
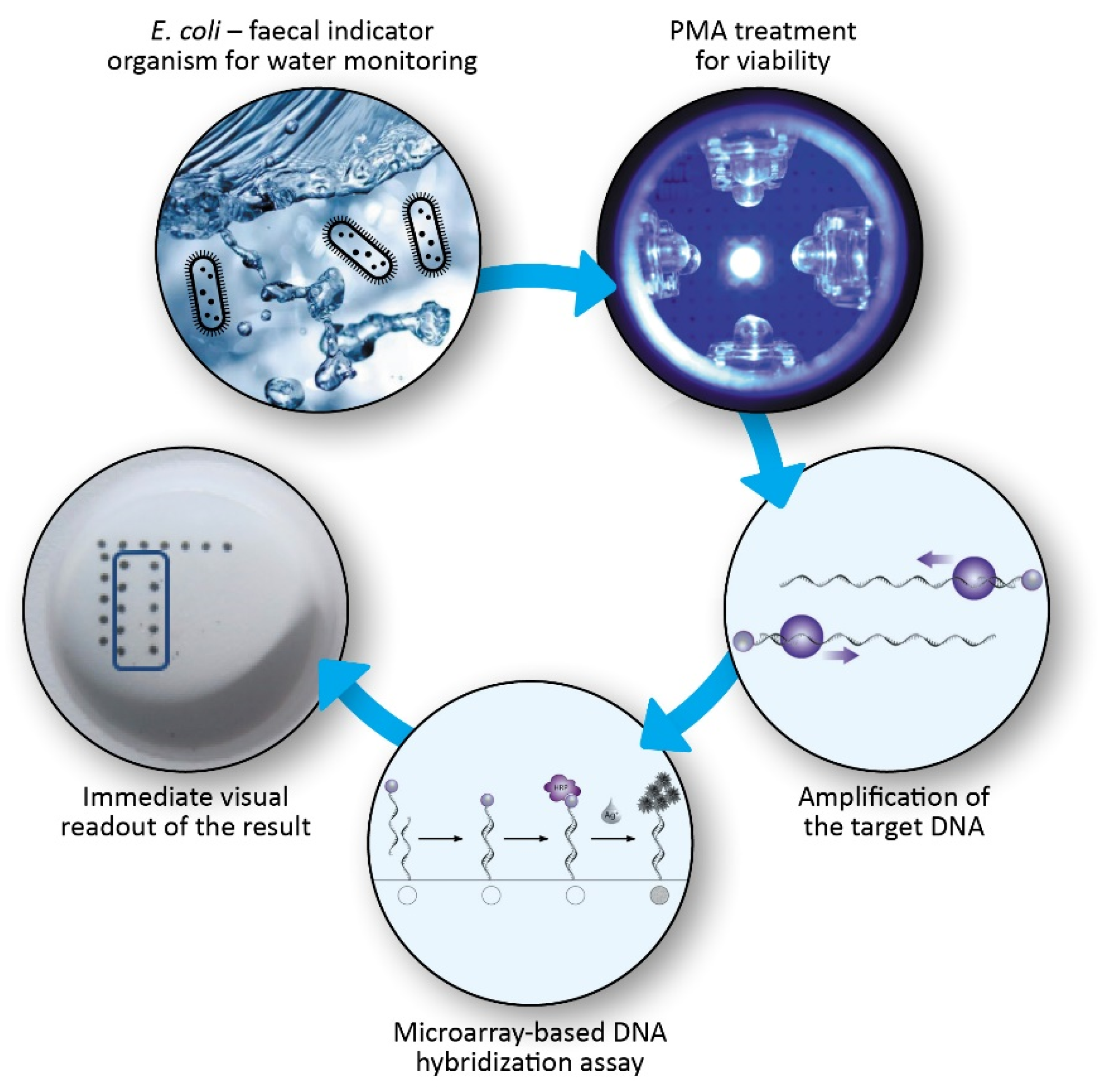
A Model System for Sensitive Detection of Viable E. coli Bacteria Combining Direct Viability PCR and a Novel Microarray-Based Detection Approach

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Culturing and Harvesting Defined Cell Numbers of E. coli Bacteria
2.2. PMA Treatment for Viability PCR
2.3. Thermal Cell Lysis for Direct PCR
2.4. gDNA Extraction
2.5. Direct, Viability, and Nested PCR Amplification
2.6. Generation of Single-Stranded DNA
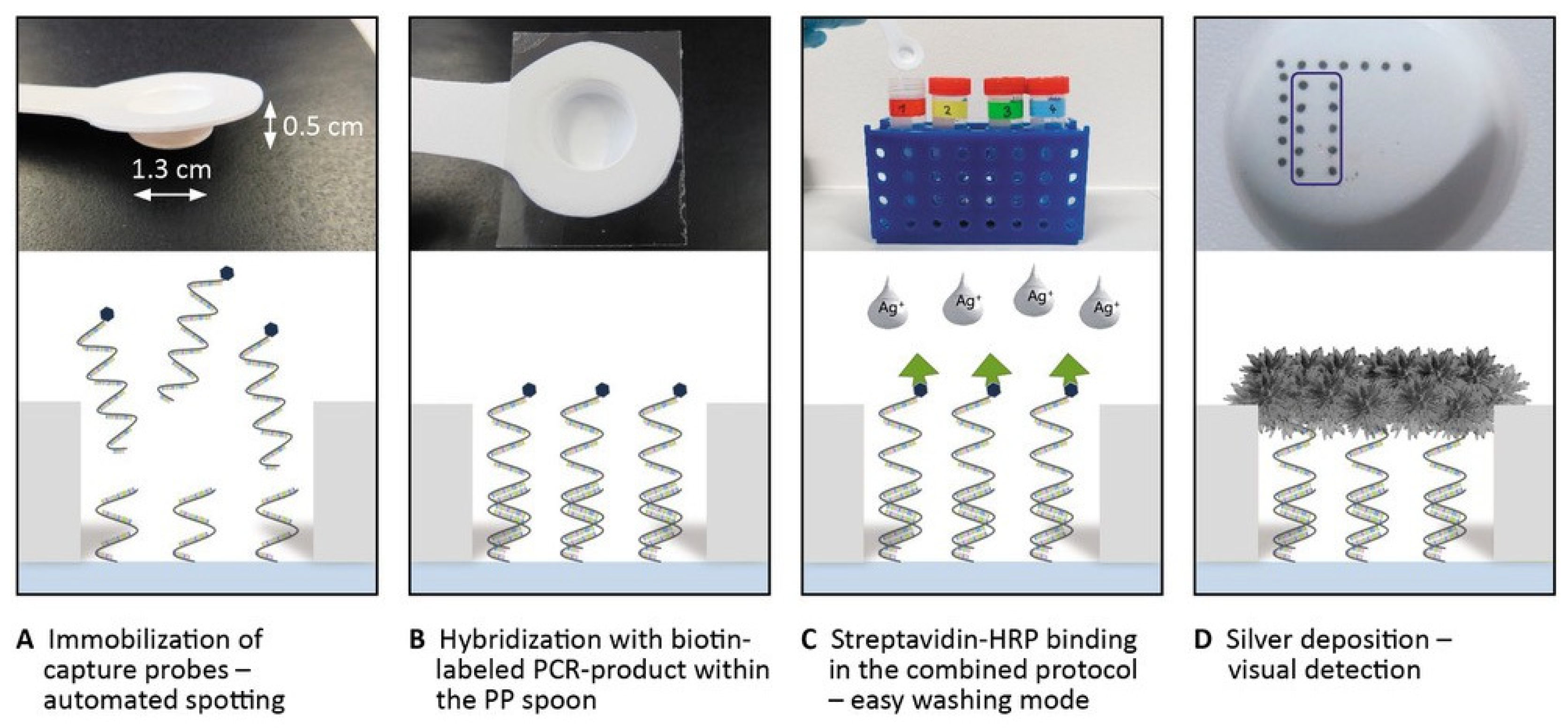
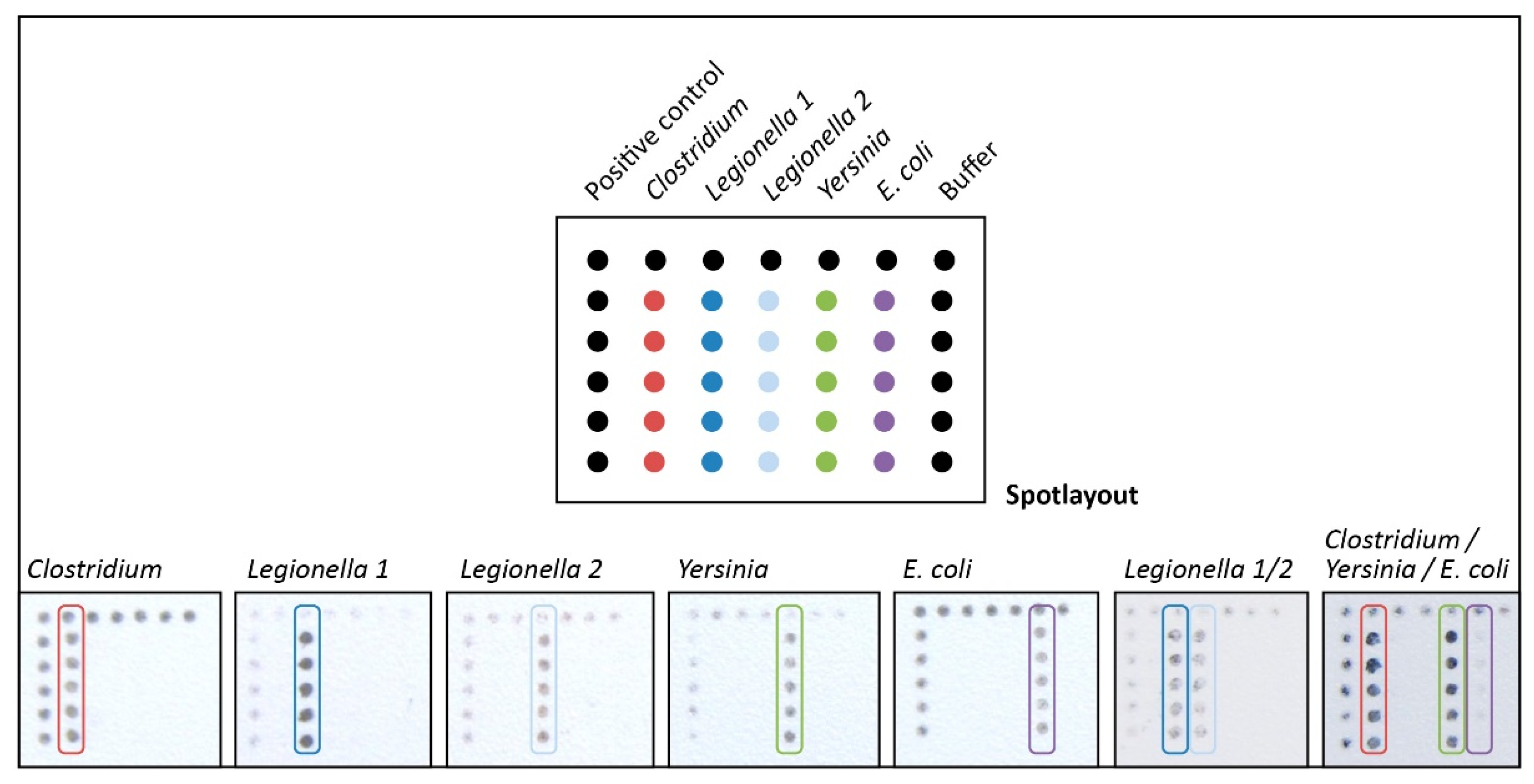
2.7. Microarray-Based DNA Hybridization and Signal Detection
3. Results
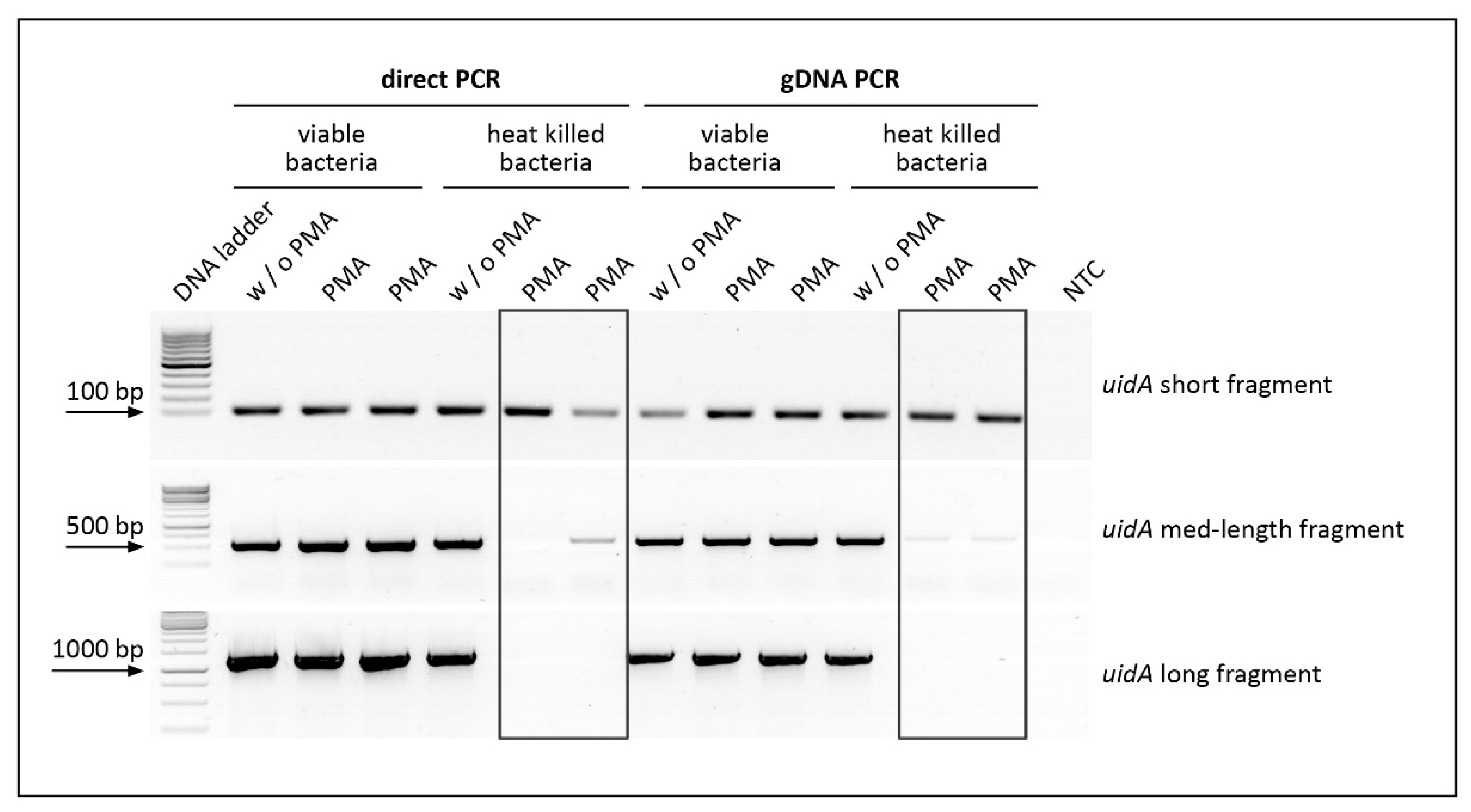
3.1. Viability PCR Using an in-House Constructed PMA-Photoactivation Device
3.2. Impact of Amplicon Length on PMA Pretreatment of Heat Killed E. coli Cells
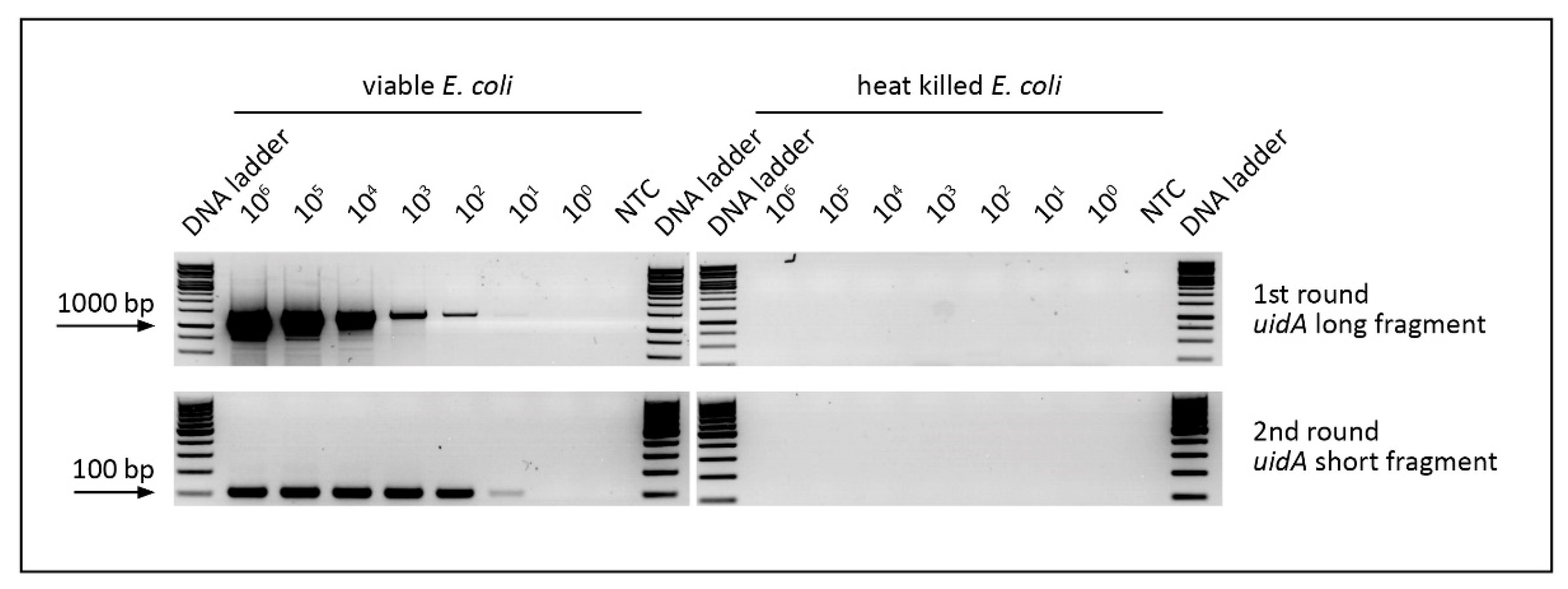
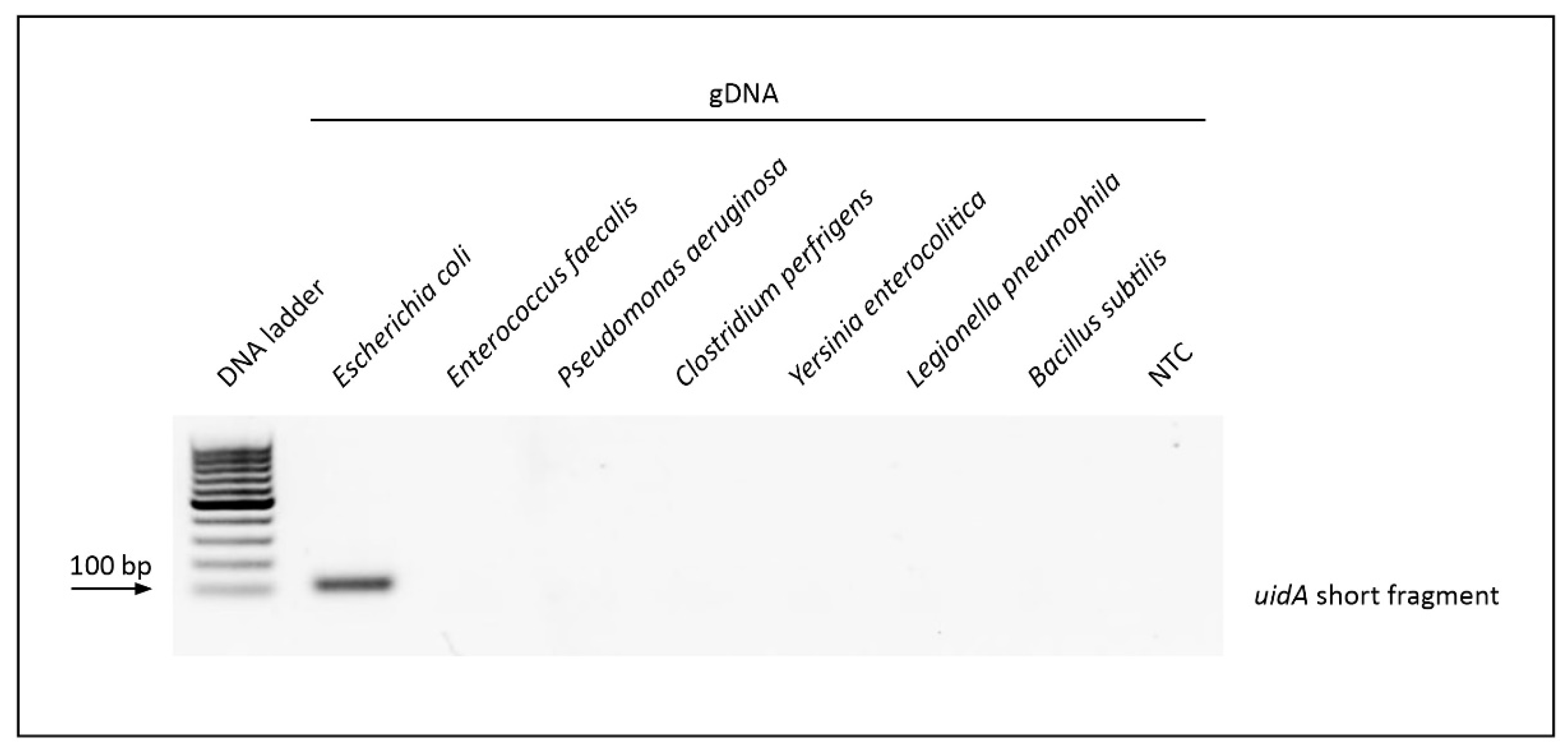
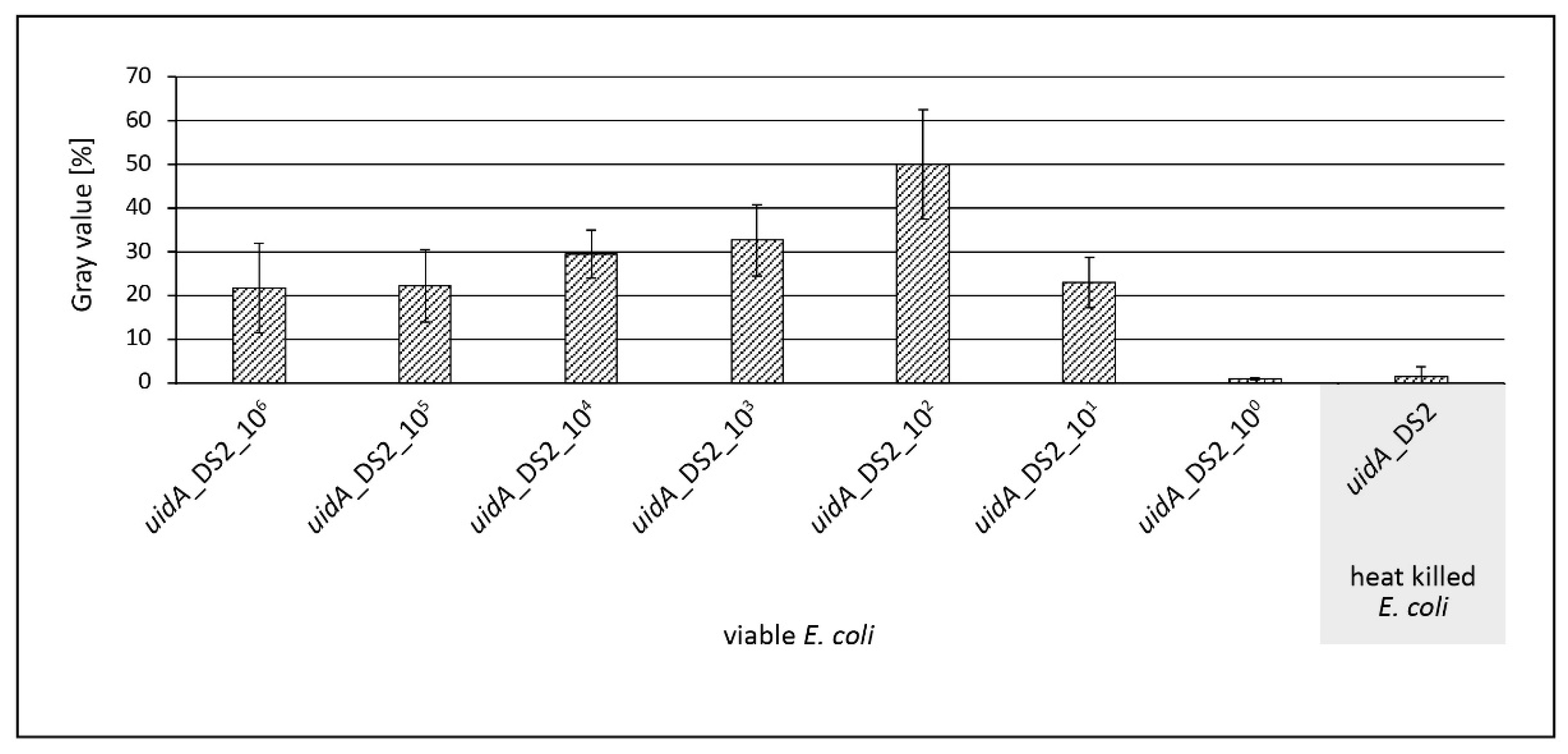
3.3. Evaluation of v-PCR
3.4. Microarray-Based DNA Detection on a Novel Designed Spoon-Shaped Substrate
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- World Health Organization. Progress on Drinking Water, Sanitation and Hygiene: 2017 Update and SDG Baselines; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Ailes, E.; Budge, P.; Shankar, M.; Collier, S.; Brinton, W.; Cronquist, A.; Chen, M.; Thornton, A.; Beach, M.J.; Brunkard, J.M. Economic and Health Impacts Associated with a Salmonella Typhimurium Drinking Water Outbreak−Alamosa, CO, 2008. PLoS ONE 2013, 8, e57439. [Google Scholar] [CrossRef]
- World Health Organization. A Global Overview of National Regulations and Standards for Drinking-Water Quality; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- Sarkar, R.; Prabhakar, A.T.; Manickam, S.; Selvapandian, D.; Raghava, M.V.; Kang, G.; Balraj, V. Epidemiological investigation of an outbreak of acute diarrhoeal disease using geographic information systems. Trans. R. Soc. Trop. Med. Hyg. 2007, 101, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Edberg, S.; Rice, E.; Karlin, R.; Allen, M. Escherichia coli: The best biological drinking water indicator for public health protection. J. Appl. Microbiol. 2000, 88, 106S–116S. [Google Scholar] [CrossRef]
