Fecal Microbiota Transplantation Controls Murine Chronic Intestinal Inflammation by Modulating Immune Cell Functions and Gut Microbiota Composition

Abstract
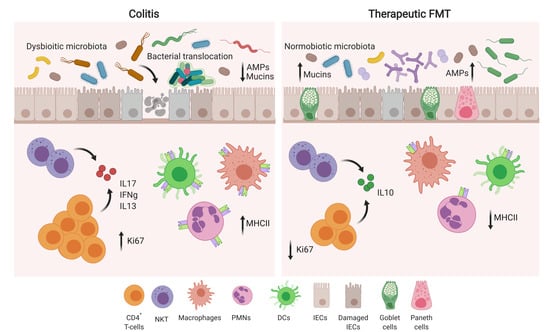
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Mice
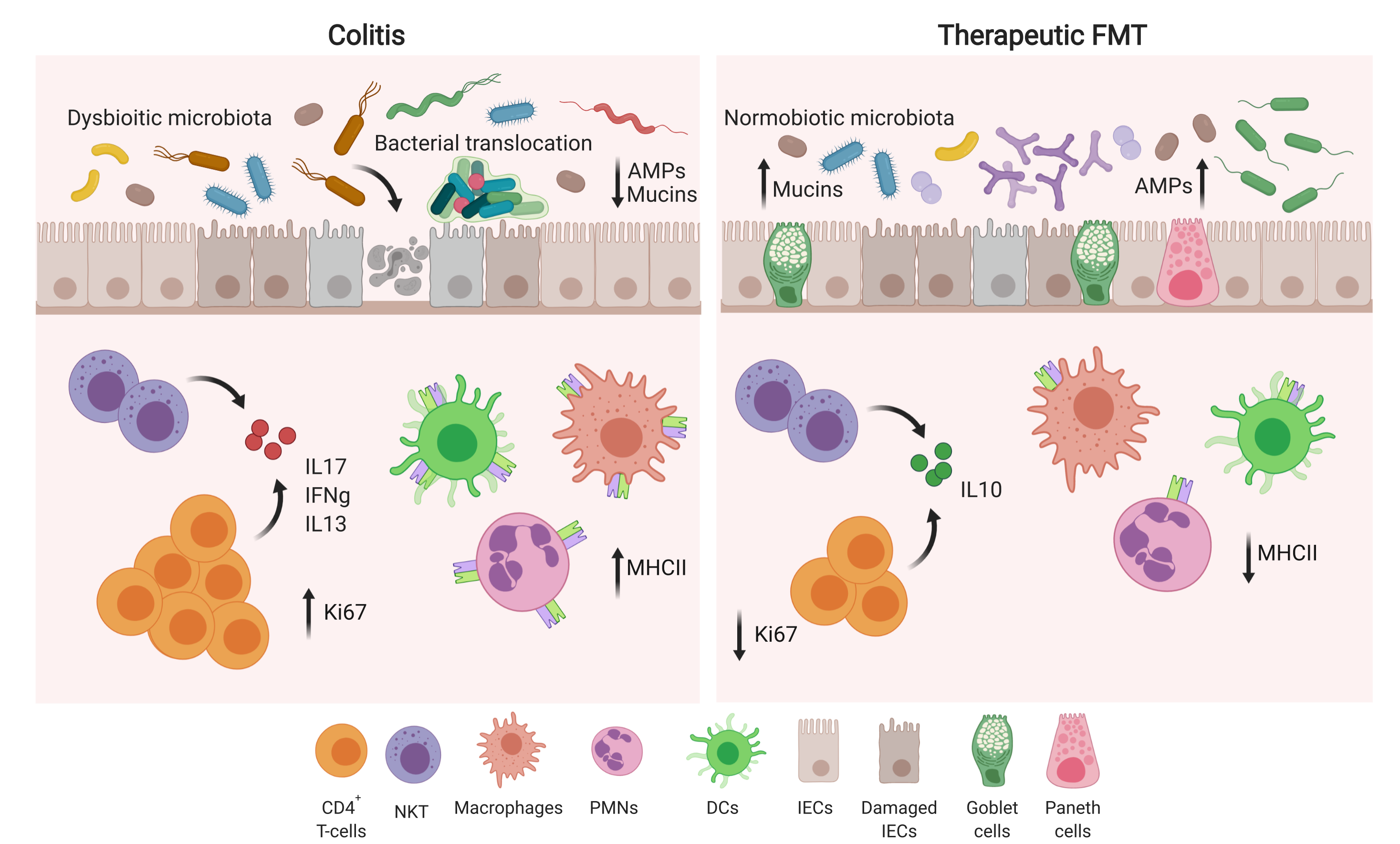
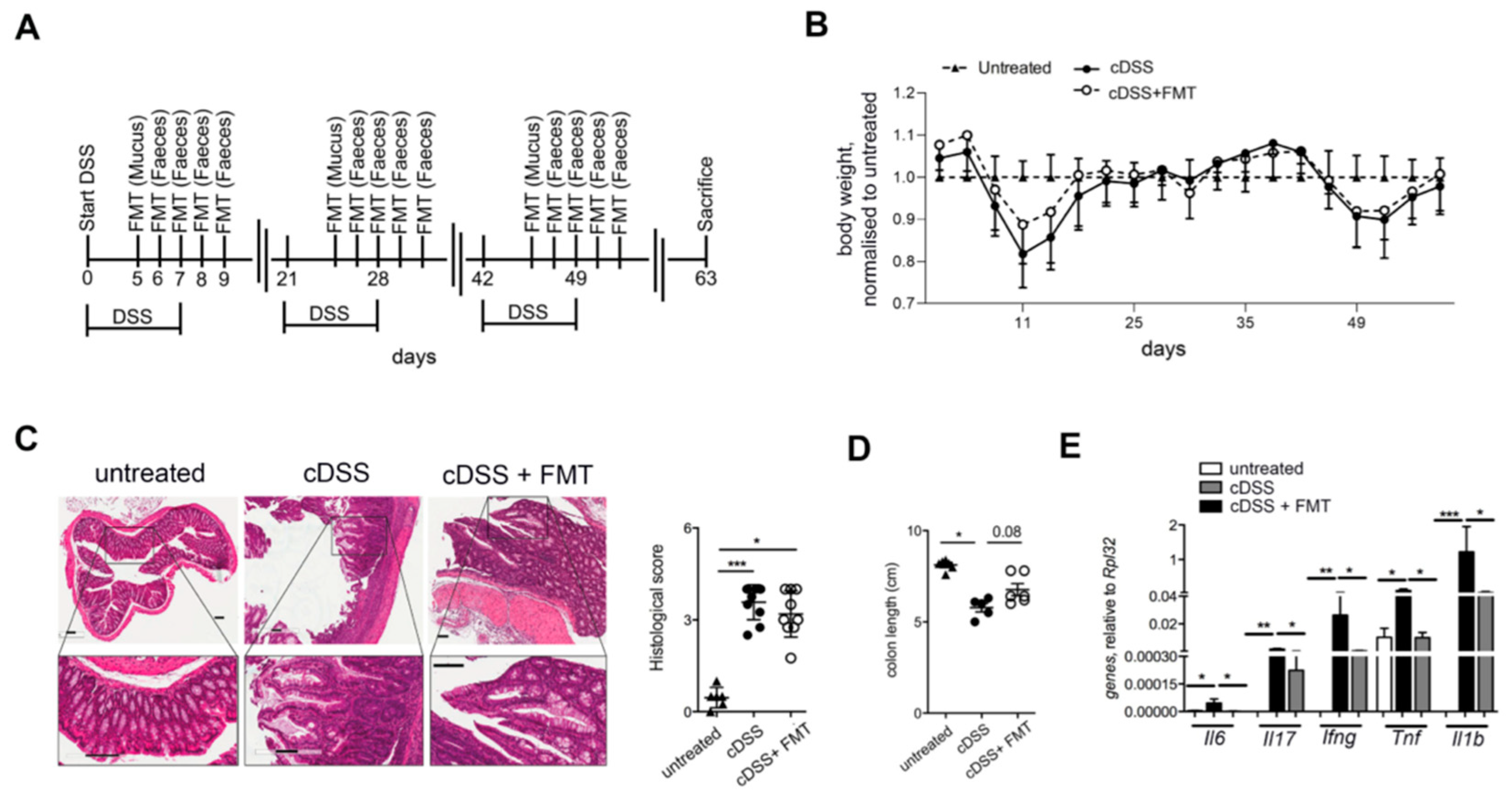
2.2. Experimental Colitis Models
2.3. Fecal Microbiota Transplantation (FMT)
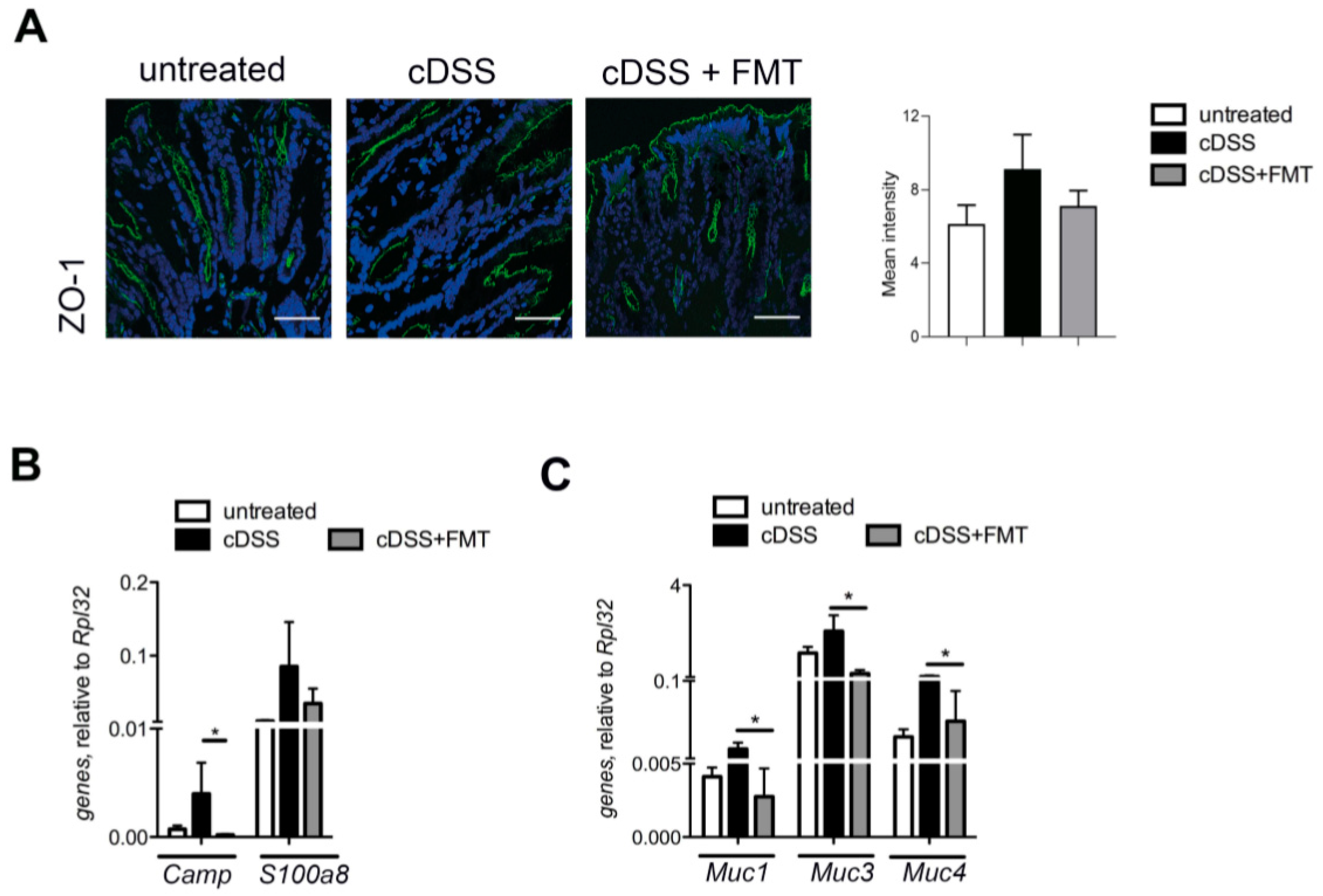
2.4. Histological Analysis
2.5. Quantitative Reverse Transcription PCR (RT-qPCR) of Tissue mRNA
2.6. Tissue ELISA of Murine IL-10
2.7. Murine Cell Isolation
2.8. Flow Cytometry Analysis
2.9. Immunofluorescence
2.10. Bacterial DNA Extraction and 16S rRNA Gene Sequencing
2.11. Statistical Analysis
2.11.1. Data Analysis
2.11.2. Microbiota Sequence Analysis
2.11.3. PICRUSt Analysis
3. Results
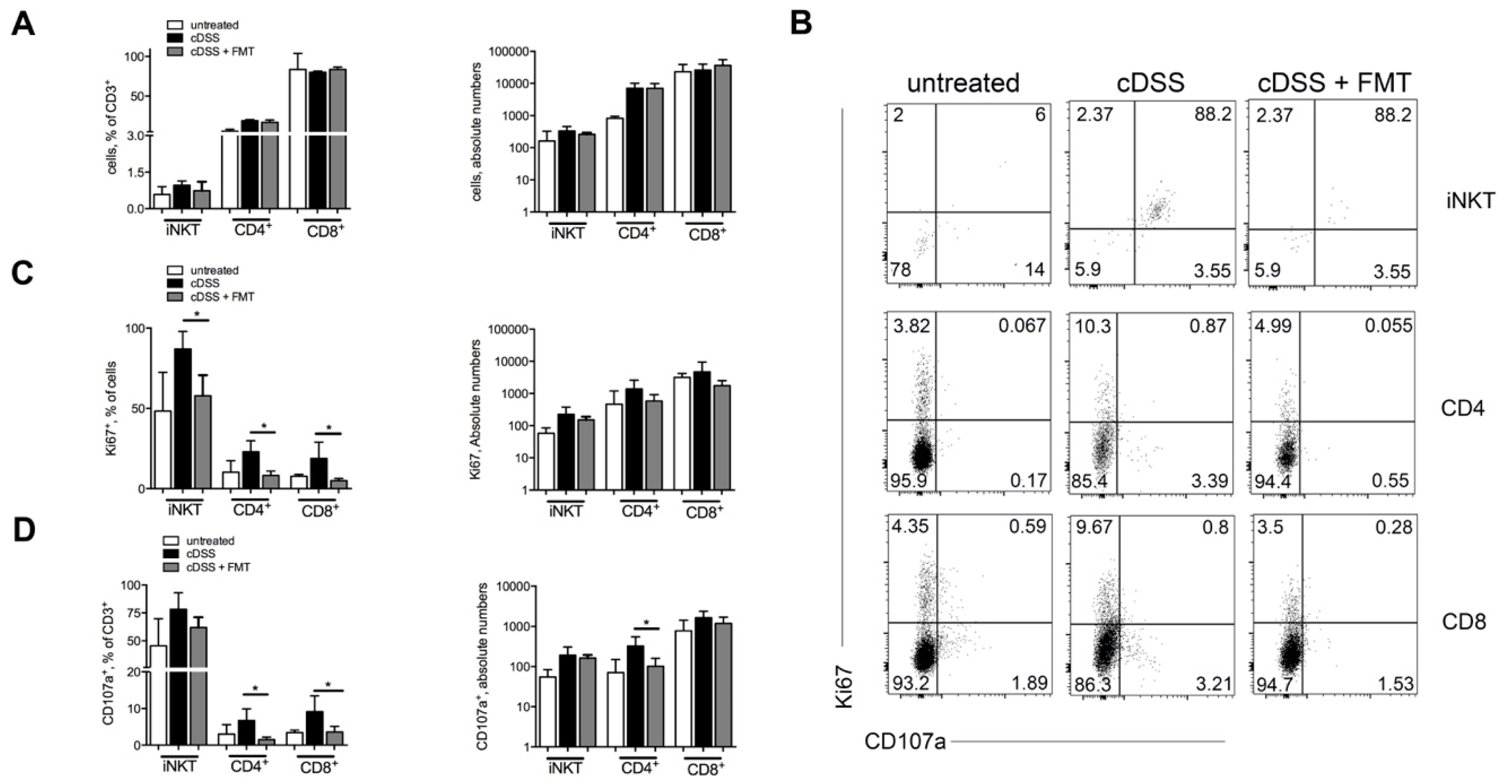
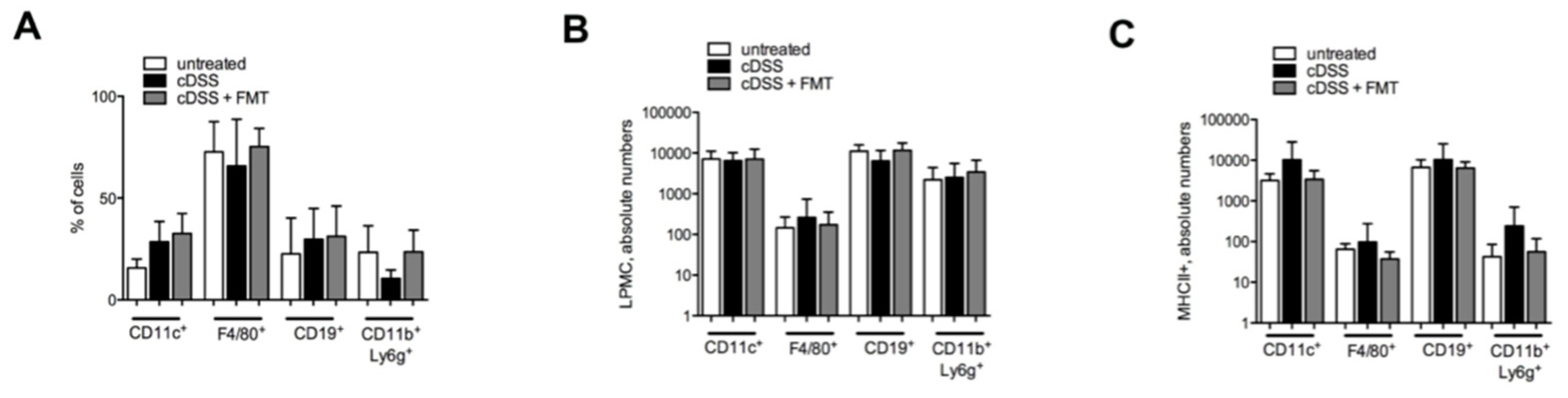
3.1. Therapeutic FMT Influences the Activation Status of Colonic T Cell Populations
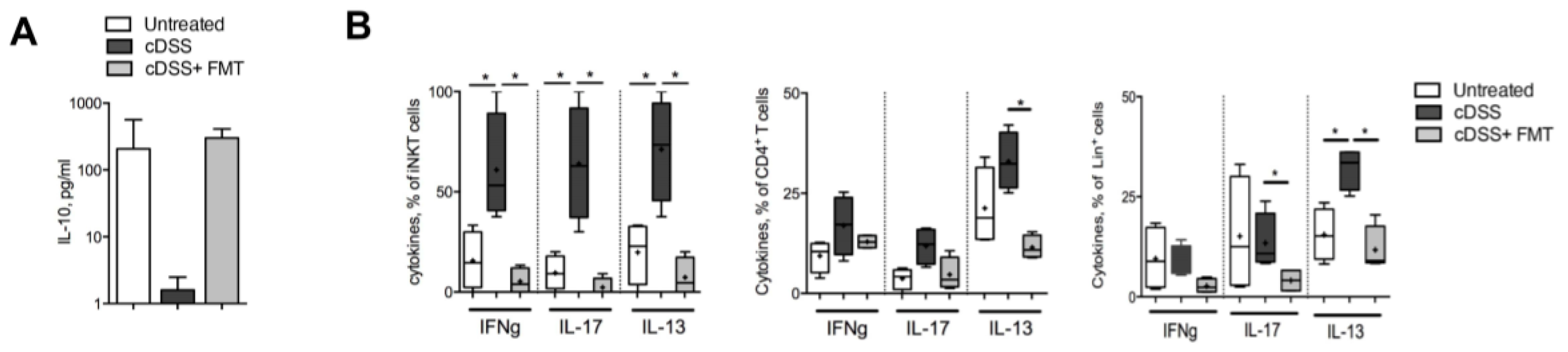
3.2. Therapeutic FMT Targets the Cytokine Profile of Colonic Immune Cell Populations
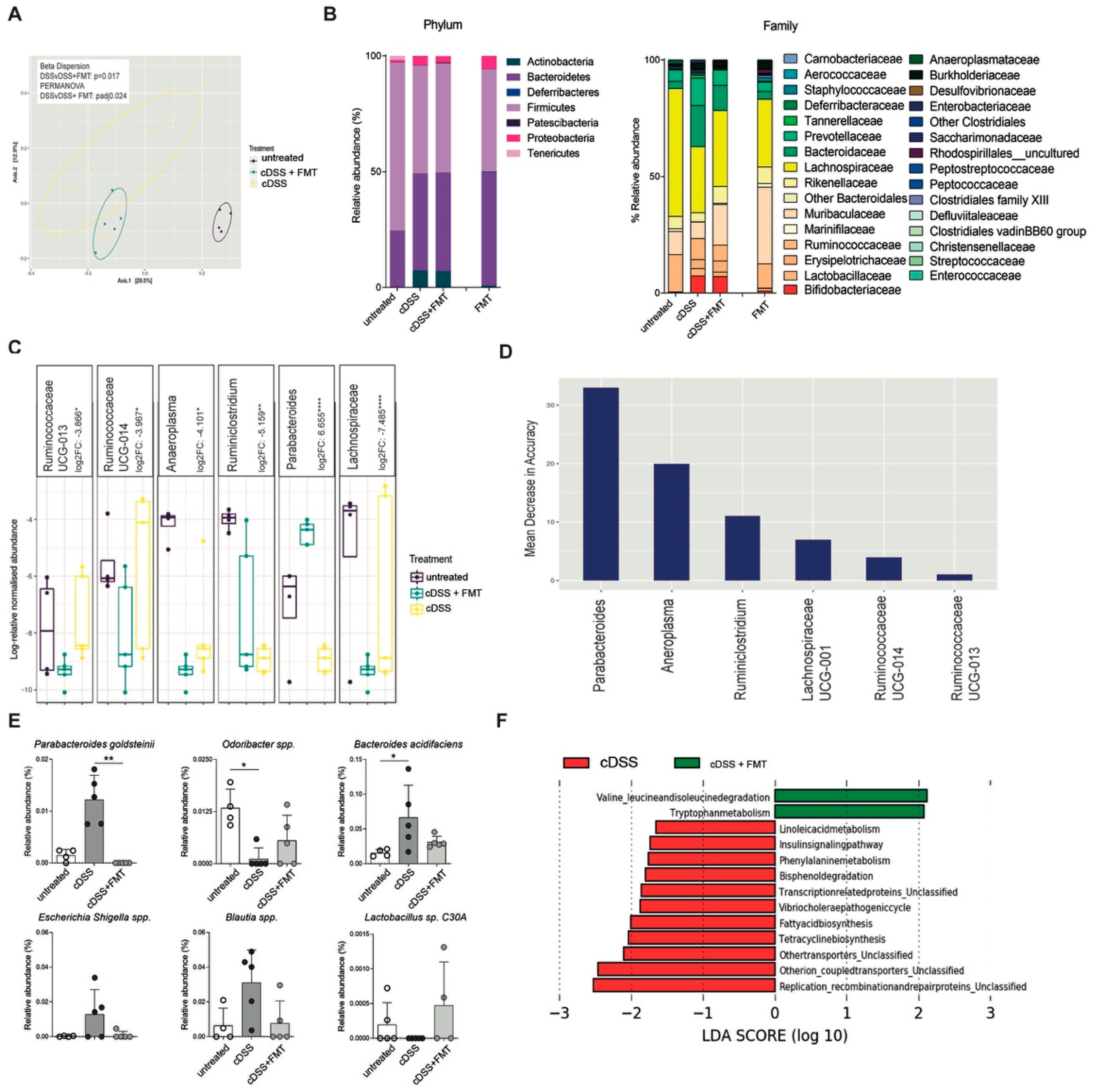
3.3. Therapeutic FMT Restores Normobiotic Microbial Ecologies
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| FMT | fecal microbiota transplantation |
| iNKT | invariant Natural Killer T cells |
| GF | Germ Free |
| Th | Thelper |
| IBD | Inflammatory Bowel Diseases |
| LP | lamina propria |
| LPMC | lamina propria mononuclear cells |
References
