
Epigallocatechine-3-gallate Inhibits the Adipogenesis of Human Mesenchymal Stem Cells via the Regulation of Protein Phosphatase-2A and Myosin Phosphatase


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Isolation and Phenotyping of Mesenchymal Stem Cells
2.3. MSC Culture and Differentiation
2.4. Nile Red Staining and Quantification
2.5. Immunofluorescence Staining
2.6. Protein Extraction and Western Blotting
2.7. Assay of Phosphatase Activity
2.8. Cell Fractionation
2.9. Reverse Transcription–Polymerase Chain Reaction
2.10. Statistical Analysis
3. Results
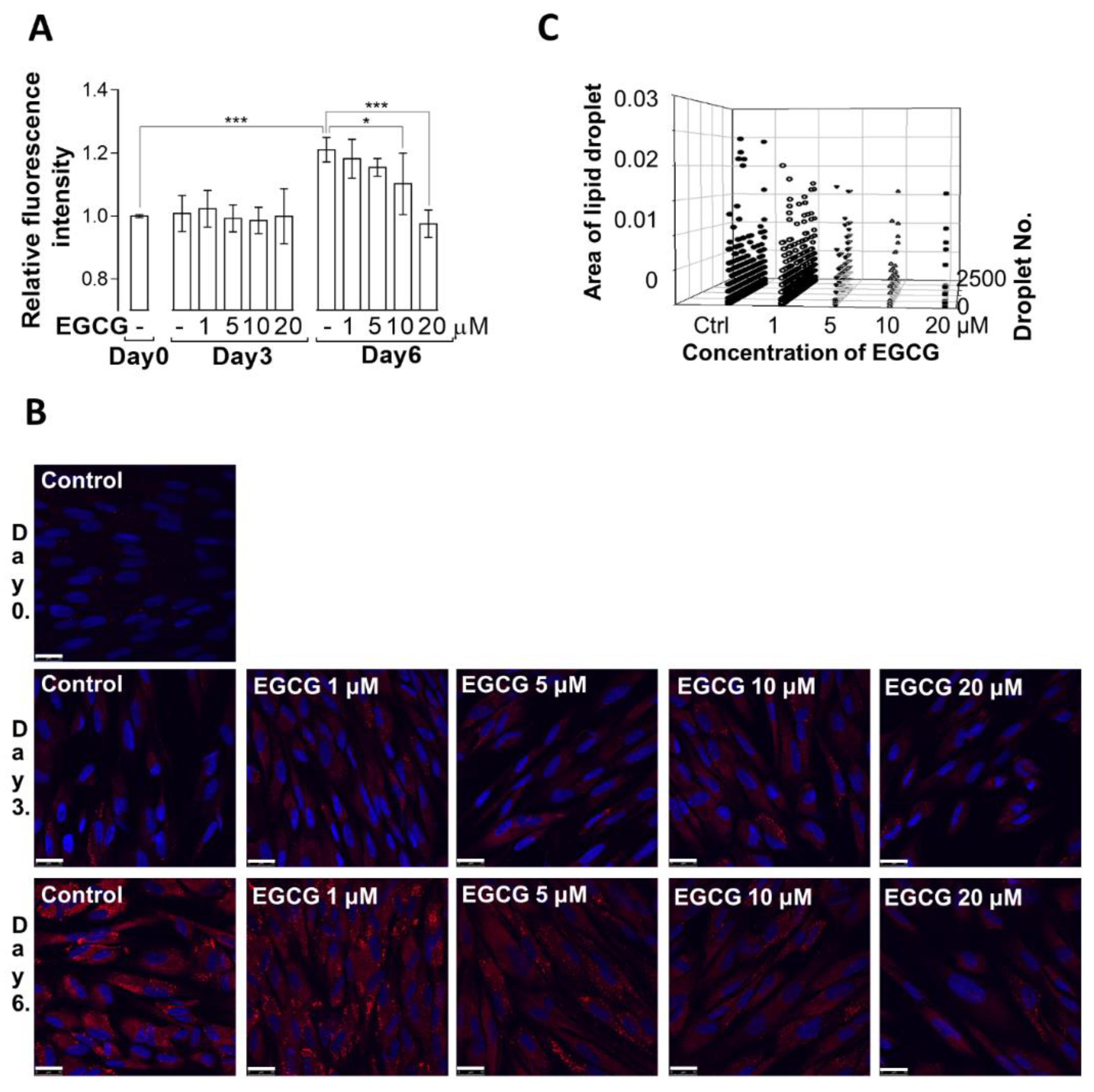
3.1. Effect of EGCG on Lipid Droplet Formation during the Adipogenic Differentiation of MSCs
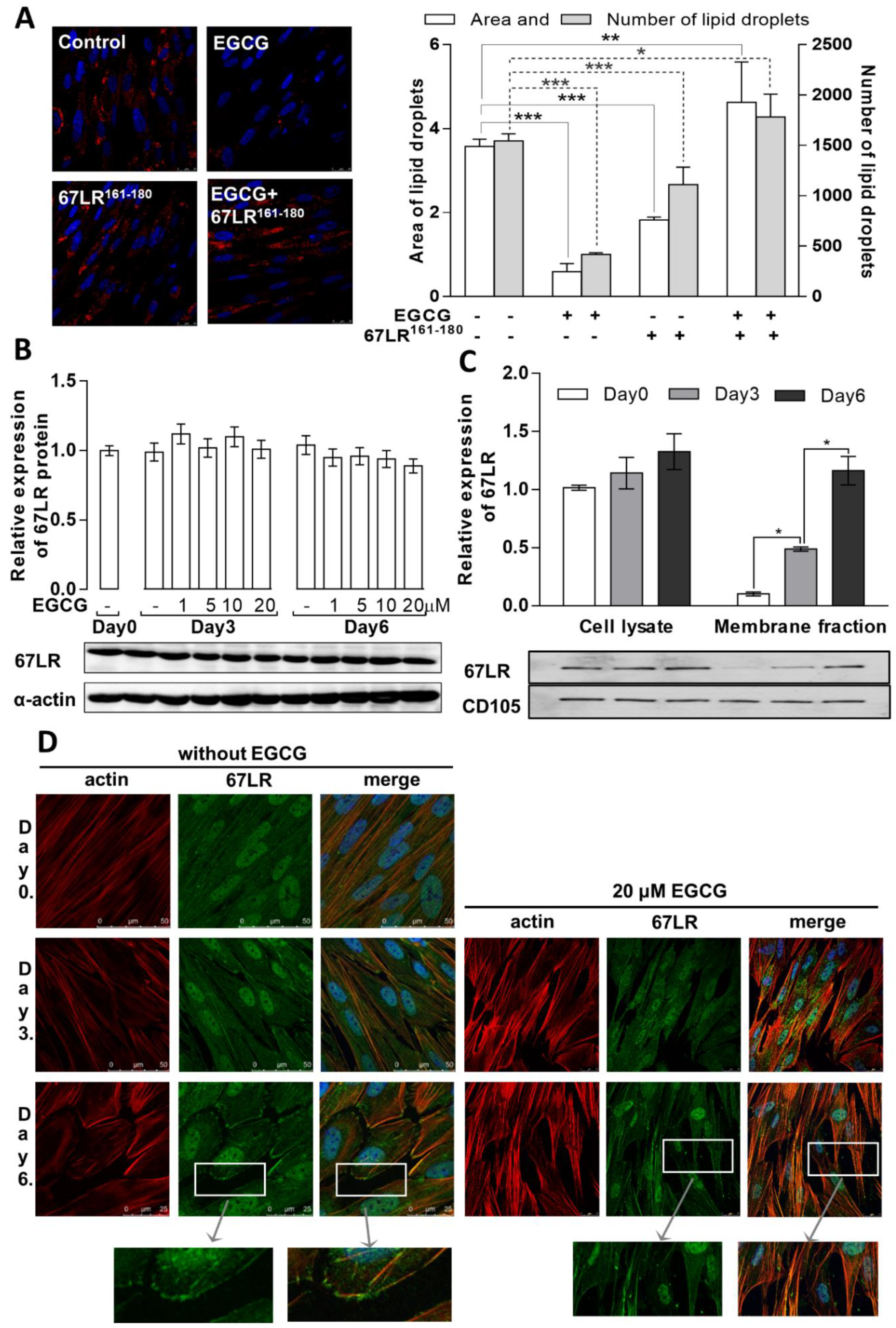
3.2. Distinct Localization of 67LR during Differentiation May Influence the Effects of EGCG on Adipogenesis
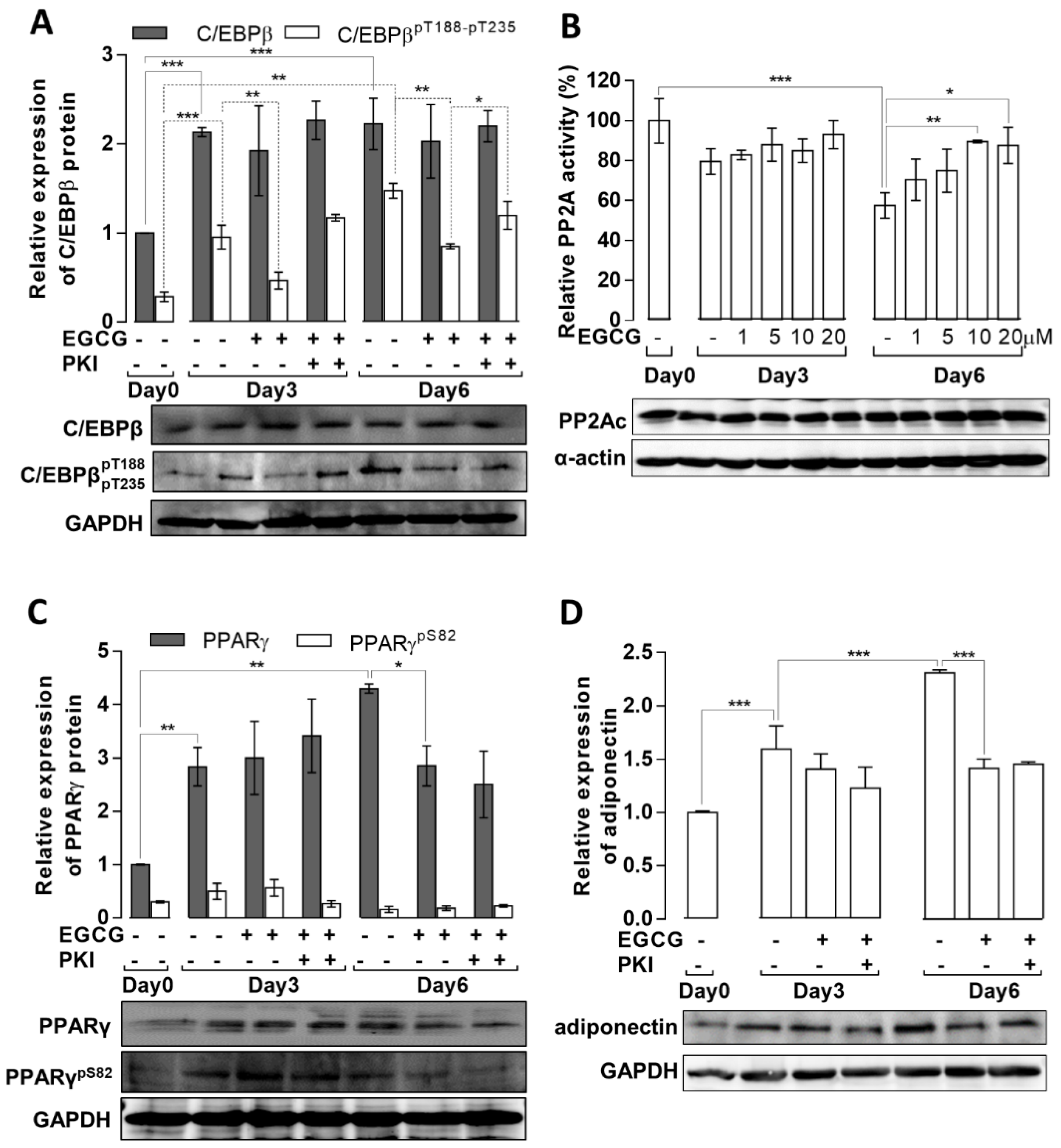
3.3. Effects of EGCG on the Expression and Phosphorylation of Key Transcription Factors, Adiponectin, and PP2A Activity
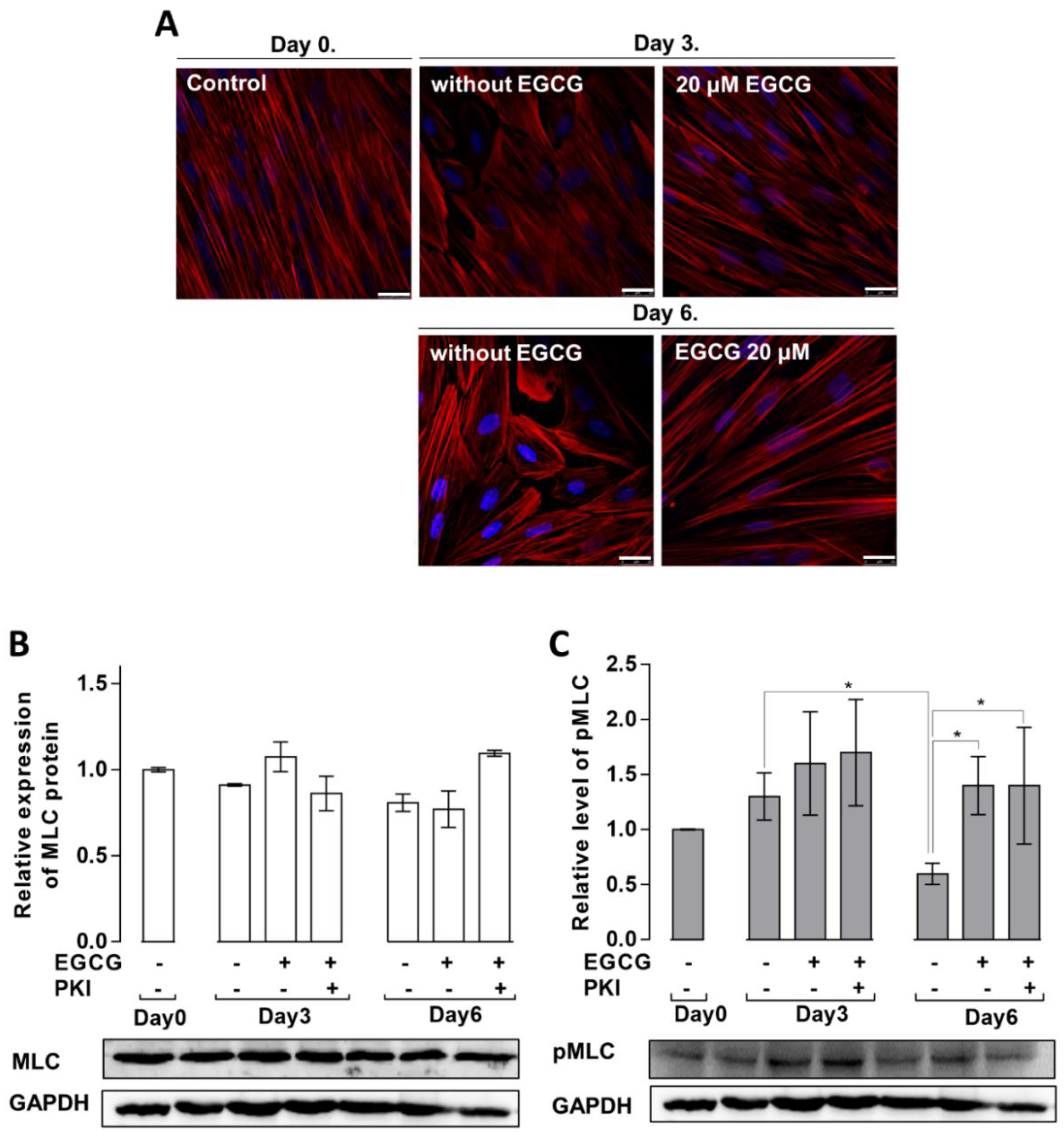
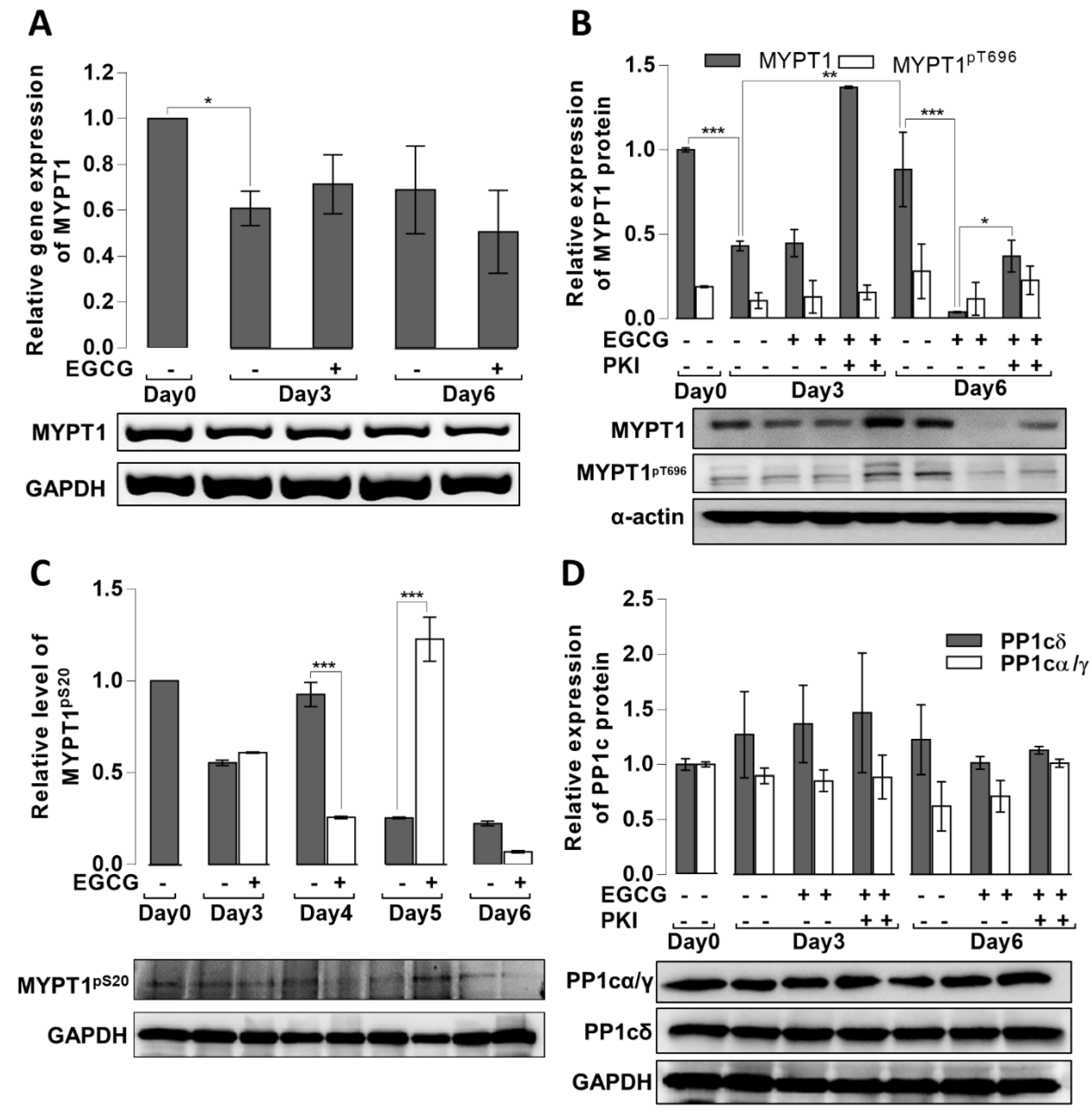
3.4. Effects of EGCG on the MSC Cytoskeletal Rearrangement and Phosphorylation of MLC20 during Adipogenesis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Castaner, O.; Goday, A.; Park, Y.M.; Lee, S.H.; Magkos, F.; Shiow, S.T.E.; Schroder, H. The Gut Microbiome Profile in Obesity: A Systematic Review. Int. J. Endocrinol. 2018, 2018, 4095789. [Google Scholar] [CrossRef] [PubMed]
- Ghaben, A.L.; Scherer, P.E. Adipogenesis and metabolic health. Nat. Rev. Mol. Cell Biol. 2019, 20, 242–258. [Google Scholar] [CrossRef] [PubMed]
- Kopelman, P.G. Obesity as a medical problem. Nature 2000, 404, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Spalding, K.L.; Arner, E.; Westermark, P.O.; Bernard, S.; Buchholz, B.A.; Bergmann, O.; Blomqvist, L.; Hoffstedt, J.; Naslund, E.; Britton, T.; et al. Dynamics of fat cell turnover in humans. Nature 2008, 453, 783–787. [Google Scholar] [CrossRef]
- Cristancho, A.G.; Lazar, M.A. Forming functional fat: A growing understanding of adipocyte differentiation. Nat. Rev. Mol. Cell Biol. 2011, 12, 722–734. [Google Scholar] [CrossRef]