- Gibson, C.; Maritim, A.; Marion, J. Comparison of the ColiPlate™ Kit with Two Common E. coli Enumeration Methods for Water. Water 2021, 13, 1804. [Google Scholar] [CrossRef]
- Nogva, H.K.; Drømtorp, S.M.; Nissen, H.; Rudi, K. Ethidium Monoazide for DNA-Based Differentiation of Viable and Dead Bacteria by 5′-Nuclease PCR. Biotechniques 2003, 34, 804–813. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.; Wuertz, S. Discrimination of Viable and Dead Fecal Bacteroidales Bacteria by Quantitative PCR with Propidium Monoazide. Appl. Environ. Microbiol. 2009, 75, 2940–2944. [Google Scholar] [CrossRef] [Green Version]
- Cawthorn, D.-M.; Witthuhn, R. Selective PCR detection of viableEnterobacter sakazakiicells utilizing propidium monoazide or ethidium bromide monoazide. J. Appl. Microbiol. 2008, 105, 1178–1185. [Google Scholar] [CrossRef]
- Delgado-Viscogliosi, P.; Solignac, L.; Delattre, J.-M. Viability PCR, a Culture-Independent Method for Rapid and Selective Quantification of Viable Legionella pneumophila Cells in Environmental Water Samples. Appl. Environ. Microbiol. 2009, 75, 3502–3512. [Google Scholar] [CrossRef] [Green Version]
- Nocker, A.; Mazza, A.; Masson, L.; Camper, A.K.; Brousseau, R. Selective detection of live bacteria combining propidium monoazide sample treatment with microarray technology. J. Microbiol. Methods 2009, 76, 253–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Breidt, F. Enumeration of Viable Listeria monocytogenes Cells by Real-Time PCR with Propidium Monoazide and Ethidium Monoazide in the Presence of Dead Cells. Appl. Environ. Microbiol. 2007, 73, 8028–8031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudi, K.; Moen, B.; Drømtorp, S.M.; Holck, A.L. Use of Ethidium Monoazide and PCR in Combination for Quantification of Viable and Dead Cells in Complex Samples. Appl. Environ. Microbiol. 2005, 71, 1018–1024. [Google Scholar] [CrossRef] [Green Version]
- Soejima, T.; Iida, K.-I.; Qin, T.; Taniai, H.; Seki, M.; Yoshida, S.-I. Method to Detect Only Live Bacteria during PCR Amplification. J. Clin. Microbiol. 2008, 46, 2305–2313. [Google Scholar] [CrossRef] [Green Version]