- Honda, K.; Littman, D.R. The microbiota in adaptive immune homeostasis and disease. Nat. Cell Boil. 2016, 535, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Gevers, D.; Kugathasan, S.; Denson, L.A.; Vázquez-Baeza, Y.; Van Treuren, W.; Ren, B.; Schwager, E.; Knights, D.; Song, S.J.; Yassour, M.; et al. The Treatment-Naive Microbiome in New-Onset Crohn’s Disease. Cell Host. Microbe 2014, 15, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Lepage, P.; Häsler, R.; Spehlmann, M.E.; Rehman, A.; Žvirbliene, A.; Begun, A.; Ott, S.; Kupcinskas, L.; Dore, J.; Raedler, A.; et al. Twin Study Indicates Loss of Interaction Between Microbiota and Mucosa of Patients With Ulcerative Colitis. Gastroenterology 2011, 141, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Hedin, C.; van der Gast, C.J.; Rogers, G.B.; Cuthbertson, L.; McCartney, S.; Stagg, A.J.; Lindsay, J.O.; Whelan, K. Siblings of patients with Crohn’s disease exhibit a biologically relevant dysbiosis in mucosal microbial metacommunities. Gut 2016, 65, 944–953. [Google Scholar] [CrossRef]
- Shah, R.; Cope, J.L.; Nagy-Szakal, D.; Dowd, S.; Versalovic, J.; Hollister, E.B.; Kellermayer, R. Composition and function of the pediatric colonic mucosal microbiome in untreated patients with ulcerative colitis. Gut Microbes 2016, 7, 384–396. [Google Scholar] [CrossRef] [Green Version]
- Kostic, A.D.; Xavier, R.J.; Gevers, D. The Microbiome in Inflammatory Bowel Diseases: Current Status and the Future Ahead. Gastroenterology 2014, 146, 1489–1499. [Google Scholar] [CrossRef]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, Z.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef]
- Hugot, J.P.; Shammaillard, M.; Zouali, H.; Lesage, S.; Cezard, J.P.; Belaiche, J.; Almer, S.; Tysk, C.; O’Morain, C.A.; Gassul, M.; et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 2001, 411, 599–603. [Google Scholar] [CrossRef]
