1. Introduction
The study of weak van der Waals (vdW) interactions has always been one of the most challenging applications of theoretical calculations of electron structure. Thus, methods based on Kohn–Sham (KS) density functional theory (DFT) have shown limitations for including weak and long-range interactions in the exchange-correlation term of the KS equation. In particular, standard functionals of DFT fail to explain these interactions because the stabilization is determined by dispersion interactions, and is not explained by these functionals. Exchange-correlation potentials, derived from local and semi-local models, often exhibit artifacts when applied to systems with large non-local correlation effects. Nevertheless, there is a continuous effort to include vdW interactions within the framework of KS theory [
1], and remarkable progress on corrections to this fact has been made, such as the exchange-hole dipole moment (XDM) model [
2,
3].
In the long range, vdW interactions are dominated by dispersion and permanent multipole moment interaction and include superposition and exchange contributions. The behavior of an exchange functional in the region of small density and large density gradients plays a very important role. From a topological point of view, the presence of vdW interactions can be identified by reduced density gradient (RDG) analysis. In this respect, a novel procedure for studying van der Waals interactions has been developed as an extension of Bader’s QTAIM in combination with RDG analysis in which a volumetric source function is used for describing the atomic composition of vdW interactions [
4].
The above considerations reinforce that the well known fact is that to reliably study vdW interactions, not only large basis sets (BS) are needed, but also fine electron correlation treatments are required to reproduce experimental data. In particular, noble gases have been the focus of much research as a starting point for the study of rare gas dimer interactions [
5], a criterion for the performance of basis sets and correlation methods in vdW studies.
Among the systems linked by vdW dispersion interactions, helium occupies an outstanding position. The properties of gaseous helium are close to those of the ideal gas because the interactions between helium atoms are extremely weak, and its behavior at very low temperatures makes helium a paramount system on its own, with unusual physicochemical properties under these conditions. Furthermore, vdW interaction in helium dimer, He
, is a touchstone for testing the capabilities of the most precise theoretical procedures available [
6], as its existence is due to purely electronic correlation effects.
The accurate computation of the He
potential energy curve is a big challenge, and a great amount of work has been devoted to this task. Ab initio studies on He
interactions have been published by Van Mourik et al., who report dimer calculations using different methods and computational levels (MPn, CCSD(T), FCI, …) with large basis sets, including polarization [
7,
8,
9]. In the late 20th and 21st centuries, many other authors have published works on He
, using various sets of a consistent basis for correlation and a high level of theory: SAPT [
10], CCSD(T) [
11], r12-MR-ACPF [
12,
13,
14], MRCI [
15], CCSD(T) [
16], Monte Carlo [
17], Compton profiles [
18] and Gaussian geminal theory [
19]. The most accurate results were obtained with BS supplemented with an additional set of bond functions [
20], obtaining better results when compared with larger BS without bond functions.
Because of the high accuracy intended and the feeble interaction involved, the question about the full elimination of basis set superposition error (BSSE) in the calculations of He
energy has been a matter of discussion [
21], which still remains today. This problem comes from the difficulty of saturating the dispersion energy in calculations with conventional basis sets, and it is especially relevant in weakly interacting species. A good representation of the dispersion energy requires polarization functions with small exponents, and extrapolation methods from the raw energies without Counterpoise (CP) correction have been proposed to reduce the BSSE [
22,
23,
24]. In particular, extrapolation to complete basis set (CBS) limit for the helium dimer was studied by A. Varandas [
22,
25], who carried out calculations with size-consistent methods such as Hartree–Fock (HF) and full configuration interaction (FCI) using Dunning BS.
As a consequence of this extraordinary effort, the accuracy achieved is certainly impressive and, according to the best estimations, the potential energy curve of He
has a minimum at a distance
Å, with an energy of
E
with respect to the limit of separated atoms [
20,
26]. This minimum in the potential energy curve is remarkably shallow and has been found to admit only one vibrational state, the mean distance between nuclei in this state being ca. ten times the equilibrium distance [
27].
In this work, we report a way of constructing new basis sets for molecular calculations which overcome the variational performance of the existing ones of equivalent composition. Furthermore, we apply the procedure to the development of basis sets, hereafter named sigma (BS), for helium atoms and use them for the study of helium atom and dimer.