- Hamm, J.K.; Park, B.H.; Farmer, S.R. A role for C/EBPbeta in regulating peroxisome proliferator-activated receptor gamma activity during adipogenesis in 3T3-L1 preadipocytes. J. Biol. Chem. 2001, 276, 18464–18471. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.E.; Schmidt, H.; Lai, B.; Ge, K. Transcriptional and Epigenomic Regulation of Adipogenesis. Mol. Cell Biol. 2019, 39. [Google Scholar] [CrossRef] [Green Version]
- Moseti, D.; Regassa, A.; Kim, W.K. Molecular Regulation of Adipogenesis and Potential Anti-Adipogenic Bioactive Molecules. Int. J. Mol. Sci. 2016, 17, 124. [Google Scholar] [CrossRef] [Green Version]
- Burns, K.A.; Vanden Heuvel, J.P. Modulation of PPAR activity via phosphorylation. Biochim. Biophys. Acta 2007, 1771, 952–960. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Li, X.; Tang, Q.Q. Transcriptional regulation of adipocyte differentiation: A central role for CCAAT/enhancer-binding protein (C/EBP) beta. J. Biol. Chem. 2015, 290, 755–761. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.L.; Min, J.K.; Roh, K.M.; Kim, W.K.; Han, B.S.; Bae, K.H.; Lee, S.C.; Chung, S.J.; Kang, H.J. Phosphoprotein phosphatase 1CB (PPP1CB), a novel adipogenic activator, promotes 3T3-L1 adipogenesis. Biochem. Biophys. Res. Commun. 2015, 467, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Choi, Y.A.; Lee, H.G.; Kim, K.I.; Lim, J.S.; Lee, M.S.; Oh, K.S.; Yang, Y. Dephosphorylation of CCAAT/enhancer-binding protein beta by protein phosphatase 2A containing B56delta is required at the early time of adipogenesis. Biochim. Biophys. Acta 2014, 1841, 1608–1618. [Google Scholar] [CrossRef] [PubMed]
- Hinds, T.D., Jr.; Stechschulte, L.A.; Cash, H.A.; Whisler, D.; Banerjee, A.; Yong, W.; Khuder, S.S.; Kaw, M.K.; Shou, W.; Najjar, S.M.; et al. Protein phosphatase 5 mediates lipid metabolism through reciprocal control of glucocorticoid receptor and peroxisome proliferator-activated receptor-gamma (PPARgamma). J. Biol. Chem. 2011, 286, 42911–42922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Zhang, L.; Xu, L.; Liu, L.; He, Y.; Zhang, Y.; Huang, X.; Zhao, T.; Wu, L.; Zhao, Y.; et al. WIP1 phosphatase is a critical regulator of adipogenesis through dephosphorylating PPARgamma serine 112. Cell Mol. Life Sci. 2017, 74, 2067–2079. [Google Scholar] [CrossRef] [PubMed]
