The Landscape of Pseudomonas aeruginosa Membrane-Associated Proteins

 , , , and
, , , and
Abstract
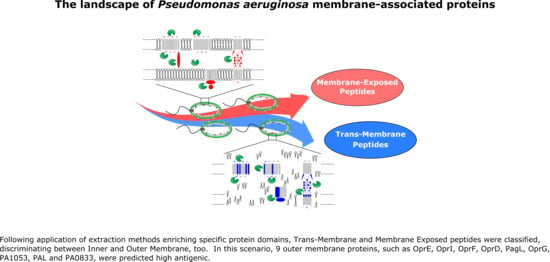
:
1. Introduction
2. Material and Methods
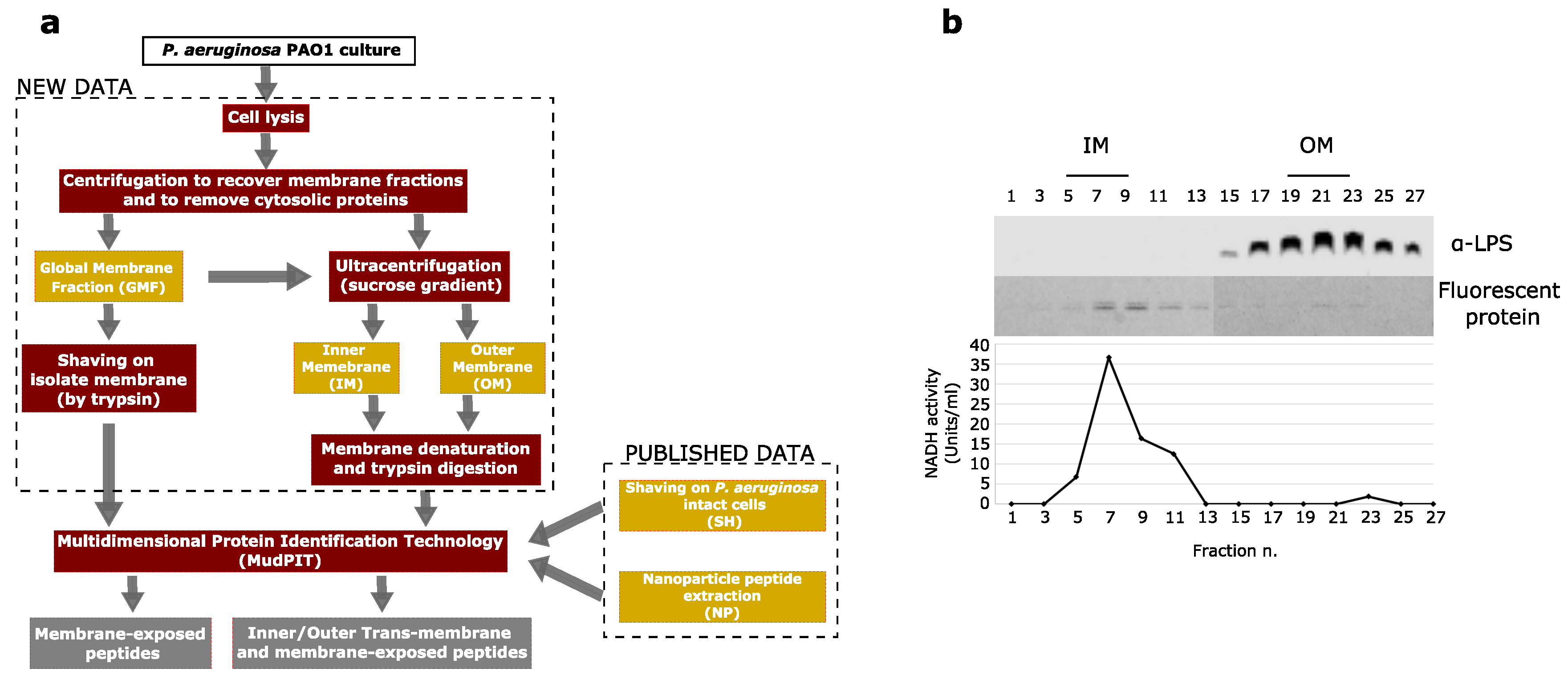
2.1. Bacterial Growth Conditions and Membrane Fractionation
2.2. Membrane Proteins Trypsin Digestion
2.3. MudPIT Analysis
2.4. Processing of Experimental Tandem Mass Spectra
2.5. CHARACTERIZATION of Trans-Membrane and Membrane-Exposed peptides
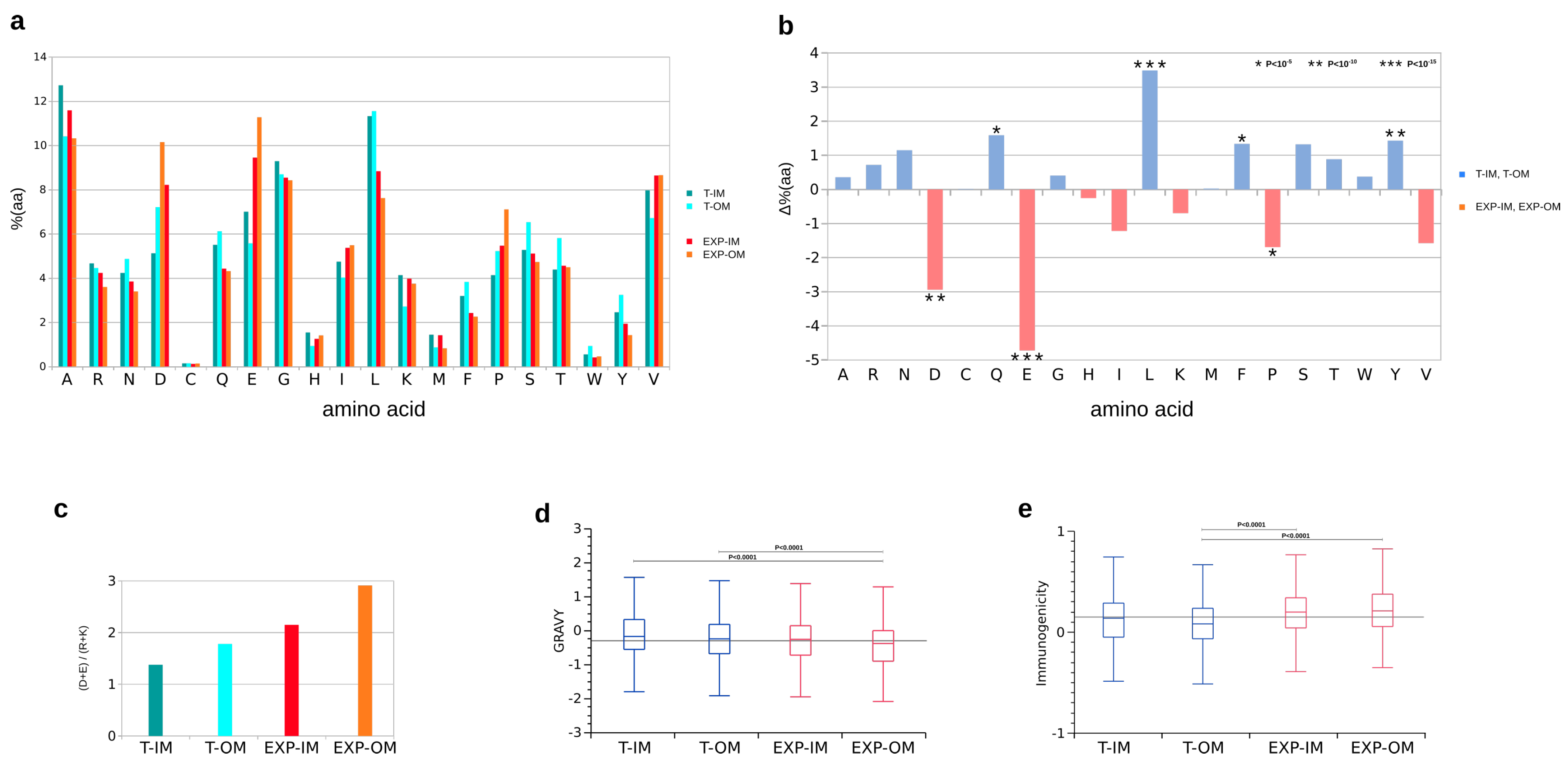
2.6. Functional Classification, Amino Acid Distribution, Gravy Index, Immunogenicity and Antigenicity
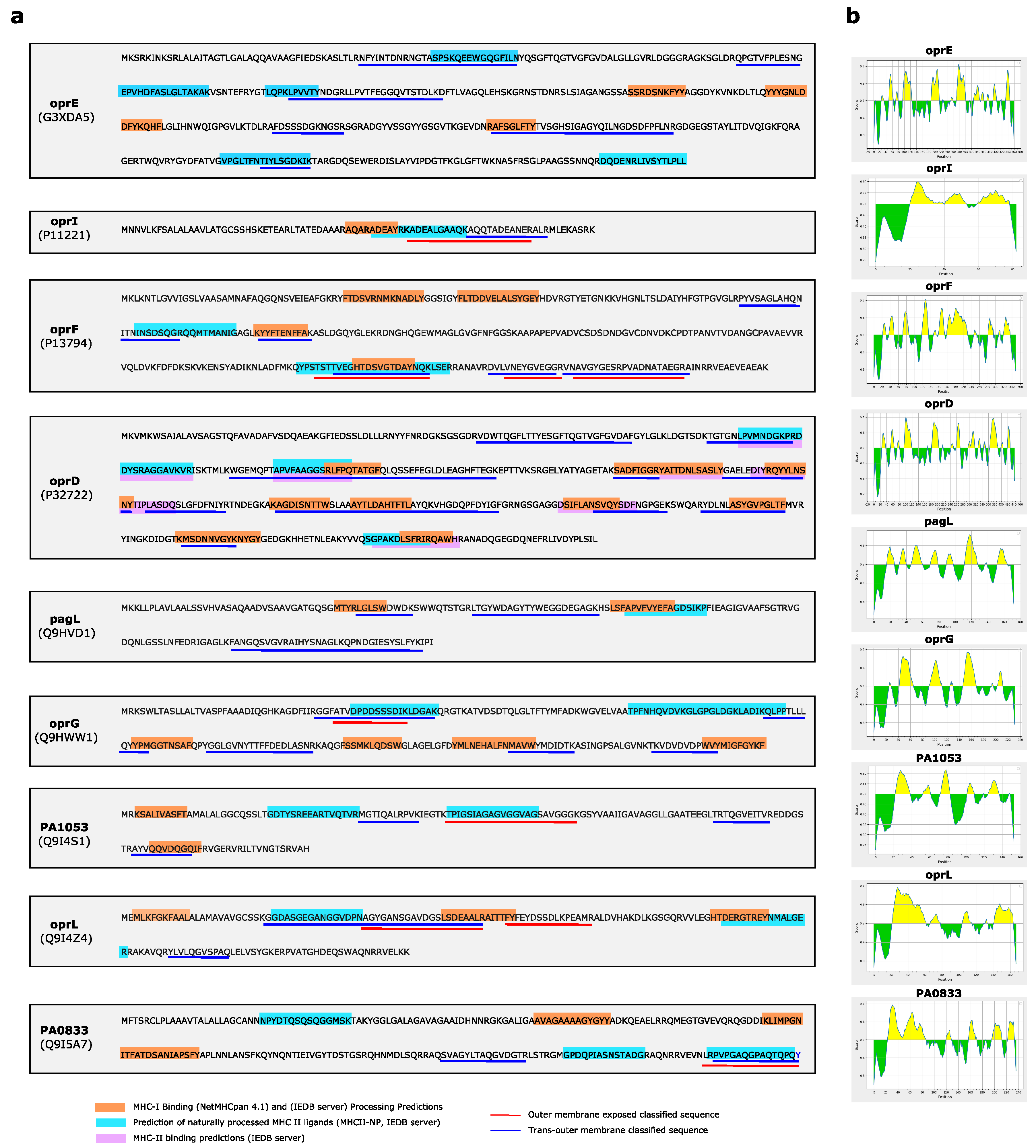
2.7. T Cell MHC (Class I and Class II) Epitope Binding and Processing Prediction
3. Results
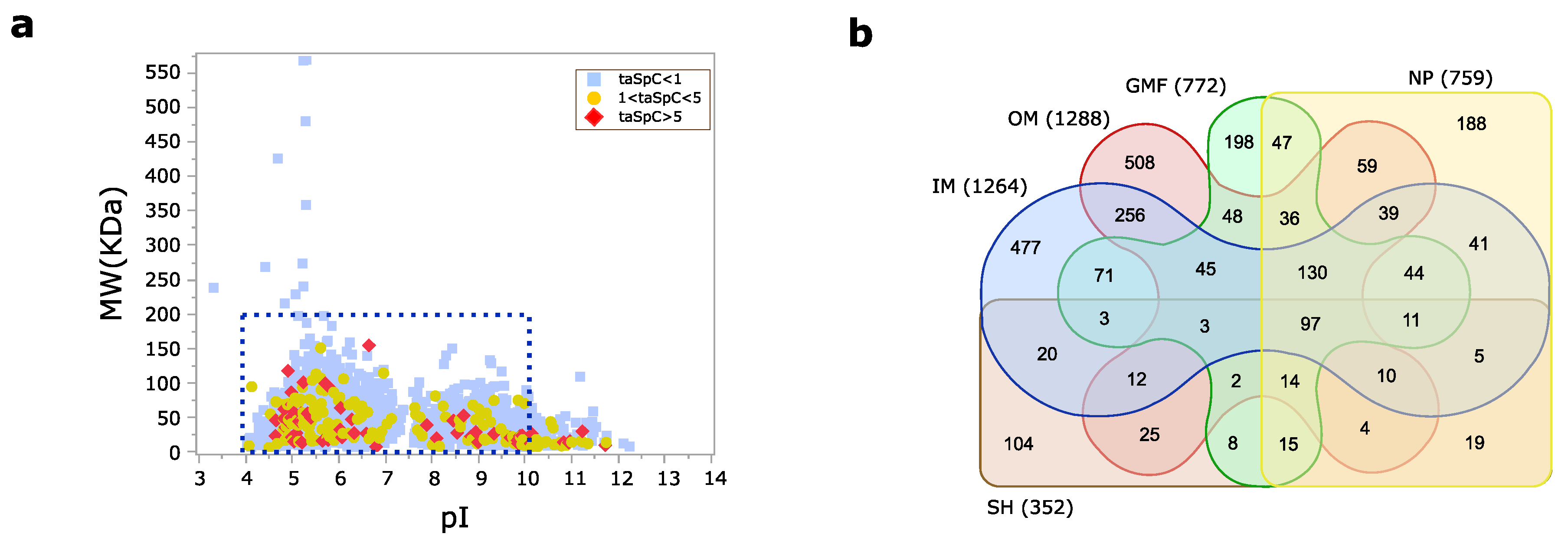
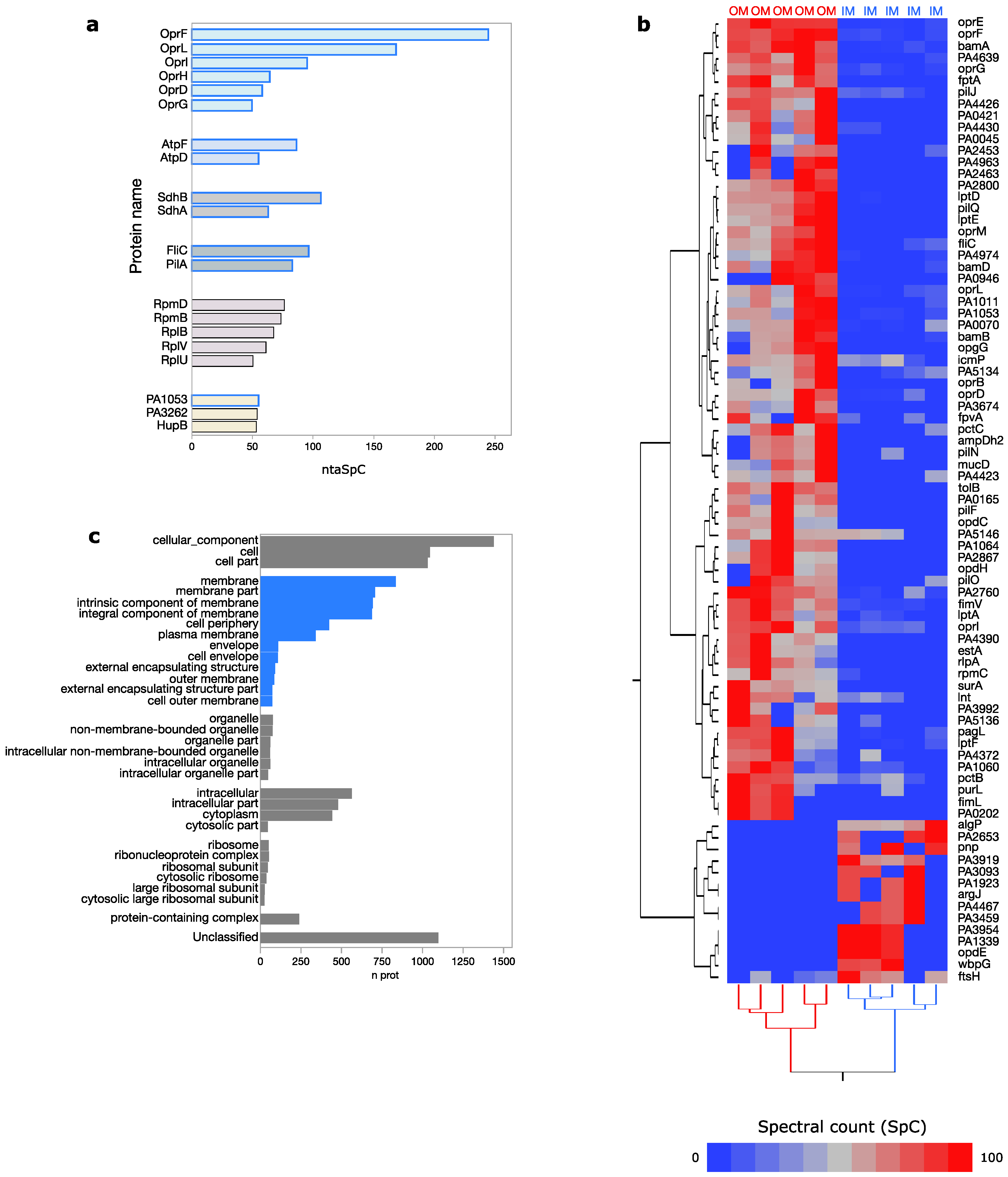
3.1. Pseudomonas aeruginosa Membrane Proteome
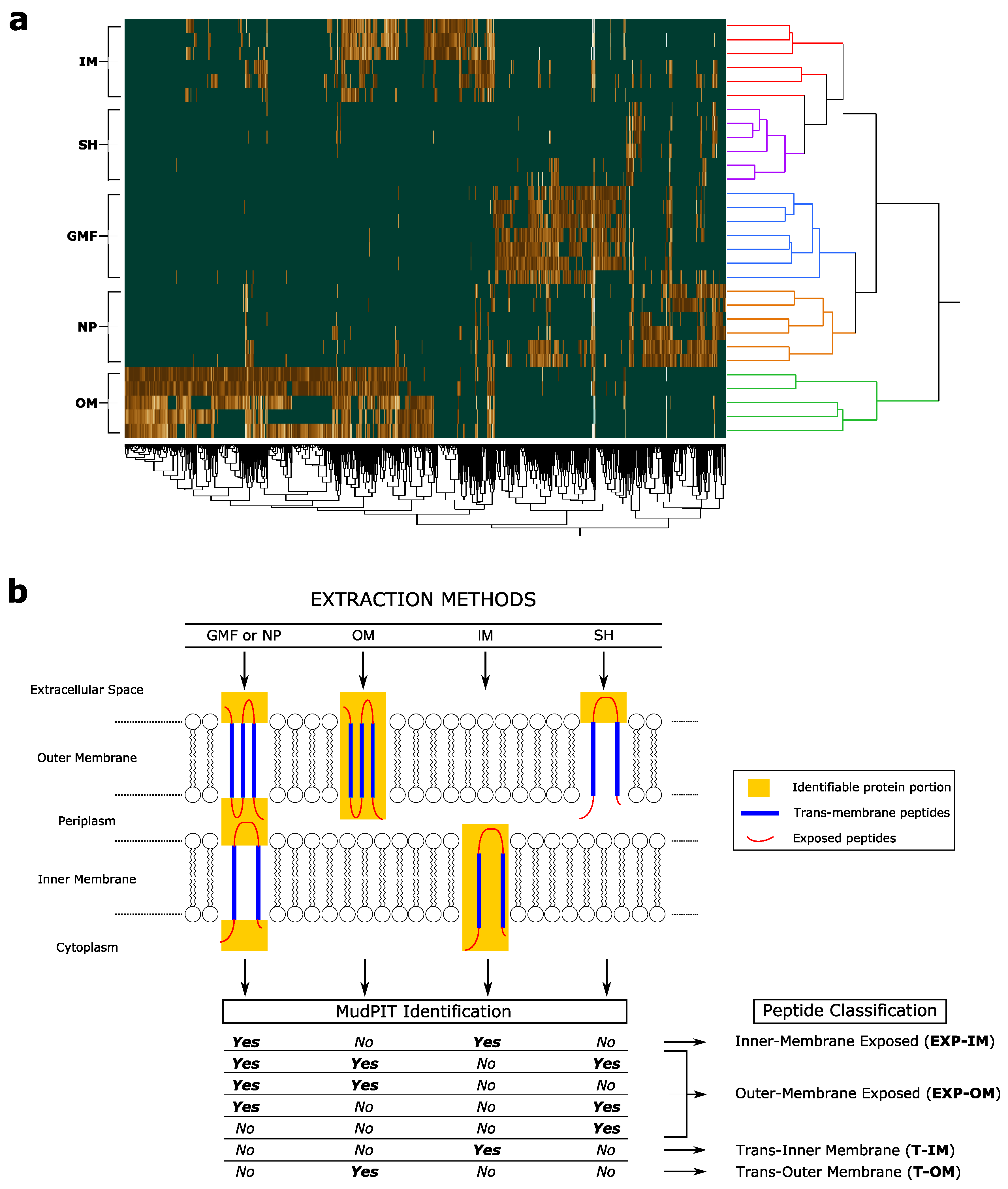
3.2. Trans-Membrane and Membrane-Exposed Peptides Classification
3.3. Inner and Outer Membrane Classified Peptides and P. aeruginosa Protein Models
3.4. Antigenicity, Immunogenicity and Toxicity of Selected Outer Membrane Proteins
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| IM | Inner Membrane |
| OM | Outer Memebrane |
| LC | Liquid Chromatography |
| MS | Mass Spectrometry |
| GFM | Global Membrane Fraction |
| MudPIT | Liquid Chromatography |
| SH | Shaving |
| NP | Carboxymethyl-dextran coated magnetic nanoparticles |
| SpC | Spectral Count |
| ntaSpC | total average Spectral Count normalized on protein length |
| LDA | Linear Discriminant Analysis |
| FDR | False Discovery Rate |
| T-IM | Trans Inner Membrane peptide |
| EXP-IM | Inner Membrane Exposed peptide |
| T-OM | Trans Outer Membrane peptide |
| EXP-OM | Outer Membrane Exposed peptide |
| MH | Mueller Hinton |
| OD | Optical Density |
| SCX | Strong Cation Exchange |
| RP | Reverse Phase |
| OMP | Outer Membrane Proteins |
| SVM | Support Vector Machine |
| TBDTs | TonB-dependent transporters |
References