The article is organized as follows. In
Section 2, the procedure for developing sigma BS is explained, and the contraction scheme and composition are described.
Supplementary Materials are included, which contain the link for the sigma basis sets for He. In
Section 3, a brief summary of computational procedures, methods, bases and programs is given. The results on the He atom and the He
dimer are reported in
Section 4, in which the the precision of the total and dissociation energies as well as the equilibrium distances is discussed. Results on the CBS extrapolation of correlation energies are included in a subsection of
Section 4. Finally, conclusions are drawn from these results in
Section 5.
2. Criteria for Basis Set Optimization and Contraction Scheme: Size and Composition
The
BS consists of linear combinations (contractions) of radial primitive Gaussians aimed at providing highly accurate energy values at different computational levels. In the construction of
BS, we have exploited our previous experience in the development of exponential type BS [
28,
29,
30,
31]. Guided by this experience, we have decided to design
BS with the characteristic that, if a given primitive contains a spherical harmonic of quantum number
, all the primitives with the same exponent and
are also present in the set. For instance, if there is a
d function with exponent
, then
s and
p functions with this exponent will appear in the BS. This characteristic was thought to reduce the computation cost of integrals involving primitives, although most standard packages are usually not prepared to profit from it. Another feature of
BS is that all primitives in a given shell, i.e., with the same angular part, participate in all contractions of the same shell. The combination