- Kilian, K.A.; Bugarija, B.; Lahn, B.T.; Mrksich, M. Geometric cues for directing the differentiation of mesenchymal stem cells. Proc. Natl. Acad. Sci. USA 2010, 107, 4872–4877. [Google Scholar] [CrossRef] [Green Version]
- Mathieu, P.S.; Loboa, E.G. Cytoskeletal and focal adhesion influences on mesenchymal stem cell shape, mechanical properties, and differentiation down osteogenic, adipogenic, and chondrogenic pathways. Tissue Eng. Part B Rev. 2012, 18, 436–444. [Google Scholar] [CrossRef] [Green Version]
- McBeath, R.; Pirone, D.M.; Nelson, C.M.; Bhadriraju, K.; Chen, C.S. Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev. Cell 2004, 6, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Hartshorne, D.J.; Ito, M.; Erdodi, F. Role of protein phosphatase type 1 in contractile functions: Myosin phosphatase. J. Biol. Chem. 2004, 279, 37211–37214. [Google Scholar] [CrossRef] [Green Version]
- Kiss, A.; Erdődi, F.; Lontay, B. Myosin phosphatase: Unexpected functions of a long-known enzyme. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 2–15. [Google Scholar] [CrossRef]
- Shin, D.W.; Kim, S.N.; Lee, S.M.; Lee, W.; Song, M.J.; Park, S.M.; Lee, T.R.; Baik, J.H.; Kim, H.K.; Hong, J.H.; et al. (-)-Catechin promotes adipocyte differentiation in human bone marrow mesenchymal stem cells through PPAR gamma transactivation. Biochem. Pharmacol. 2009, 77, 125–133. [Google Scholar] [CrossRef]
- Yang, X.; Yin, L.; Li, T.; Chen, Z. Green tea extracts reduce adipogenesis by decreasing expression of transcription factors C/EBPalpha and PPARgamma. Int. J. Clin. Exp. Med. 2014, 7, 4906–4914. [Google Scholar] [PubMed]
- Klaus, S.; Pultz, S.; Thone-Reineke, C.; Wolfram, S. Epigallocatechin gallate attenuates diet-induced obesity in mice by decreasing energy absorption and increasing fat oxidation. Int. J. Obes. 2005, 29, 615–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ku, H.C.; Liu, H.S.; Hung, P.F.; Chen, C.L.; Liu, H.C.; Chang, H.H.; Tsuei, Y.W.; Shih, L.J.; Lin, C.L.; Lin, C.M.; et al. Green tea (-)-epigallocatechin gallate inhibits IGF-I and IGF-II stimulation of 3T3-L1 preadipocyte mitogenesis via the 67-kDa laminin receptor, but