- Gellatly, S.L.; Hancock, R.E. Pseudomonas aeruginosa: New insights into pathogenesis and host defenses. Pathog. Dis. 2013, 67, 159–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, Z.; Raudonis, R.; Glick, B.R.; Lin, T.J.; Cheng, Z. Antibiotic resistance in Pseudomonas aeruginosa: Mechanisms and alternative therapeutic strategies. Biotechnol. Adv. 2018, 37, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Huber, P.; Basso, P.; Reboud, E.; Attrée, I. Pseudomonas aeruginosa renews its virulence factors. Environ. Microbiol. Rep. 2016, 8, 564–571. [Google Scholar] [CrossRef]
- Azam, M.W.; Khan, A.U. Updates on the pathogenicity status of Pseudomonas aeruginosa. Drug Discov. Today 2018, 24, 350–359. [Google Scholar] [CrossRef]
- Krishnamoorthy, G.; Leus, I.V.; Weeks, J.W.; Wolloscheck, D.; Rybenkov, V.V.; Zgurskaya, H.I. Synergy between active efflux and outer membrane diffusion defines rules of antibiotic permeation into Gram-negative bacteria. MBio 2017, 8, e01172-17. [Google Scholar] [CrossRef] [Green Version]
- Choi, U.; Lee, C.R. Antimicrobial Agents That Inhibit the Outer Membrane Assembly Machines of Gram-Negative Bacteria. J. Microbiol. Biotechnol. 2019, 29, 1–10. [Google Scholar] [CrossRef]
- Nouwens, A.S.; Willcox, M.D.; Walsh, B.J.; Cordwell, S.J. Proteomic comparison of membrane and extracellular proteins from invasive (PAO1) and cytotoxic (6206) strains of Pseudomonas aeruginosa. Proteom. Int. Ed. 2002, 2, 1325–1346. [Google Scholar] [CrossRef]
- Duchesne, R.; Bouffartigues, E.; Oxaran, V.; Maillot, O.; Bénard, M.; Feuilloley, M.G.; Orange, N.; Chevalier, S. A proteomic approach of SigX function in Pseudomonas aeruginosa outer membrane composition. J. Proteom. 2013, 94, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Vecchietti, D.; Di Silvestre, D.; Miriani, M.; Bonomi, F.; Marengo, M.; Bragonzi, A.; Cova, L.; Franceschi, E.; Mauri, P.; Bertoni, G. Analysis of Pseudomonas aeruginosa cell envelope proteome by capture of surface-exposed proteins on activated magnetic nanoparticles. PLoS ONE 2012, 7, e51062. [Google Scholar] [CrossRef] [PubMed]
- Mlouka, M.A.B.; Khemiri, A.; Seyer, D.; Hardouin, J.; Song, P.C.T.; Dé, E.; Jouenne, T.; Cosette, P. Characterization of new outer membrane proteins of Pseudomonas aeruginosa using a combinatorial peptide ligand library. Anal. Bioanal. Chem. 2015, 407, 1513–1518. [Google Scholar] [CrossRef] [PubMed]