- Duchmann, R.; Kaiser, I.; Hermann, E.; Mayet, W.; Ewe, K.; Büschenfelde, K.H.M.Z. Tolerance exists towards resident intestinal flora but is broken in active inflammatory bowel disease (IBD). Clin. Exp. Immunol. 1995, 102, 448–455. [Google Scholar] [CrossRef]
- Kaser, A.; Zeissig, S.; Blumberg, R.S. Inflammatory bowel disease. Annu. Rev. Immunol. 2010, 28, 573–621. [Google Scholar] [CrossRef] [PubMed]
- Caprioli, F.; Pallone, F.; Monteleone, G. Th17 immune response in IBD: A new pathogenic mechanism. J. Crohns Colitis 2008, 2, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Nizzoli, G.; Burrello, C.; Cribiù, F.M.; Lovati, G.; Ercoli, G.; Botti, F.; Trombetta, E.; Porretti, L.; Todoerti, K.; Neri, A.; et al. Pathogenicity of In Vivo Generated Intestinal Th17 Lymphocytes is IFNgamma Dependent. J. Crohns Colitis 2018, 12, 981–992. [Google Scholar] [CrossRef]
- Burrello, C.; Pellegrino, G.; Giuffrè, M.R.; Lovati, G.; Magagna, I.; Bertocchi, A.; Cribiù, F.M.; Boggio, F.; Botti, F.; Trombetta, E.; et al. Mucosa-associated microbiota drives pathogenic functions in IBD-derived intestinal iNKT cells. Life Sci. Alliance 2019, 2, e201800229. [Google Scholar] [CrossRef] [PubMed]
- Heller, F.; Fuss, I.J.; Nieuwenhuis, E.E.; Blumberg, R.S.; Strober, W. Oxazolone Colitis, a Th2 Colitis Model Resembling Ulcerative Colitis, Is Mediated by IL-13-Producing NK-T Cells. Immunity 2002, 17, 629–638. [Google Scholar] [CrossRef] [Green Version]
- Fuss, I.J.; Heller, F.; Boirivant, M.; León, F.; Yoshida, M.; Fichtner-Feigl, S.; Yang, Z.; Exley, M.; Kitani, A.; Blumberg, R.S.; et al. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J. Clin. Investig. 