not AMP-activated protein kinase pathway. Mol. Nutr. Food Res. 2012, 56, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Song, H.; Cai, W.; Shen, X. Real Time Monitoring of Inhibition of Adipogenesis and Angiogenesis by (-)-Epigallocatechin-3-Gallate in 3T3-L1 Adipocytes and Human Umbilical Vein Endothelial Cells. Nutrients 2015, 7, 8871–8886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.; Zou, B.; Nie, R.; Zhang, Y.; Li, C.M. A-type ECG and EGCG dimers disturb the structure of 3T3-L1 cell membrane and strongly inhibit its differentiation by targeting peroxisome proliferator-activated receptor gamma with miR-27 involved mechanism. J. Nutr. Biochem. 2015, 26, 1124–1135. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.Y.; Suresh, S.; Jang, M.; Park, M.N.; Gobianand, K.; You, S.; Yeon, S.H.; Lee, H.J. Epigallocatechin-3-gallate suppresses the lipid deposition through the apoptosis during differentiation in bovine bone marrow mesenchymal stem cells. Cell Biol. Int. 2015, 39, 52–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, J.Y.; Park, M.N.; Cho, E.S.; Jang, H.J.; Park, S.; Lee, H.J. Epigallocatechin-3-gallate-induced free-radical production upon adipogenic differentiation in bovine bone-marrow mesenchymal stem cells. Cell Tissue Res. 2015, 362, 87–96. [Google Scholar] [CrossRef]
- Choi, C.; Song, H.D.; Son, Y.; Cho, Y.K.; Ahn, S.Y.; Jung, Y.S.; Yoon, Y.C.; Kwon, S.W.; Lee, Y.H. Epigallocatechin-3-Gallate Reduces Visceral Adiposity Partly through the Regulation of Beclin1-Dependent Autophagy in White Adipose Tissues. Nutrients 2020, 12, 3072. [Google Scholar] [CrossRef]
- Li, F.; Gao, C.; Yan, P.; Zhang, M.; Wang, Y.; Hu, Y.; Wu, X.; Wang, X.; Sheng, J. EGCG Reduces Obesity and White Adipose Tissue Gain Partly Through AMPK Activation in Mice. Front. Pharmacol. 2018, 9, 1366. [Google Scholar] [CrossRef] [Green Version]
- Tachibana, H.; Koga, K.; Fujimura, Y.; Yamada, K. A receptor for green tea polyphenol EGCG. Nat. Struct. Mol. Biol. 2004, 11, 380–381. [Google Scholar] [CrossRef]