- Blonder, J.; Goshe, M.B.; Xiao, W.; Camp, D.G.; Wingerd, M.; Davis, R.W.; Smith, R.D. Global analysis of the membrane subproteome of Pseudomonas aeruginosa using liquid chromatography-tandem mass spectrometry. J. Proteome Res. 2004, 3, 434–444. [Google Scholar] [CrossRef]
- Kamath, K.S.; Pascovici, D.; Penesyan, A.; Goel, A.; Venkatakrishnan, V.; Paulsen, I.T.; Packer, N.H.; Molloy, M.P. Pseudomonas aeruginosa cell membrane protein expression from phenotypically diverse cystic fibrosis isolates demonstrates host-specific adaptations. J. Proteome Res. 2016, 15, 2152–2163. [Google Scholar] [CrossRef]
- Wang, A.Y.; Thuy-Boun, P.S.; Stupp, G.S.; Su, A.I.; Wolan, D.W. Triflic acid treatment enables LC-MS/MS analysis of insoluble bacterial biomass. J. Proteome Res. 2018, 17, 2978–2986. [Google Scholar] [CrossRef]
- Imperi, F.; Ciccosanti, F.; Perdomo, A.B.; Tiburzi, F.; Mancone, C.; Alonzi, T.; Ascenzi, P.; Piacentini, M.; Visca, P.; Fimia, G.M. Analysis of the periplasmic proteome of Pseudomonas aeruginosa, a metabolically versatile opportunistic pathogen. Proteomics 2009, 9, 1901–1915. [Google Scholar] [CrossRef] [PubMed]
- Casabona, M.G.; Vandenbrouck, Y.; Attree, I.; Couté, Y. Proteomic characterization of Pseudomonas aeruginosa PAO1 inner membrane. Proteomics 2013, 13, 2419–2423. [Google Scholar] [CrossRef]
- Martorana, A.M.; Motta, S.; Sperandeo, P.; Mauri, P.; Polissi, A. Differential Proteomics Based on Multidimensional Protein Identification Technology to Understand the Biogenesis of Outer Membrane of Escherichia coli. In Bacterial Cell Wall Homeostasis; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2016; Volume 1440, pp. 57–70. [Google Scholar] [CrossRef]
- Leal, T.; Bergamini, G.; Huaux, F.; Panin, N.; Noel, S.; Dhooghe, B.; Haaf, J.B.; Mauri, P.; Motta, S.; Di Silvestre, D.; et al. Azithromycin attenuates Pseudomonas-induced lung inflammation by Targeting Bacterial Proteins secreted in the cultured Medium. Front. Immunol. 2016, 7, 499. [Google Scholar] [CrossRef] [Green Version]