2004, 113, 1490–1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhry, A.; Samstein, R.M.; Treuting, P.; Liang, Y.; Pils, M.C.; Heinrich, J.-M.; Jack, R.S.; Wunderlich, F.T.; Brüning, J.C.; Müller, W.; et al. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity 2011, 34, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Alfen, J.S.; Larghi, P.; Facciotti, F.; Gagliani, N.; Bosotti, R.; Paroni, M.; Maglie, S.; Gruarin, P.; Vasco, C.M.; Ranzani, V.; et al. Intestinal IFN-gamma-producing type 1 regulatory T cells coexpress CCR5 and programmed cell death protein 1 and downregulate IL-10 in the inflamed guts of patients with inflammatory bowel disease. J. Allergy Clin. Immunol. 2018, 142, 1537–1547. [Google Scholar] [CrossRef] [PubMed]
- Burrello, C.; Garavaglia, F.; Cribiù, F.M.; Ercoli, G.; Lopez, G.; Troisi, J.; Colucci, A.; Guglietta, S.; Carloni, S.; Guglielmetti, S.; et al. Therapeutic faecal microbiota transplantation controls intestinal inflammation through IL10 secretion by immune cells. Nat. Commun. 2018, 9, 5184. [Google Scholar] [CrossRef] [PubMed]
- Burrello, C.; Garavaglia, F.; Cribiu’, F.M.; Ercoli, G.; Bosari, S.; Caprioli, F.; Facciotti, F. Short-term Oral Antibiotics Treatment Promotes Inflammatory Activation of Colonic Invariant Natural Killer T and Conventional CD4(+) T Cells. Front. Med. 2018, 5, 21. [Google Scholar] [CrossRef]
- McIlroy, J.; Ianiro, G.; Mukhopadhya, I.; Hansen, R.; Hold, G.L. Review article: the gut microbiome in inflammatory bowel disease-avenues for microbial management. Aliment. Pharmacol. Ther. 2017, 47, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Cammarota, G.; Ianiro, G.; Tilg, H.; Rajilić-Stojanović, M.; Kump, P.; Satokari, R.; Sokol, H.; Arkkila, P.; Pintus, C.; Hart, A.; et al. European consensus conference on faecal microbiota transplantation in clinical practice. Gut 2017, 66, 569–580. [Google Scholar] [CrossRef]