- Tsukamoto, S.; Huang, Y.; Umeda, D.; Yamada, S.; Yamashita, S.; Kumazoe, M.; Kim, Y.; Murata, M.; Yamada, K.; Tachibana, H. 67-kDa laminin receptor-dependent protein phosphatase 2A (PP2A) activation elicits melanoma-specific antitumor activity overcoming drug resistance. J. Biol. Chem. 2014, 289, 32671–32681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, J.H.; McAvoy, T.; Rakhilin, S.V.; Nishi, A.; Greengard, P.; Nairn, A.C. Protein kinase A activates protein phosphatase 2A by phosphorylation of the B56delta subunit. Proc. Natl. Acad. Sci. USA 2007, 104, 2979–2984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bátori, R.; Bécsi, B.; Nagy, D.; Kónya, Z.; Hegedűs, C.; Bordán, Z.; Verin, A.; Lontay, B.; Erdődi, F. Interplay of myosin phosphatase and protein phosphatase-2A in the regulation of endothelial nitric-oxide synthase phosphorylation and nitric oxide production. Sci. Rep. 2017, 7, 44698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tóth, E.; Erdődi, F.; Kiss, A. Activation of Myosin Phosphatase by Epigallocatechin-Gallate Sensitizes THP-1 Leukemic Cells to Daunorubicin. Anticancer Agents Med. Chem. 2021, 21, 1092–1098. [Google Scholar] [CrossRef]
- Tóth, E.; Erdődi, F.; Kiss, A. Myosin Phosphatase Is Implicated in the Control of THP-1 Monocyte to Macrophage Differentiation. Int. J. Mol. Sci. 2021, 22, 2516. [Google Scholar] [CrossRef]
- Hirschi, A.; Cecchini, M.; Steinhardt, R.C.; Schamber, M.R.; Dick, F.A.; Rubin, S.M. An overlapping kinase and phosphatase docking site regulates activity of the retinoblastoma protein. Nat. Struct. Mol. Biol. 2010, 17, 1051–1057. [Google Scholar] [CrossRef]
- Lontay, B.; Serfozo, Z.; Gergely, P.; Ito, M.; Hartshorne, D.J.; Erdodi, F. Localization of myosin phosphatase target subunit 1 in rat brain and in primary cultures of neuronal cells. J. Comp. Neurol. 2004, 478, 72–87. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Robaszkiewicz, A.; Erdelyi, K.; Kovacs, K.; Kovacs, I.; Bai, P.; Rajnavolgyi, E.; Virag, L. Hydrogen peroxide-induced poly(ADP-ribosyl)ation regulates osteogenic differentiation-associated cell death. Free Radic. Biol. Med. 2012, 53, 1552–1564. [Google Scholar] [CrossRef]
- Fujimura, Y.; Sumida, M.; Sugihara, K.; Tsukamoto, S.; Yamada, K.; Tachibana, H. Green tea polyphenol EGCG sensing motif on the 67-kDa laminin receptor. PLoS ONE 2012, 7, e37942. [Google Scholar] [CrossRef] [Green Version]