- Hilario, M.; Kalousis, A. Approaches to dimensionality reduction in proteomic biomarker studies. Brief. Bioinform. 2008, 9, 102–118. [Google Scholar] [CrossRef] [Green Version]
- Lo Sciuto, A.; Martorana, A.M.; Fernández-Piñar, R.; Mancone, C.; Polissi, A.; Imperi, F. Pseudomonas aeruginosa LptE is crucial for LptD assembly, cell envelope integrity, antibiotic resistance and virulence. Virulence 2018, 9, 1718–1733. [Google Scholar] [CrossRef] [Green Version]
- Tabb, D.L. The SEQUEST family tree. J. Am. Soc. Mass Spectrom. 2015, 26, 1814–1819. [Google Scholar] [CrossRef] [Green Version]
- Thomas, P.D.; Campbell, M.J.; Kejariwal, A.; Mi, H.; Karlak, B.; Daverman, R.; Diemer, K.; Muruganujan, A.; Narechania, A. PANTHER: A library of protein families and subfamilies indexed by function. Genome Res. 2003, 13, 2129–2141. [Google Scholar] [CrossRef] [Green Version]
- Cock, P.J.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef]
- Calis, J.J.; Maybeno, M.; Greenbaum, J.A.; Weiskopf, D.; De Silva, A.D.; Sette, A.; Keşmir, C.; Peters, B. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput. Biol. 2013, 9, e1003266. [Google Scholar] [CrossRef] [Green Version]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Reynisson, B.; Alvarez, B.; Paul, S.; Peters, B.; Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020, 48, W449–W454. [Google Scholar] [CrossRef]
- Tenzer, S.; Peters, B.; Bulik, S.; Schoor, O.; Lemmel, C.; Schatz, M.M.; Kloetzel, P.M.; Rammensee, H.G.; Schild, H.; Holzhütter, H.G. Modeling the MHC class I pathway by combining predictions of proteasomal cleavage, TAP transport and MHC class I binding. Cell. Mol. Life Sci. CMLS 2005, 62, 1025–1037. [Google Scholar] [CrossRef]
- Wang, P.; Sidney, J.; Kim, Y.; Sette, A.; Lund, O.; Nielsen, M.; Peters, B. Peptide binding predictions for HLA DR, DP and DQ molecules. BMC Bioinform. 2010, 11, 568. [Google Scholar] [CrossRef] [Green Version]
- Paul, S.; Karosiene, E.; Dhanda, S.K.; Jurtz, V.; Edwards, L.; Nielsen, M.; Sette, A.; Peters, B. Determination of a Predictive Cleavage Motif for Eluted Major Histocompatibility Complex Class II Ligands. Front. Immunol. 2018, 9, 1795. [Google Scholar] [CrossRef]