- Van Nood, E.; Vrieze, A.; Nieuwdorp, M.; Fuentes, S.; Zoetendal, E.G.; de Vos, W.M.; Visser, C.E.; Kuijper, E.J.; Bartelsman, J.F.; Tijssen, J.G.; et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. N. Engl. J. Med. 2013, 368, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Paramsothy, S.; Kamm, M.A.; Kaakoush, N.O.; Walsh, A.J.; Bogaerde, J.V.D.; Samuel, D.; Leong, R.W.L.; Connor, S.; Ng, W.; Paramsothy, R.; et al. Multidonor intensive faecal microbiota transplantation for active ulcerative colitis: A randomised placebo-controlled trial. Lancet 2017, 389, 1218–1228. [Google Scholar] [CrossRef]
- Rossen, N.G.; Fuentes, S.; Van Der Spek, M.J.; Tijssen, J.G.; Hartman, J.H.; Duflou, A.; Lowenberg, M.; Brink, G.R.V.D.; Mathus-Vliegen, E.M.; De Vos, W.M.; et al. Findings From a Randomized Controlled Trial of Fecal Transplantation for Patients With Ulcerative Colitis. Gastroenterology 2015, 149, 110–118. [Google Scholar] [CrossRef]
- Moayyedi, P.; Surette, M.G.; Kim, P.T.; Libertucci, J.; Wolfe, M.; Onischi, C.; Armstrong, D.; Marshall, J.K.; Kassam, Z.; Reinisch, W.; et al. Fecal Microbiota Transplantation Induces Remission in Patients with Active Ulcerative Colitis in a Randomized Controlled Trial. Gastroenterology 2015, 149, 102–109. [Google Scholar] [CrossRef]
- Tian, Z.; Liu, J.; Liao, M.; Li, W.; Zou, J.; Han, X.; Kuang, M.; Shen, W. Beneficial Effects of Fecal Microbiota Transplantation on Ulcerative Colitis in Mice. Am. J. Dig. Dis. 2016, 61, 2262–2271. [Google Scholar] [CrossRef]