of both features makes it possible for the number of primitives in the contractions can be increased, and the quality of the BS functions is thus improved, without penalizing the computational cost. In summary, contractions in
BS are built from the same set of exponentials combined with different angular functions. Furthermore, whereas polarization functions of Dunning BS consist of single Gaussian functions (one primitive per function without contraction), in sigma basis sets they are true contractions, yielding significant improvements in the results on energy.
As a rule of thumb, the number of primitives included in each shell of polarization functions is equal to the number of contractions in the shell plus two. As mentioned before, the radial parts of the primitives used in the polarization functions are also present in the functions of the core shells. The choice of primitives for polarization functions, among those of core shells functions, is not obvious and must be accomplished by optimizing the exponents together for both types of functions. It is also noticeable that, albeit not specifically intended, the exponents of the primitives thus obtained almost follow an even tempered sequence, with slight variations and covering a wide range of values.
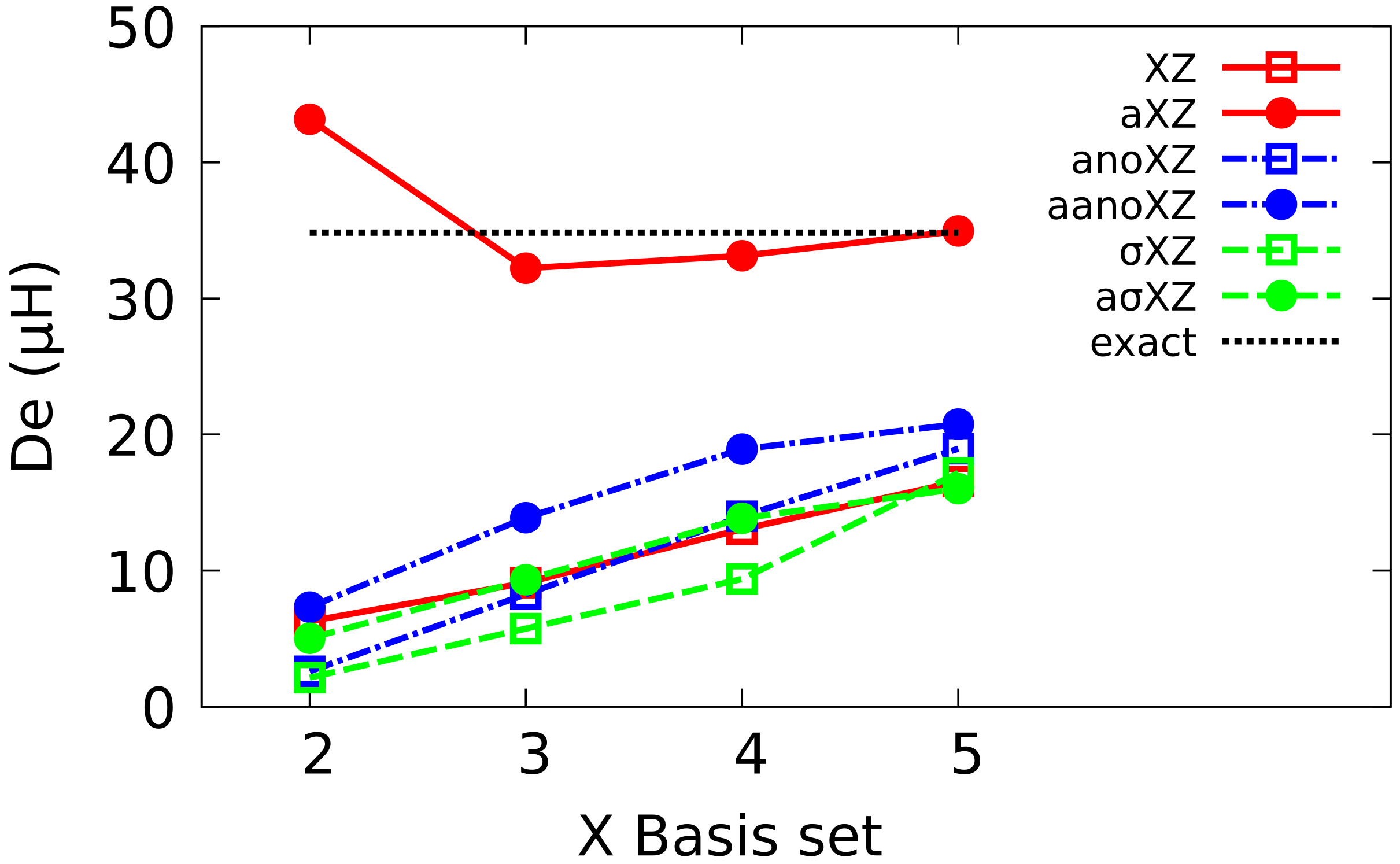
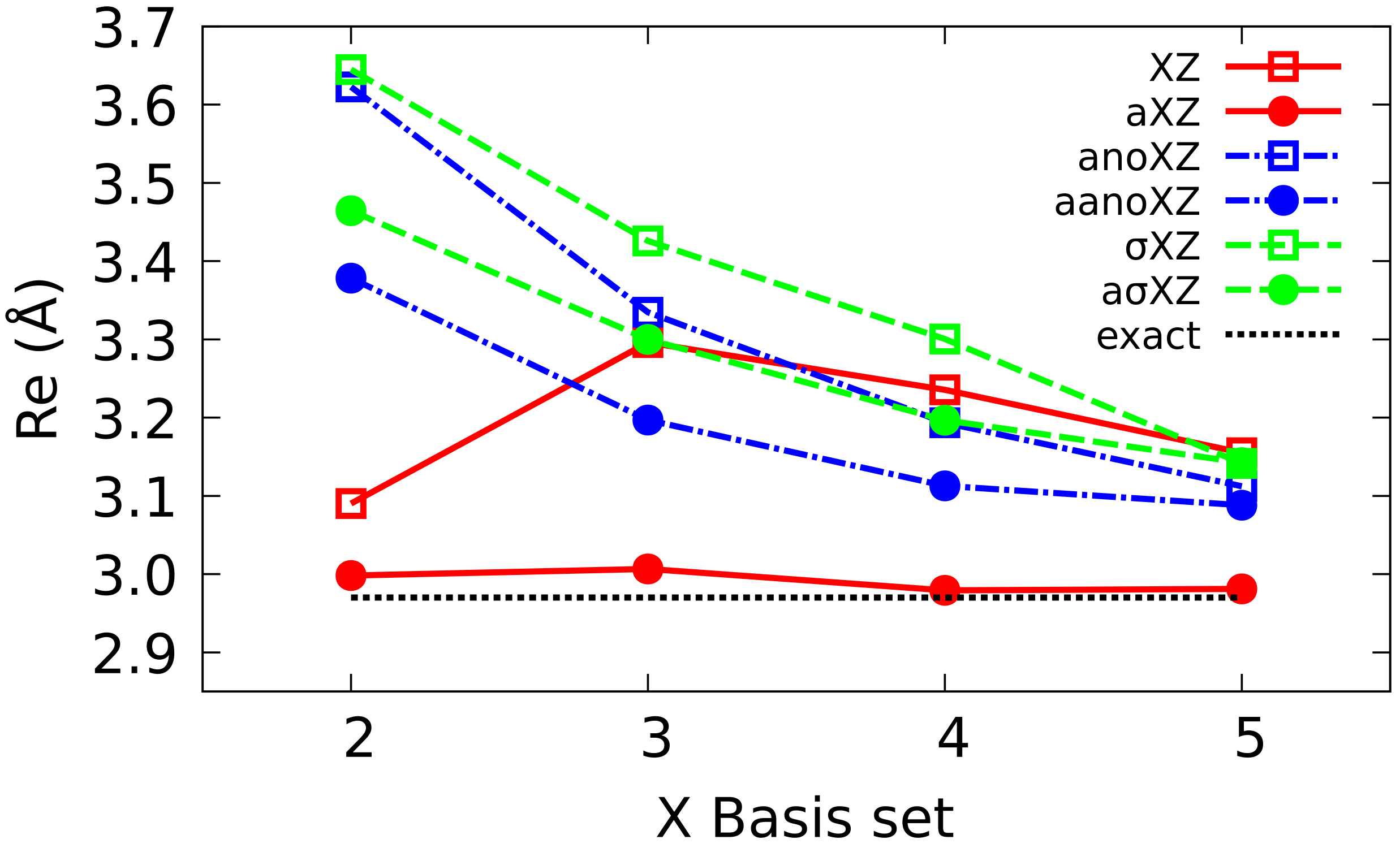
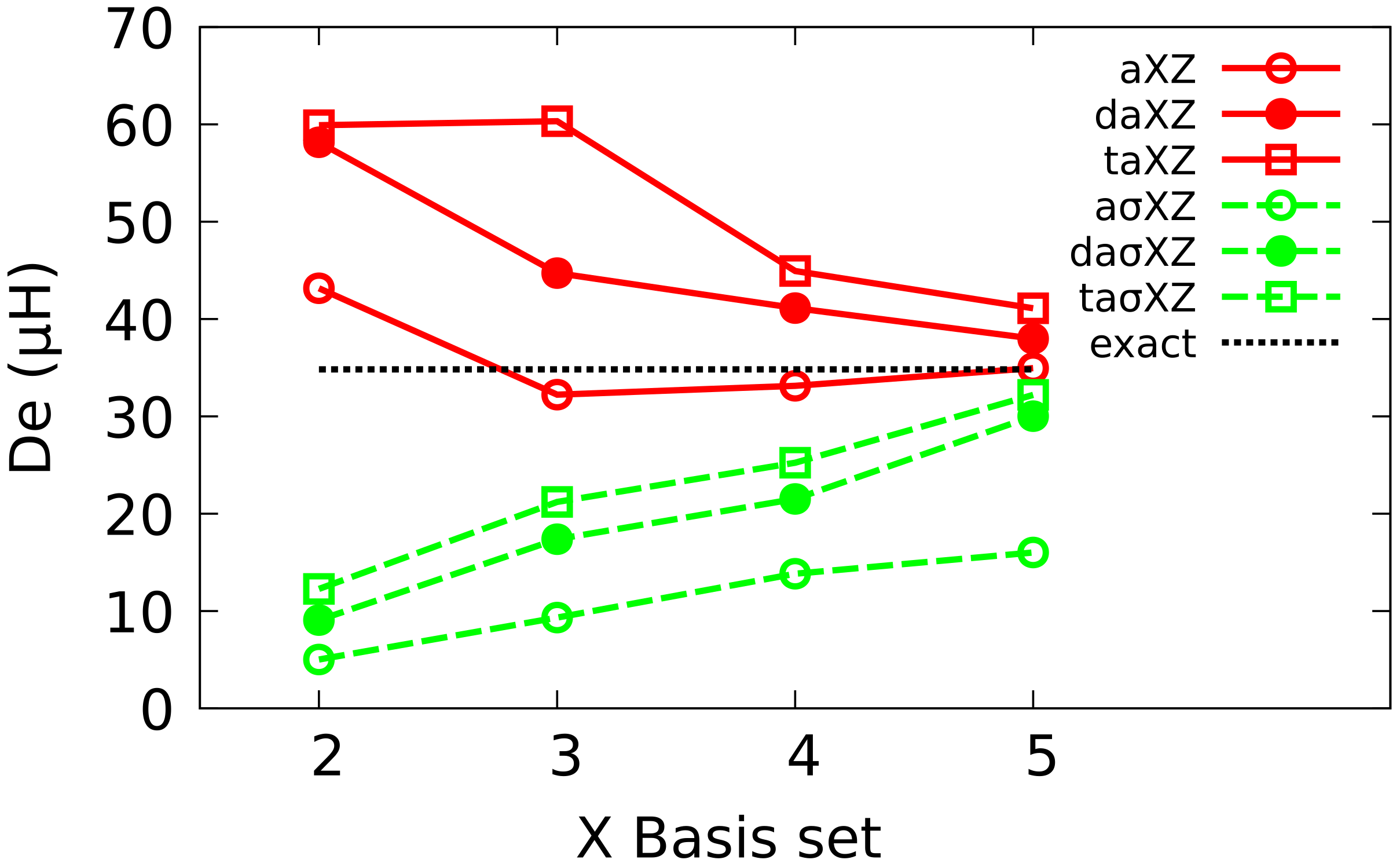
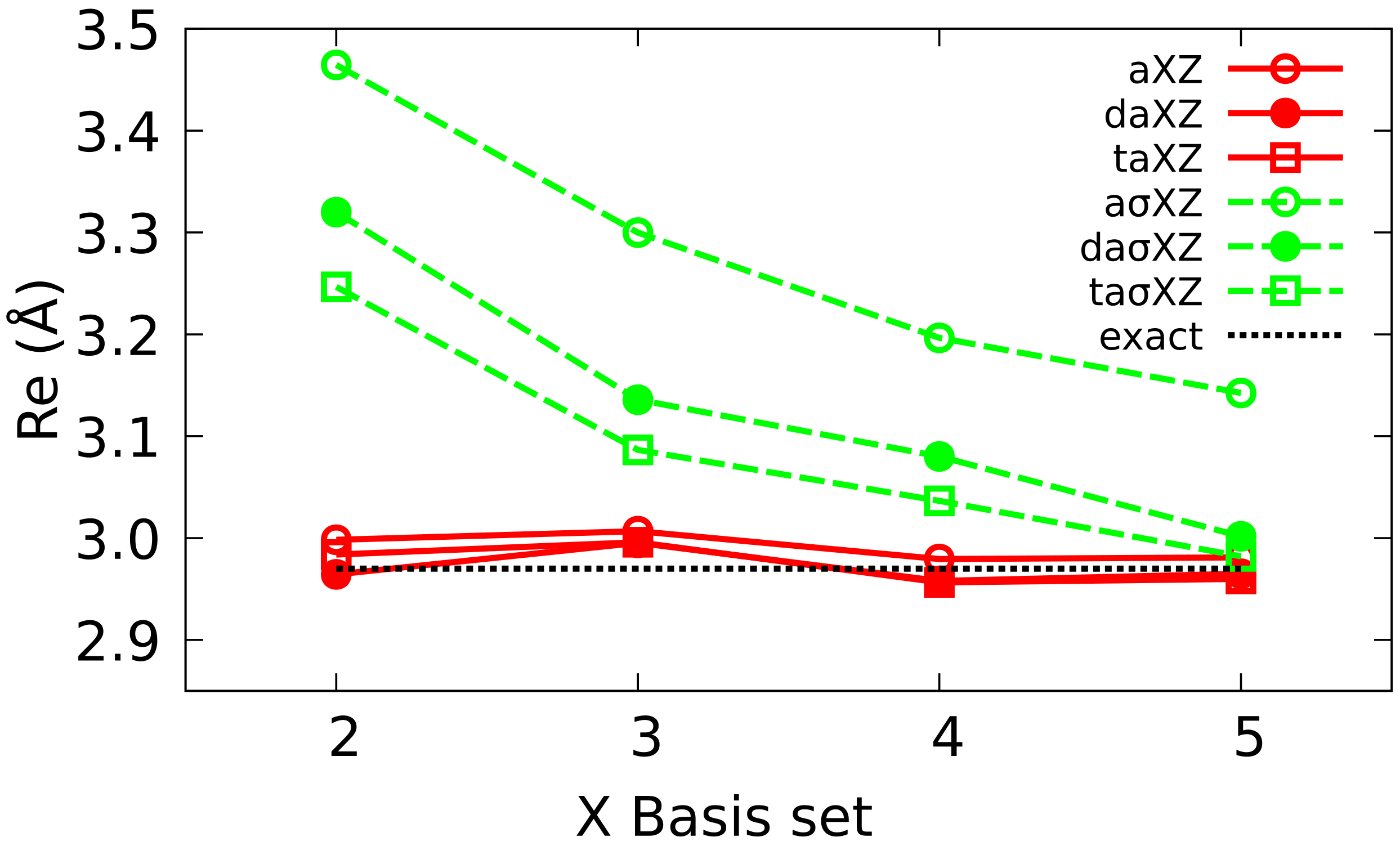
To simplify the notation, XZ will be used in the following as an abbreviation of cc-pVXZ, and aXZ of aug-cc-pcVXZ families. The equivalent Atomic Natural Orbitals (ANO) and BS will be denoted as anoXZ, aanoXZ, XZ and aXZ, respectively. In each of the six families, we have considered basis sets ranging from X = 2 (DZ) up to 5 (5Z).