- Soejima, T.; Schlitt-Dittrich, F.; Yoshida, S.-I. Polymerase chain reaction amplification length-dependent ethidium monoazide suppression power for heat-killed cells of Enterobacteriaceae. Anal. Biochem. 2011, 418, 37–43. [Google Scholar] [CrossRef]
- Agustí, G.; Codony, F.; Fittipaldi, M.; Adrados, B.; Morató, J. Viability Determination of Helicobacter pylori Using Propidium Monoazide Quantitative PCR. Helicobacter 2010, 15, 473–476. [Google Scholar] [CrossRef]
- Rawsthorne, H.; Dock, C.N.; Jaykus, L.A. PCR-Based Method Using Propidium Monoazide to Distinguish Viable from Nonviable Bacillus subtilis Spores. Appl. Environ. Microbiol. 2009, 75, 2936–2939. [Google Scholar] [CrossRef] [Green Version]
- Brescia, C.C.; Griffin, S.M.; Ware, M.W.; Varughese, E.A.; Egorov, A.I.; Villegas, E.N. Cryptosporidium Propidium Monoazide-PCR, a Molecular Biology-Based Technique for Genotyping of Viable Cryptosporidium Oocysts. Appl. Environ. Microbiol. 2009, 75, 6856–6863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fittipaldi, M.; Rodriguez, N.J.P.; Adrados, B.; Agustí, G.; Peñuela, G.; Morató, J.; Codony, F. Discrimination of Viable Acanthamoeba castellani Trophozoites and Cysts by Propidium Monoazide Real-Time Polymerase Chain Reaction. J. Eukaryot. Microbiol. 2011, 58, 359–364. [Google Scholar] [CrossRef]
- Graiver, D.A.; Saunders, S.E.; Topliff, C.L.; Kelling, C.L.; Bartelt-Hunt, S.L. Ethidium monoazide does not inhibit RT-PCR amplification of nonviable avian influenza RNA. J. Virol. Methods 2010, 164, 51–54. [Google Scholar] [CrossRef]
- Fittipaldi, M.; Rodriguez, N.J.P.; Codony, F.; Adrados, B.; Peñuela, G.A.; Morató, J. Discrimination of infectious bacteriophage T4 virus by propidium monoazide real-time PCR. J. Virol. Methods 2010, 168, 228–232. [Google Scholar] [CrossRef]
- Sánchez, G.; Elizaquível, P.; Aznar, R. Discrimination of Infectious Hepatitis A Viruses by Propidium Monoazide Real-Time RT-PCR. Food Environ. Virol. 2012, 4, 21–25. [Google Scholar] [CrossRef]
- Vesper, S.; McKinstry, C.; Hartmann, C.; Neace, M.; Yoder, S.; Vesper, A. Quantifying fungal viability in air and water samples using quantitative PCR after treatment with propidium monoazide (PMA). J. Microbiol. Methods 2008, 72, 180–184. [Google Scholar] [CrossRef]