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: Improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017, 45, W24–W29. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Consortium, O.S.D.D.; Raghava, G.P.S. In silico approach for predicting toxicity of peptides and proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef] [Green Version]
- Chevalier, S.; Bouffartigues, E.; Bodilis, J.; Maillot, O.; Lesouhaitier, O.; Feuilloley, M.G.J.; Orange, N.; Dufour, A.; Cornelis, P. Structure, function and regulation of Pseudomonas aeruginosa porins. FEMS Microbiol. Rev. 2017, 41, 698–722. [Google Scholar] [CrossRef] [PubMed]
- Knowles, T.J.; Scott-Tucker, A.; Overduin, M.; Henderson, I.R. Membrane protein architects: The role of the BAM complex in outer membrane protein assembly. Nat. Rev. Microbiol. 2009, 7, 206. [Google Scholar] [CrossRef]
- Sperandeo, P.; Martorana, A.M.; Polissi, A. The lipopolysaccharide transport (Lpt) machinery: A nonconventional transporter for lipopolysaccharide assembly at the outer membrane of Gram-negative bacteria. J. Biol. Chem. 2017, 292, 17981–17990. [Google Scholar] [CrossRef] [Green Version]
- Weber, J. ATP synthase: Subunit–subunit interactions in the stator stalk. Biochim. Biophys. Acta (BBA) Bioenergy 2006, 1757, 1162–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, K.M.; Anselmi, C.; Wittig, I.; Faraldo-Gómez, J.D.; Kühlbrandt, W. Structure of the yeast F1Fo-ATP synthase dimer and its role in shaping the mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2012, 109, 13602–13607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugawara, E.; Nagano, K.; Nikaido, H. Factors affecting the folding of Pseudomonas aeruginosa OprF porin into the one-domain open conformer. MBio 2010, 1, e00228-10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugawara, E.; Nagano, K.; Nikaido, H. Alternative folding pathways of the major porin OprF of Pseudomonas aeruginosa. FEBS J. 2012, 279, 910–918. [Google Scholar] [CrossRef] [Green Version]
- Tjalsma, H.; Lambooy, L.; Hermans, P.W.; Swinkels, D.W. Shedding & shaving: Disclosure of proteomic expressions on a bacterial face. Proteomics 2008, 8, 1415–1428. [Google Scholar]