The optimization of helium BS follows the general lines of Dunning’s procedure which relies on the minimization of CISD energy in He atoms. Starting with a given set of exponents in the primitives, the contractions are constructed in a stepwise way. In the case of He, the (1s) contraction of the DZ is determined by minimizing the HF energy for the ground state. Next, the CISD is used to add a new shell and one more contraction per shell, but keeping unchanged the contraction previously optimized. These two steps yield the DZ basis set. This procedure is repeated, changing the primitive exponents until their optimum values are obtained.
To build the TZ, we proceed as in the case of the DZ. After optimizing the 2(s) + 1(p) contractions, a new shell and a new contraction per shell, 1(s) + 1(p) + 1(d), are added and optimized with CISD of the He atom. The scheme for all BS follows the procedure described but repeating the steps of contraction/optimization as many times as required according to the BS level. Proceeding in this way, and taking into account that the number of primitives is increased according to the rule mentioned, BS tend to saturate one-electron space per shell, yielding energies as close as possible to the best attainable values according to their size.
In general, these
BS give energy values for He atoms lower than those of Dunning BS and similar to those of ANO BS, as we will see the next section. Unlike in Dunning and ANO BS, in the case of the augmented
BS (
BS), the CISD energy of the dimer at the best available equilibrium distance (BED)
Å [
26] is considered for the optimization of
BS exponents.
3. Computational Details
As it is well known, basis sets augmented with polarization and diffuse functions can adequately reproduce weak dispersion interactions [
9,
25,
32]. Bearing this in mind, to study He
interactions in a systematic way, we have carried out calculations using atom-centered basis sets. In particular, in this work we use correlation consistent basis sets developed by Dunning et al. [
32,
33,
34,
35] as a reference for testing the performance of
BS reported herein. Dunning BS are widely used, and they are especially well suited for our purposes because they incorporate polarization functions and, in the case of augmented versions, diffuse functions. In addition, it has been shown that correlation-consistent basis sets doubly augmented with diffuse functions can be used to nearly saturate the radial contribution to the dispersion energy in rare gas dimers [
8,
9,
36]. Furthermore, these basis sets are grouped in families whose members are ranked in size (and quality), and this facilitates the extrapolation of results to the CBS limit.
For testing the accuracy of energy values attained with the
BS, we have also used the ANO BS, which have proved to be able to exhaust the capabilities of the underlying Gaussian expansion basis (to minimize the contraction error) and provide a highly accurate reference [
37,
38].
Electronic structure calculations of He and He
have been carried out at HF, CISD and FCI levels, using MOLPRO suite [
39].
5. Conclusions
A new scheme for developing basis sets is reported and applied to helium atom and dimer. The BS thus developed are configured by combining the same set of exponentials with different angular functions and saturating the corresponding one-electron spaces. The new family ranges from DZ to 5Z, with the same composition as Dunning BS, and also includes, simply, double and triple augmented versions. Energy values for helium atom and diatom computed with the BS are lower than those obtained with their partners in Dunning BS, and similar to ANO BS, both at HF and FCI levels.