- Nocker, A.; Cheung, C.-Y.; Camper, A.K. Comparison of propidium monoazide with ethidium monoazide for differentiation of live vs. dead bacteria by selective removal of DNA from dead cells. J. Microbiol. Methods 2006, 67, 310–320. [Google Scholar] [CrossRef]
- Loaiza, Ó.A.; Campuzano, S.; Pedrero, M.; García, P.; Pingarrón, J.M. Ultrasensitive detection of coliforms by means of direct asymmetric PCR combined with disposable magnetic amperometric genosensors. Analyst 2009, 134, 34–37. [Google Scholar] [CrossRef]
- Maheux, A.F.; Boudreau, D.K.; Bisson, M.-A.; Dion-Dupont, V.; Bouchard, S.; Nkuranga, M.; Bergeron, M.G.; Rodriguez, M.J. Molecular Method for Detection of Total Coliforms in Drinking Water Samples. Appl. Environ. Microbiol. 2014, 80, 4074–4084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, T.; Trabbic-Carlson, K.; Liu, W.; Chilkoti, A. Purification of recombinant proteins from Escherichia coli at low expression levels by inverse transition cycling. Anal. Biochem. 2007, 360, 166–168. [Google Scholar] [CrossRef] [Green Version]
- Beyer, A.; Pollok, S.; Rudloff, A.; Cialla-May, D.; Weber, K.; Popp, J. Fast-Track, One-Step E. coli Detection: A Miniaturized Hydrogel Array Permits Specific Direct PCR and DNA Hybridization while Amplification. Macromol. Biosci. 2016, 16, 1325–1333. [Google Scholar] [CrossRef]
- Schwenkbier, L.; König, S.; Wagner, S.; Pollok, S.; Weber, J.; Hentschel, M.; Popp, J.; Werres, S.; Weber, K. On-site detection of Phytophthora spp.—single-stranded target DNA as the limiting factor to improve on-chip hybridization. Microchim. Acta 2014, 181, 1669–1679. [Google Scholar] [CrossRef]
- Schüler, T.; Nykytenko, A.; Csaki, A.; Möller, R.; Fritzsche, W.; Popp, J. UV cross-linking of unmodified DNA on glass surfaces. Anal. Bioanal. Chem. 2009, 395, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Schüler, T.; Asmus, T.; Fritzsche, W.; Möller, R. Screen printing as cost-efficient fabrication method for DNA-chips with electrical readout for detection of viral DNA. Biosens. Bioelectron. 2009, 24, 2077–2084. [Google Scholar] [CrossRef] [PubMed]