- Qi, Y.; Rao, J.; Shen, W.; Li, J.; Zeng, W.; Zheng, S.; Liu, S.; Li, Y.; Wang, B.; Wu, F.; et al. Identification of a Ribosomal Protein RpsB as a Surface-Exposed Protein and Adhesin of Rickettsia heilongjiangensis. BioMed Res. Int. 2019, 2019. [Google Scholar] [CrossRef]
- Ulmschneider, M.B.; Sansom, M.S. Amino acid distributions in integral membrane protein structures. Biochim. Biophys. Acta (BBA) Biomembr. 2001, 1512, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Marothy, M.T.D.; Elofsson, A. Marginally hydrophobic transmembrane a-helices shaping membrane protein folding. Protein Sci. 2015, 24, 1057–1074. [Google Scholar] [CrossRef] [Green Version]
- Rost, U.; Steinem, C.; Diederichsen, U. b-Glutamine-mediated self-association of transmembrane b-peptides within lipid bilayers. Chem. Sci. 2016, 7, 5900–5907. [Google Scholar] [CrossRef] [Green Version]
- Barnes, M.R.; Gray, I.C. Bioinformatics for Geneticists; John Wiley & Sons: Hoboken, NJ, USA, 2003. [Google Scholar]
- Berisio, R.; Vitagliano, L. Polyproline and triple helix motifs in host-pathogen recognition. Curr. Protein Pept. Sci. 2012, 13, 855–865. [Google Scholar] [CrossRef] [Green Version]
- Vitali, A. Proline-rich peptides: Multifunctional bioactive molecules as new potential therapeutic drugs. Curr. Protein Pept. Sci. 2015, 16, 147–162. [Google Scholar] [CrossRef]
- Solanki, V.; Tiwari, M.; Tiwari, V. Host-bacteria interaction and adhesin study for development of therapeutics. Int. J. Biol. Macromol. 2018, 112, 54–64. [Google Scholar] [CrossRef]
- Solanki, V.; Tiwari, M.; Tiwari, V. Prioritization of potential vaccine targets using comparative proteomics and designing of the chimeric multi-epitope vaccine against Pseudomonas aeruginosa. Sci. Rep. 2019, 9, 5240. [Google Scholar] [CrossRef] [PubMed]
- Hoggarth, A.; Weaver, A.; Pu, Q.; Huang, T.; Schettler, J.; Chen, F.; Yuan, X.; Wu, M. Mechanistic research holds promise for bacterial vaccines and phage therapies for Pseudomonas aeruginosa. Drug Des. Dev. Ther. 2019, 13, 909–924. [Google Scholar] [CrossRef] [Green Version]
- Gong, Q.; Ruan, M.D.; Niu, M.F.; Qin, C.L.; Hou, Y.; Guo, J.Z. Immune efficacy of DNA vaccines based on oprL and oprF genes of Pseudomonas aeruginosa in chickens. Poult. Sci. 2018, 97, 4219–4227. [Google Scholar] [CrossRef]