- Selvanantham, T.; Lin, Q.; Guo, C.X.; Surendra, A.; Fieve, S.; Escalante, N.K.; Guttman, D.S.; Streutker, C.J.; Robertson, S.J.; Philpott, D.J.; et al. NKT Cell–Deficient Mice Harbor an Altered Microbiota That Fuels Intestinal Inflammation during Chemically Induced Colitis. J. Immunol. 2016, 197, 4464–4472. [Google Scholar] [CrossRef]
- Cribiù, F.M.; Burrello, C.; Ercoli, G.; Garavaglia, F.; Villanacci, V.; Caprioli, F.; Bosari, S.; Facciotti, F. Implementation of an automated inclusion system for the histological analysis of murine tissue samples: A feasibility study in DSS-induced chronic colitis. Eur. J. Inflamm. 2018. [Google Scholar] [CrossRef]
- Cribiu, F.M.; Burrello, C.; Tacchi, R.; Boggio, F.; Ricca, D.; Caprioli, F.; Ferrero, S.; Facciotti, F. Using Robotic Systems to Process and Embed Colonic Murine Samples for Histological Analyses. J. Vis. Exp. 2019, 143, e58654. [Google Scholar] [CrossRef]
- Borgo, F.; Garbossa, S.; Riva, A.; Severgnini, M.; Luigiano, C.; Benetti, A.; Pontiroli, A.E.; Morace, G.; Borghi, E. Body Mass Index and Sex Affect Diverse Microbial Niches within the Gut. Front. Microbiol. 2018, 9, 213. [Google Scholar] [CrossRef] [Green Version]
- Boylen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.; Al-Ghalith, G.A.; Alexander, A.; Alm, E.J.; Arumugam, A.; Asnicar, F.; et al. QIIME 2: Reproducible, interactive, scalable, and extensible microbiome data science. Peer J. Preprints 2018, 6, e27295v2. [Google Scholar]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; von Haesseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- McMurdie, P.J.; Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. R Package Version 2.2-0. 2014. Available online: http://ww1.cran.rproject.org/package=vegan (accessed on 1 April 2019).