- Schüler, T.; Kretschmer, R.; Jessing, S.; Urban, M.; Fritzsche, W.; Möller, R.; Popp, J. A disposable and cost efficient microfluidic device for the rapid chip-based electrical detection of DNA. Biosens. Bioelectron. 2009, 25, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Seise, B.; Brinker, A.; Kretschmer, R.; Schwarz, M.; Rudolph, B.; Kaulfuß, T.; Urban, M.; Henkel, T.; Popp, J.; Möller, R. Chip-based detection system for the on-site analysis of animal diseases. Eng. Life Sci. 2011, 11, 148–156. [Google Scholar] [CrossRef]
- Fittipaldi, M.; Nocker, A.; Codony, F. Progress in understanding preferential detection of live cells using viability dyes in combination with DNA amplification. J. Microbiol. Methods 2012, 91, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Vondrakova, L.; Turonova, H.; Scholtz, V.; Pazlarova, J.; Demnerova, K. Impact of various killing methods on EMA/PMA-qPCR efficacy. Food Control. 2018, 85, 23–28. [Google Scholar] [CrossRef]
- Contreras, P.J.; Urrutia, H.; Sossa, K.; Nocker, A. Effect of PCR amplicon length on suppressing signals from membrane-compromised cells by propidium monoazide treatment. J. Microbiol. Methods 2011, 87, 89–95. [Google Scholar] [CrossRef]
- Nocker, A.; Sossa-Fernandez, P.; Burr, M.D.; Camper, A.K. Use of Propidium Monoazide for Live/Dead Distinction in Microbial Ecology. Appl. Environ. Microbiol. 2007, 73, 5111–5117. [Google Scholar] [CrossRef] [Green Version]
- Kralik, P.; Nocker, A.; Pavlik, I. Mycobacterium avium subsp. paratuberculosis viability determination using F57 quantitative PCR in combination with propidium monoazide treatment. Int. J. Food Microbiol. 2010, 141, S80–S86. [Google Scholar] [CrossRef]
- Banihashemi, A.; Van Dyke, M.; Huck, P. Long-amplicon propidium monoazide-PCR enumeration assay to detect viable Campylobacter and Salmonella. J. Appl. Microbiol. 2012, 113, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.-F.; Lin, W.-T.; Guo, Y. Method to detect only viable cells in microbial ecology. Appl. Microbiol. Biotechnol. 2010, 86, 377–384. [Google Scholar] [CrossRef]
- Juck, D.; Ingram, J.; Prévost, M.; Coallier, J.; Greer, C. Nested PCR protocol for the rapid detection of Escherichia coli in potable water. Can. J. Microbiol. 1996, 42, 862–866. [Google Scholar] [CrossRef] [PubMed]
- Brinker, A.; Schulze, H.; Bachmann, T.; Möller, R. Lambda exonuclease pre-treatment for improved DNA-chip performance depends on the relative probe-target position. Biosens. Bioelectron. 2010, 26, 898–902. [Google Scholar] [CrossRef]