- Ingebritsen, T.S.; Stewart, A.A.; Cohen, P. The protein phosphatases involved in cellular regulation. 6. Measurement of type-1 and type-2 protein phosphatases in extracts of mammalian tissues; an assessment of their physiological roles. Eur. J. Biochem. 1983, 132, 297–307. [Google Scholar] [CrossRef]
- Kloeker, S.; Reed, R.; McConnell, J.L.; Chang, D.; Tran, K.; Westphal, R.S.; Law, B.K.; Colbran, R.J.; Kamoun, M.; Campbell, K.S.; et al. Parallel purification of three catalytic subunits of the protein serine/threonine phosphatase 2A family (PP2A(C), PP4(C), and PP6(C)) and analysis of the interaction of PP2A(C) with alpha4 protein. Protein Expr. Purif. 2003, 31, 19–33. [Google Scholar] [CrossRef]
- Hu, X.; Li, Z.; Ding, Y.; Geng, Q.; Xiahou, Z.; Ru, H.; Dong, M.Q.; Xu, X.; Li, J. Chk1 modulates the interaction between myosin phosphatase targeting protein 1 (MYPT1) and protein phosphatase 1cbeta (PP1cbeta). Cell Cycle 2018, 17, 421–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, C.Y.; Wei, L.; Castro-Munozledo, F.; Koo, W.L. (-)-Epigallocatechin-3-gallate blocks 3T3-L1 adipose conversion by inhibition of cell proliferation and suppression of adipose phenotype expression. Life Sci. 2011, 89, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Kumazoe, M.; Sugihara, K.; Tsukamoto, S.; Huang, Y.; Tsurudome, Y.; Suzuki, T.; Suemasu, Y.; Ueda, N.; Yamashita, S.; Kim, Y.; et al. 67-kDa laminin receptor increases cGMP to induce cancer-selective apoptosis. J. Clin. Investig. 2013, 123, 787–799. [Google Scholar] [CrossRef]
- Nelson, J.; McFerran, N.V.; Pivato, G.; Chambers, E.; Doherty, C.; Steele, D.; Timson, D.J. The 67 kDa laminin receptor: Structure, function and role in disease. Biosci. Rep. 2008, 28, 33–48. [Google Scholar] [CrossRef] [Green Version]
- Buto, S.; Tagliabue, E.; Ardini, E.; Magnifico, A.; Ghirelli, C.; van den Brule, F.; Castronovo, V.; Colnaghi, M.I.; Sobel, M.E.; Menard, S. Formation of the 67-kDa laminin receptor by acylation of the precursor. J. Cell Biochem. 1998, 69, 244–251. [Google Scholar] [CrossRef]
