yang Q, Delafontaine P. Multiomics Approach Reveals an Important Role of BNIP3 in Myocardial Remodeling and the Pathogenesis of Heart Failure with Reduced Ejection Fraction. Cells. 2022; 11(9):1572. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11091572
Chicago/Turabian StyleChaanine, Antoine H., LeeAnn Higgins, Lothar Lauterboeck, Todd Markowski, Qinglin Yang, and Patrice Delafontaine. 2022. "Multiomics Approach Reveals an Important Role of BNIP3 in Myocardial Remodeling and the Pathogenesis of Heart Failure with Reduced Ejection Fraction" Cells 11, no. 9: 1572. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11091572