- Farnleitner, A.H.; Kreuzinger, N.; Kavka, G.G.; Grillenberger, S.; Rath, J.; Mach, R.L. Simultaneous Detection and Differentiation of Escherichia coli Populations from Environmental Freshwaters by Means of Sequence Variations in a Fragment of the β- d -Glucuronidase Gene. Appl. Environ. Microbiol. 2000, 66, 1340–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, I.U.; Gannon, V.; Kent, R.; Koning, W.; Lapen, D.R.; Miller, J.; Neumann, N.; Phillips, R.; Robertson, W.; Topp, E.; et al. Development of a rapid quantitative PCR assay for direct detection and quantification of culturable and non-culturable Escherichia coli from agriculture watersheds. J. Microbiol. Methods 2007, 69, 480–488. [Google Scholar] [CrossRef]
- Maheux, A.F.; Picard, F.J.; Boissinot, M.; Bissonnette, L.; Paradis, S.; Bergeron, M.G. Analytical comparison of nine PCR primer sets designed to detect the presence of Escherichia coli/Shigella in water samples. Water Res. 2009, 43, 3019–3028. [Google Scholar] [CrossRef] [PubMed]
- Tryland, I.; Fiksdal, L. Enzyme characteristics of beta-D-galactosidase- and beta-D-glucuronidase-positive bacteria and their interference in rapid methods for detection of waterborne coliforms and Escherichia coli. Appl. Environ. Micro-Biol. 1998, 64, 1018–1023. [Google Scholar] [CrossRef] [Green Version]
- Schwenkbier, L.; Pollok, S.; Rudloff, A.; Sailer, S.; Cialla-May, D.; Weber, K.; Popp, J. Non-instrumented DNA isolation, amplification and microarray-based hybridization for a rapid on-site detection of devastating Phytophthora kernoviae. Analyst 2015, 140, 6610–6618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wünscher, S.; Seise, B.; Pretzel, D.; Pollok, S.; Perelaer, J.; Weber, K.; Popp, J.; Schubert, U.S. Chip-on-foil devices for DNA analysis based on inkjet-printed silver electrodes. Lab Chip 2014, 14, 392–401. [Google Scholar] [CrossRef] [Green Version]
- Beckman, A.K.; Ferrieri, P. Prospective Investigation of an Automated PCR/Nucleic Acid Microarray-Based Platform for Enteric Pathogen Testing. Lab. Med. 2019, 50, 390–395. [Google Scholar] [CrossRef]