- Birsoy, K.; Chen, Z.; Friedman, J. Transcriptional regulation of adipogenesis by KLF4. Cell Metab. 2008, 7, 339–347. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.C.; Chuang, W.T.; Lin, A.H.; Tsai, C.W.; Huang, C.S.; Chen, Y.T.; Chen, H.W.; Lii, C.K. Andrographolide inhibits adipogenesis of 3T3-L1 cells by suppressing C/EBPbeta expression and activation. Toxicol. Appl. Pharmacol. 2016, 307, 115–122. [Google Scholar] [CrossRef]
- Park, B.H.; Qiang, L.; Farmer, S.R. Phosphorylation of C/EBPbeta at a consensus extracellular signal-regulated kinase/glycogen synthase kinase 3 site is required for the induction of adiponectin gene expression during the differentiation of mouse fibroblasts into adipocytes. Mol. Cell Biol. 2004, 24, 8671–8680. [Google Scholar] [CrossRef] [Green Version]
- Park, H.S.; Ju, U.I.; Park, J.W.; Song, J.Y.; Shin, D.H.; Lee, K.H.; Jeong, L.S.; Yu, J.; Lee, H.W.; Cho, J.Y.; et al. PPARgamma neddylation essential for adipogenesis is a potential target for treating obesity. Cell Death Differ. 2016, 23, 1296–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seenprachawong, K.; Tawornsawutruk, T.; Nantasenamat, C.; Nuchnoi, P.; Hongeng, S.; Supokawej, A. miR-130a and miR-27b Enhance Osteogenesis in Human Bone Marrow Mesenchymal Stem Cells via Specific Down-Regulation of Peroxisome Proliferator-Activated Receptor gamma. Front. Genet. 2018, 9, 543. [Google Scholar] [CrossRef] [PubMed]
- Schwind, L.; Schetting, S.; Montenarh, M. Inhibition of Protein Kinase CK2 Prevents Adipogenic Differentiation of Mesenchymal Stem Cells Like C3H/10T1/2 Cells. Pharmaceuticals 2017, 10, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwasaki, T.; Katayama, T.; Kohama, K.; Endo, Y.; Sawasaki, T. Myosin phosphatase is inactivated by caspase-3 cleavage and phosphorylation of myosin phosphatase targeting subunit 1 during apoptosis. Mol. Biol. Cell 2013, 24, 748–756. [Google Scholar] [CrossRef]
- Twomey, E.; Li, Y.; Lei, J.; Sodja, C.; Ribecco-Lutkiewicz, M.; Smith, B.; Fang, H.; Bani-Yaghoub, M.; McKinnell, I.; Sikorska, M. Regulation of MYPT1 stability by the E3 ubiquitin ligase SIAH2. Exp. Cell Res. 2010, 316, 68–77. [Google Scholar] [CrossRef]
- Kalfon, L.; Youdim, M.B.; Mandel, S.A. Green tea polyphenol (-)-epigallocatechin-3-gallate promotes the rapid protein kinase C- and proteasome-mediated degradation of Bad: Implications for neuroprotection. J. Neurochem. 2007, 100, 992–1002. [Google Scholar] [CrossRef]