- Yang, F.; Gu, J.; Zou, J.; Lei, L.; Jing, H.; Zhang, J.; Zeng, H.; Zou, Q.; Lv, F.; Zhang, J. PA0833 Is an OmpA C-Like Protein That Confers Protection Against Pseudomonas aeruginosa Infection. Front. Microbiol. 2018, 9, 1062. [Google Scholar] [CrossRef]
- Perraud, Q.; Cantero, P.; Roche, B.; Gasser, V.; Normant, V.P.; Kuhn, L.; Hammann, P.; Mislin, G.L.A.; Ehret-Sabatier, L.; Schalk, I.J. Phenotypic Adaption of Pseudomonas aeruginosa by Hacking Siderophores Produced by Other Microorganisms. Mol. Cell. Proteom. MCP 2020, 19, 589–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noinaj, N.; Guillier, M.; Barnard, T.J.; Buchanan, S.K. TonB-dependent transporters: Regulation, structure, and function. Annu. Rev. Microbiol. 2010, 64, 43–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savoia, D. New perspectives in the management of Pseudomonas aeruginosa infections. Future Microbiol. 2014, 9, 917–928. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UNIPROT ID | Protein Name | Gene Name | EXP-IM | T-IM | EXP-OM | T-OM | Sub. Localization |
|---|---|---|---|---|---|---|---|
| Q51485 | Porin B | oprB | 4 | plasma membrane [GO:0005886] | |||
| G3XD89 | Putative copper transport outer membrane porin OprC | oprC | - | 5 | plasma membrane [GO:0005886] | ||
| P32722 | Porin D | oprD | 18 | plasma membrane [GO:0005886] | |||
| G3XDA5 | Anaerobically-induced outer membrane porin OprE | oprE | 14 | plasma membrane [GO:0005886] | |||
| P13794 | Outer membrane porin F | oprF | 1 | 5 | 11 | plasma membrane [GO:0005886] | |
| Q9HWW1 | Outer membrane protein OprG | oprG | 1 | 10 | plasma membrane [GO:0005886] | ||
| G3XD11 | PhoP/Q and low Mg2+ inducible outer membrane protein H1 | oprH | 2 | 3 | 1 | 1 | plasma membrane [GO:0005886] |
| P11221 | Major outer membrane lipoprotein | oprI | 1 | 2 | plasma membrane [GO:0005886] | ||
| Q9I4Z4 | Peptidoglycan-associated lipoprotein | oprL | 1 | 2 | 3 | 9 | plasma membrane [GO:0005886] |
| Q51487 | Outer membrane protein OprM | oprM | - | - | 15 | plasma membrane [GO:0005886] | |