- Brennecke, J.; Kraut, S.; Zwadlo, K.; Gandi, S.K.; Pritchard, D.; Templeton, K.; Bachmann, T. High-yield extraction of Escherichia coli RNA from human whole blood. J. Med。 Microbiol. 2017, 66, 301–311. [Google Scholar] [CrossRef]
- Krapf, T.; Kuhn, R.M.; Kauf, P.; Gantenbein-Demarchi, C.H.; Fieseler, L. Quantitative real-time PCR does not reliably detect single fecal indicator bacteria in drinking water. Water Supply 2016, 16, 1674–1682. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, F.; Stedtfeld, R.D.; Waseem, H.; Williams, M.R.; Cupples, A.M.; Tiedje, J.M.; Hashsham, S.A. Most probable number—Loop mediated isothermal amplification (MPN-LAMP) for quantifying waterborne pathogens in <25 min. J. Microbiol. Methods 2017, 132, 27–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duarte-Guevara, P.; Duarte-Guevara, C.; Ornob, A.; Bashir, R. On-chip PMA labeling of foodborne pathogenic bacteria for viable qPCR and qLAMP detection. Microfluid. Nanofluidics 2016, 20, 114. [Google Scholar] [CrossRef]
- Kibbee, R.J.; Örmeci, B. Development of a sensitive and false-positive free PMA-qPCR viability assay to quantify VBNC Escherichia coli and evaluate disinfection performance in wastewater effluent. J. Microbiol. Methods 2017, 132, 139–147. [Google Scholar] [CrossRef]
- Salam, K.W.; El-Fadel, M.; Barbour, E.K.; Saikaly, P.E. A propidium monoazide–quantitative PCR method for the detection and quantification of viable Enterococcus faecalis in large-volume samples of marine waters. Appl. Microbiol. Biotechnol. 2014, 98, 8707–8718. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Primers and Probes | Sequence 5′ → 3′ | Tm (°C) | Modification | Amplicon Length (bp) | Target Gene |
|---|---|---|---|---|---|---|
| E. coli | uidA_long_F | ATT TGA AGC CGA TGT CAC GC | 60.01 | 1.37 kbp | uidA | |
| uidA_long_R | TCC CTT TCT TGT TAC CGC CA | 59.71 | ||||
| uidA_mid_F | CCG ACG AAA ACG GCA AGA AA | 60.15 | 5′-biotin | 556 bp | ||
| uidA_mid_R | TCA GCG TAA GGG TAA TGC GA | 59.65 | 5′-phosphate | |||
| uidA_short_F | AGT CAA CGG GGA AAC TCA GCA A | 56.43 | 5′-biotin | 96 bp | ||
| uidA_short_R | GCA ATA CTC CAC ATC ACC ACG CTT | 57.79 | 5′-phosphate | |||
| process control | AGA ATC AAG GAG CAG ATG CTG AAA AAA | 5′-NH2, 3′-biotin | ||||