- Comeau, A.M.; Douglas, G.M.; Langille, M.G.I. Microbiome Helper: A Custom and Streamlined Workflow for Microbiome Research. MSystem 2017, 2, e00127-16. [Google Scholar] [CrossRef]
- Rescigno, M. The intestinal epithelial barrier in the control of homeostasis and immunity. Trends Immunol. 2011, 32, 256–264. [Google Scholar] [CrossRef]
- Ueno, K.; Koga, T.; Kato, K.; Golenbock, D.T.; Gendler, S.J.; Kai, H.; Kim, K.C. MUC1 Mucin Is a Negative Regulator of Toll-Like Receptor Signaling. Am. J. Respir. Cell Mol. Boil. 2008, 38, 263–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blumberg, R.S.; Saubermann, L.J.; Strober, W. Animal models of mucosal inflammation and their relation to human inflammatory bowel disease. Curr. Opin. Immunol. 1999, 11, 648–656. [Google Scholar] [CrossRef]
- Aktas, E.; Kücüksezer, U.C.; Bilgic, S.; Erten, G.; Deniz, G. Relationship between CD107a expression and cytotoxic activity. Cell. Immunol. 2009, 254, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Munyaka, P.M.; Rabbi, M.F.; Khafipour, E.; Ghia, J.-E.; Ghia, J. Acute dextran sulfate sodium (DSS)-induced colitis promotes gut microbial dysbiosis in mice. J. Basic Microbiol. 2016, 56, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.; Schwab, C.; Milinovich, G.; Reichert, J.; Ben Mahfoudh, K.; Decker, T.; Engel, M.; Hai, B.; Hainzl, E.; Heider, S.; et al. Phylotype-level 16S rRNA analysis reveals new bacterial indicators of health state in acute murine colitis. ISME J. 2012, 6, 2091–2106. [Google Scholar] [CrossRef] [PubMed]
- Lamas, B.; Richard, M.L.; Leducq, V.; Pham, H.-P.; Michel, M.-L.; Da Costa, G.; Bridonneau, C.; Jegou, S.; Hoffmann, T.W.; Natividad, J.M.; et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat. Med. 2016, 22, 598–605. [Google Scholar] [CrossRef]
- Ohland, C.L.; Jobin, C. Microbial activities and intestinal homeostasis: A delicate balance between health and disease. Cell Mol. Gastroenterol. Hepatol. 2015, 1, 28–40. [Google Scholar] [CrossRef]
- IBD in EPIC Study Investigators; Tjonneland, A.; Overvad, K.; Bergmann, M.M.; Nagel, G.; Linseisen, J.; Hallmans, G.; Palmqvist, R.; Sjodin, H.; Hagglund, G.; et al. Linoleic acid, a dietary n-6 polyunsaturated fatty acid, and the aetiology of ulcerative colitis: A nested case-control study within a European prospective cohort study. Gut 2009, 58, 1606–1611. [Google Scholar]
- Paramsothy, S.; Nielsen, S.; Kamm, M.A.; Deshpande, N.P.; Faith, J.J.; Clemente, J.C.; Paramsothy, R.; Walsh, A.J.; Bogaerde, J.V.D.; Samuel, D.; et al. Specific Bacteria and Metabolites Associated With Response to Fecal Microbiota Transplantation in Patients With Ulcerative Colitis. Gastroenterology 2019, 156, 1440–1454. [Google Scholar] [CrossRef]
- Palm, N.W.; De Zoete, M.R.; Cullen, T.W.; Barry, N.A.; Stefanowski, J.; Hao, L.; Degnan, P.H.; Hu, J.; Peter, I.; Zhang, W.; et al. Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell 2014, 158, 1000–1010. [Google Scholar] [CrossRef] [PubMed]
- Nikolaus, S.; Schulte, B.; Al-Massad, N.; Thieme, F.; Schulte, D.M.; Bethge, J.; Rehman, A.; Tran, F.; Aden, K.; Häsler, R.; et al. Increased Tryptophan Metabolism Is Associated with Activity of Inflammatory Bowel Diseases. Gastroenterology 2017, 153, 1504–1516. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Perlot, T.; Rehman, A.; Trichereau, J.; Ishiguro, H.; Paolino, M.; Sigl, V.; Hanada, T.; Hanada, R.; Lipinski, S.; et al. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature 2012, 487, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.J.; Kovacs-Nolan, J.A.; Yang, C.; Archbold, T.; Fan, M.Z.; Mine, Y. l-Tryptophan exhibits therapeutic function in a porcine model of dextran sodium sulfate (DSS)-induced colitis. J. Nutr. Biochem. 2010, 21, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Monteleone, I.; Rizzo, A.; Sarra, M.; Sica, G.; Sileri, P.; Biancone, L.; Macdonald, T.T.; Pallone, F.; Monteleone, G. Aryl Hydrocarbon Receptor-Induced Signals Up-regulate IL-22 Production and Inhibit Inflammation in the Gastrointestinal Tract. Gastroenterology 2011, 141, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Brestoff, J.R.; Artis, D. Commensal bacteria at the interface of host metabolism and the immune system. Nat. Immunol. 2013, 14, 676–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, T.; Miwa, H.; Shigematsu, A.; Yamamoto, M.; Iida, M.; Fujishima, M. Increased arachidonic acid composition of phospholipids in colonic mucosa from patients with active ulcerative colitis. Gut 1987, 28, 1002–1007. [Google Scholar] [CrossRef]