- Quan, Y.; Li, L.; Dong, L.; Wang, S.; Jiang, X.; Zhang, T.; Jin, P.; Fan, J.; Mao, S.; Fan, X.; et al. Epigallocatechin-3-gallate (EGCG) inhibits aggregation of pulmonary fibrosis associated mutant surfactant protein A2 via a proteasomal degradation pathway. Int. J. Biochem. Cell Biol. 2019, 116, 105612. [Google Scholar] [CrossRef]
- Lokireddy, S.; Kukushkin, N.V.; Goldberg, A.L. cAMP-induced phosphorylation of 26S proteasomes on Rpn6/PSMD11 enhances their activity and the degradation of misfolded proteins. Proc. Natl. Acad. Sci. USA 2015, 112, E7176–E7185. [Google Scholar] [CrossRef] [Green Version]
- Wiseman, S.L.; Shimizu, Y.; Palfrey, C.; Nairn, A.C. Proteasomal degradation of eukaryotic elongation factor-2 kinase (EF2K) is regulated by cAMP-PKA signaling and the SCFbetaTRCP ubiquitin E3 ligase. J. Biol. Chem. 2013, 288, 17803–17811. [Google Scholar] [CrossRef] [Green Version]
- Cho, E.A.; Juhnn, Y.S. The cAMP signaling system inhibits the repair of gamma-ray-induced DNA damage by promoting Epac1-mediated proteasomal degradation of XRCC1 protein in human lung cancer cells. Biochem. Biophys. Res. Commun. 2012, 422, 256–262. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bécsi, B.; Kónya, Z.; Boratkó, A.; Kovács, K.; Erdődi, F. Epigallocatechine-3-gallate Inhibits the Adipogenesis of Human Mesenchymal Stem Cells via the Regulation of Protein Phosphatase-2A and Myosin Phosphatase. Cells 2022, 11, 1704. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11101704
Bécsi B, Kónya Z, Boratkó A, Kovács K, Erdődi F. Epigallocatechine-3-gallate Inhibits the Adipogenesis of Human Mesenchymal Stem Cells via the Regulation of Protein Phosphatase-2A and Myosin Phosphatase. Cells. 2022; 11(10):1704. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11101704
Chicago/Turabian StyleBécsi, Bálint, Zoltán Kónya, Anita Boratkó, Katalin Kovács, and Ferenc Erdődi. 2022. "Epigallocatechine-3-gallate Inhibits the Adipogenesis of Human Mesenchymal Stem Cells via the Regulation of Protein Phosphatase-2A and Myosin Phosphatase" Cells 11, no. 10: 1704. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11101704