| uidA_DM1 | GTC CAC CCA GGT GTT CGG C | 5′-NH2-C12 | ||||
| uidA_DM2 | TTT TTT TTT TTT TTT GTC CAC CCA GGT GTT CGG C | 5′-NH2-C12 | ||||
| uidA_DS1 | TGG TTT TTG TCA CGC GCT ATC AGC | 5′-NH2-C12 | ||||
| uidA_DS2 | TTT TTT TTT TTT TTT TGG TTT TTG TCA CGC GCT ATC AGC | 5′-NH2-C12 | ||||
| Eco4 | TTT TTT TTT TTT TTT GAATCACAAAGTGGTAAGCG | 5′-NH2-C12 | ||||
| Legionella 1 | forward | GAGGGTTGATAGGTTAAG | 52 | 5′-biotin | 167 bp | legU |
| reverse | CCAGGAATTTCACAGATA | 49 | 5′-phosphate | |||
| mdx74 | CTTAATCAACCACCTACGCAC | 68 | 5′-NH2-C12-Poly-T | |||
| Legionella 2 | forward | CCGATGCCACATCATTAGC | 57 | 5′-biotin | 150 bp | mipN |
| reverse | CCAATTGAGCGCCACTCATAG | 61 | 5′-phosphate | |||
| mdx84 | TGCCTTTAGCCATTGCTTCCG | 69 | 5′-NH2-C12-Poly-T | |||
| Clostridium | forward | ATGATTGGGATTATGCAGCAAAGGT | 63 | 5′-biotin | 112 bp | cpa |
| reverse | CCAACTGATGGATCATTACCCTCTG | 66 | 5′-phosphate | |||
| mdx40 | TCTATAAATATATCCTGCTGTTCCTT | 67 | 5′-NH2-C12-Poly-T | |||
| Yersinia | forward | AACAGTTTCAGGGCAGTTCAGTG | 63 | 5′-biotin | 128 bp | yst |
| reverse | AACATACATCGCAGCAATCCCAAT | 62 | 5′-phosphate | |||
| mdx43 | CGACACCAATAACCGCTGAG | 68 | 5′-NH2-C12-Poly-T |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lehniger, L.; Rudloff, A.; Pollok, S.; Große, N.; Wessel, K.; Brendel, M.; Popp, J.; Weber, K. A Model System for Sensitive Detection of Viable E. coli Bacteria Combining Direct Viability PCR and a Novel Microarray-Based Detection Approach. Chemosensors 2021, 9, 357. https://0-doi-org.brum.beds.ac.uk/10.3390/chemosensors9120357
Lehniger L, Rudloff A, Pollok S, Große N, Wessel K, Brendel M, Popp J, Weber K. A Model System for Sensitive Detection of Viable E. coli Bacteria Combining Direct Viability PCR and a Novel Microarray-Based Detection Approach. Chemosensors. 2021; 9(12):357. https://0-doi-org.brum.beds.ac.uk/10.3390/chemosensors9120357
Chicago/Turabian StyleLehniger, Lydia, Anne Rudloff, Sibyll Pollok, Norman Große, Kristin Wessel, Monique Brendel, Jürgen Popp, and Karina Weber. 2021. "A Model System for Sensitive Detection of Viable E. coli Bacteria Combining Direct Viability PCR and a Novel Microarray-Based Detection Approach" Chemosensors 9, no. 12: 357. https://0-doi-org.brum.beds.ac.uk/10.3390/chemosensors9120357