| Q9HXY4 | Outer membrane protein assembly factor BamA | bamA | 2 | 1 | 19 | cell outer membrane [GO:0009279] | |
| Q9HXJ7 | Outer membrane protein assembly factor BamB | bamB | 2 | 8 | cell outer membrane [GO:0009279] | ||
| P33641 | Outer membrane protein assembly factor BamD | bamD | 6 | cell outer membrane [GO:0009279] | |||
| O68562 | Outer membrane protein assembly factor BamE | bamE | 1 | 3 | cell outer membrane [GO:0009279] | ||
| Q9HVV7 | Lipopolysaccharide export system protein LptA | lptA | 1 | cell outer membrane [GO:0009279] | |||
| Q9I5U2 | LPS-assembly protein LptD | lptD | 23 | cell outer membrane [GO:0009279] | |||
| Q9HX32 | LPS-assembly lipoprotein LptE | lptE | 1 | 4 | cell outer membrane [GO:0009279] | ||
| Q9HT18 | ATP synthase subunit alpha | atpA | 10 | 1 | 2 | - | plasma membrane [GO:0005886] |
| Q9HT14 | ATP synthase subunit a | atpB | 1 | 1 | - | - | plasma membrane [GO:0005886] |
| Q9HT21 | ATP synthase epsilon | atpC | 1 | 1 | - | - | plasma membrane [GO:0005886] |
| Q9HT20 | ATP synthase subunit beta | atpD | 11 | 6 | - | 1 | plasma membrane [GO:0005886] |
| Q9HT16 | ATP synthase subunit b | atpF | - | 2 | - | - | plasma membrane [GO:0005886] |
| Q9HT19 | ATP synthase gamma | atpG | 3 | - | - | - | plasma membrane [GO:0005886] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Motta, S.; Vecchietti, D.; Martorana, A.M.; Brunetti, P.; Bertoni, G.; Polissi, A.; Mauri, P.; Di Silvestre, D. The Landscape of Pseudomonas aeruginosa Membrane-Associated Proteins. Cells 2020, 9, 2421. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9112421
Motta S, Vecchietti D, Martorana AM, Brunetti P, Bertoni G, Polissi A, Mauri P, Di Silvestre D. The Landscape of Pseudomonas aeruginosa Membrane-Associated Proteins. Cells. 2020; 9(11):2421. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9112421
Chicago/Turabian StyleMotta, Sara, Davide Vecchietti, Alessandra M. Martorana, Pietro Brunetti, Giovanni Bertoni, Alessandra Polissi, Pierluigi Mauri, and Dario Di Silvestre. 2020. "The Landscape of Pseudomonas aeruginosa Membrane-Associated Proteins" Cells 9, no. 11: 2421. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9112421