- Rampton, D.S.; Collins, C.E. Review article: Thromboxanes in inflammatory bowel disease--pathogenic and therapeutic implications. Aliment. Pharmacol. Ther. 1993, 7, 357–367. [Google Scholar] [CrossRef]
- Sharon, P.; Ligumsky, M.; Rachmilewitz, D.; Zor, U. Role of prostaglandins in ulcerative colitis. Enhanced production during active disease and inhibition by sulfasalazine. Gastroenterology 1978, 75, 638–640. [Google Scholar] [CrossRef]
- Duchmann, R.; May, E.; Heike, M.; Knolle, P.A.; Neurath, M.; Buschenfelde, K.-H.M.Z. T cell specificity and cross reactivity towards enterobacteria, Bacteroides, Bifidobacterium, and antigens from resident intestinal flora in humans. Gut 1999, 44, 812–818. [Google Scholar] [CrossRef]
- Yang, Y.; Torchinsky, M.B.; Gobert, M.; Xiong, H.; Xu, M.; Linehan, J.L.; Alonzo, F.; Ng, C.; Chen, A.; Lin, X.; et al. Focused specificity of intestinal TH17 cells towards commensal bacterial antigens. Nature 2014, 510, 152–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nistala, K.; Adams, S.; Cambrook, H.; Ursu, S.; Olivito, B.; De Jager, W.; Evans, J.G.; Cimaz, R.; Bajaj-Elliott, M.; Wedderburn, L.R. Th17 plasticity in human autoimmune arthritis is driven by the inflammatory environment. Proc. Natl. Acad. Sci. USA 2010, 107, 14751–14756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujino, S.; Andoh, A.; Bamba, S.; Ogawa, A.; Hata, K.; Araki, Y.; Bamba, T.; Fujiyama, Y. Increased expression of interleukin 17 in inflammatory bowel disease. Gut 2003, 52, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuis, E.E.; Matsumoto, T.; Lindenbergh, D.; Willemsen, R.; Kaser, A.; Simons-Oosterhuis, Y.; Brugman, S.; Yamaguchi, K.; Ishikawa, H.; Aiba, Y.; et al. Cd1d-dependent regulation of bacterial colonization in the intestine of mice. J. Clin. Investig. 2009, 119, 1241–1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, M.H.; Kamada, K.; Kim, Y.G.; Nunez, G. Microbiota-induced IL-1beta, but not IL-6, is critical for the development of steady-state TH17 cells in the intestine. J. Exp. Med. 2012, 209, 251–258. [Google Scholar] [CrossRef] [PubMed]
- MacPherson, A.J.; Harris, N.L. Interactions between commensal intestinal bacteria and the immune system. Nat. Rev. Immunol. 2004, 4, 478–485. [Google Scholar] [CrossRef]
- Moore, K.W.; de Waal Malefyt, R.; Coffman, R.L.; O’Garra, A. Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 2001, 19, 683–765. [Google Scholar] [CrossRef]
- Huber, S.; Gagliani, N.; Esplunges, E.; O’Connor, W., Jr.; Huber, F.J.; Chaudhry, A.; Kamanaka, M.; Kobayashi, Y.; Booth, C.J.; Rudensky, A.Y.; et al. Th17 cells express interleukin-10 receptor and are controlled by Foxp3(-) and Foxp3+ regulatory CD4+ T cells in an interleukin-10-dependent manner. Immunity 2011, 34, 554–565. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burrello, C.; Giuffrè, M.R.; Macandog, A.D.; Diaz-Basabe, A.; Cribiù, F.M.; Lopez, G.; Borgo, F.; Nezi, L.; Caprioli, F.; Vecchi, M.; et al. Fecal Microbiota Transplantation Controls Murine Chronic Intestinal Inflammation by Modulating Immune Cell Functions and Gut Microbiota Composition. Cells 2019, 8, 517. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8060517
Burrello C, Giuffrè MR, Macandog AD, Diaz-Basabe A, Cribiù FM, Lopez G, Borgo F, Nezi L, Caprioli F, Vecchi M, et al. Fecal Microbiota Transplantation Controls Murine Chronic Intestinal Inflammation by Modulating Immune Cell Functions and Gut Microbiota Composition. Cells. 2019; 8(6):517. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8060517
Chicago/Turabian StyleBurrello, Claudia, Maria Rita Giuffrè, Angeli Dominique Macandog, Angelica Diaz-Basabe, Fulvia Milena Cribiù, Gianluca Lopez, Francesca Borgo, Luigi Nezi, Flavio Caprioli, Maurizio Vecchi, and et al. 2019. "Fecal Microbiota Transplantation Controls Murine Chronic Intestinal Inflammation by Modulating Immune Cell Functions and Gut Microbiota Composition" Cells 8, no. 6: 517. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8060517