The analysis of the energy of He reveals the presence of a minimum in the curve at the HF level in the case of Dunning BS that does not appear with or ANO BS. In the case of FCI, all calculations yield a well whose depth tends to the reference value as the BS quality improves. Augmented BS display a good convergence which, in the case of multiple augmented BS, is even more regular than in the corresponding Dunning BS.
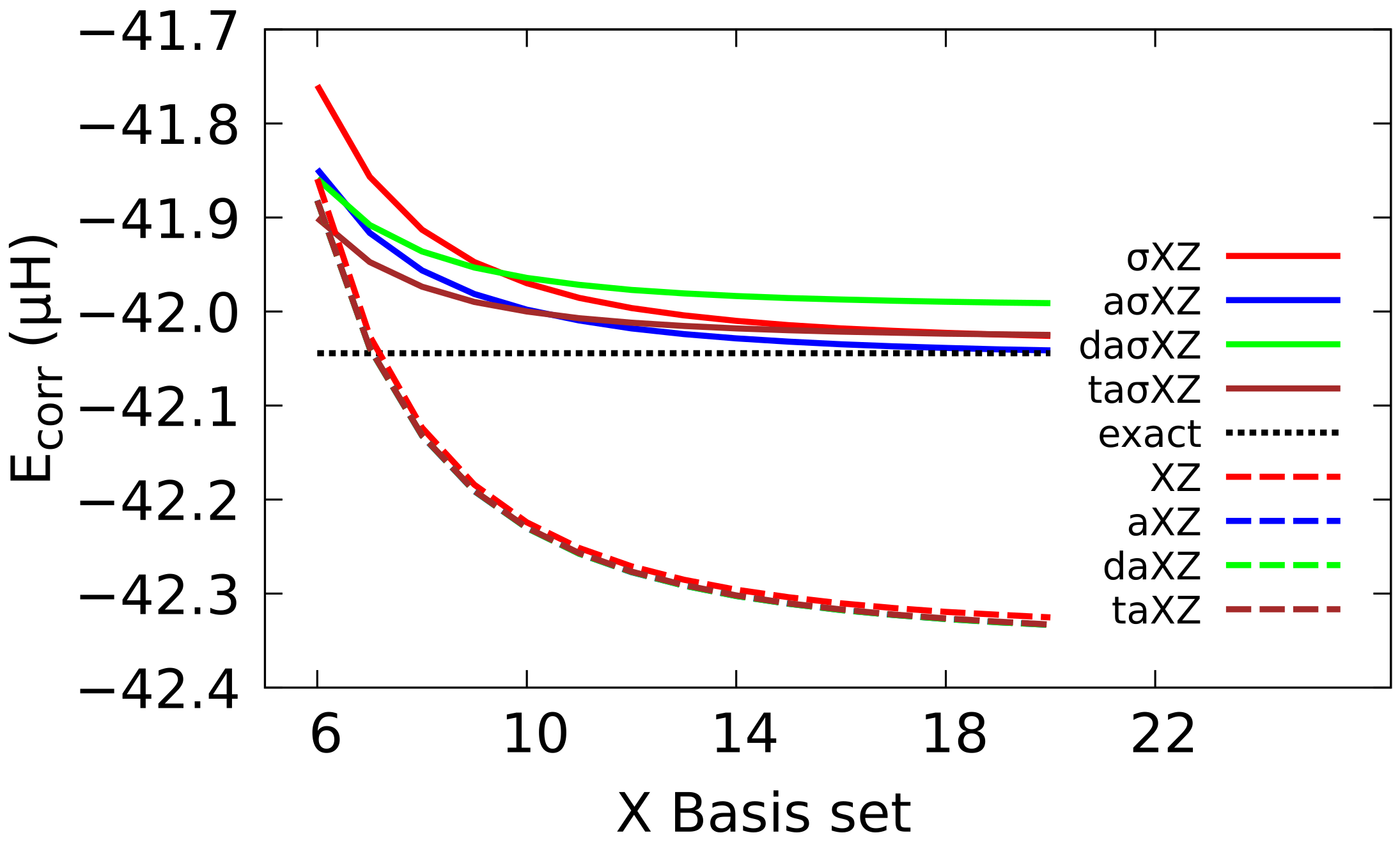
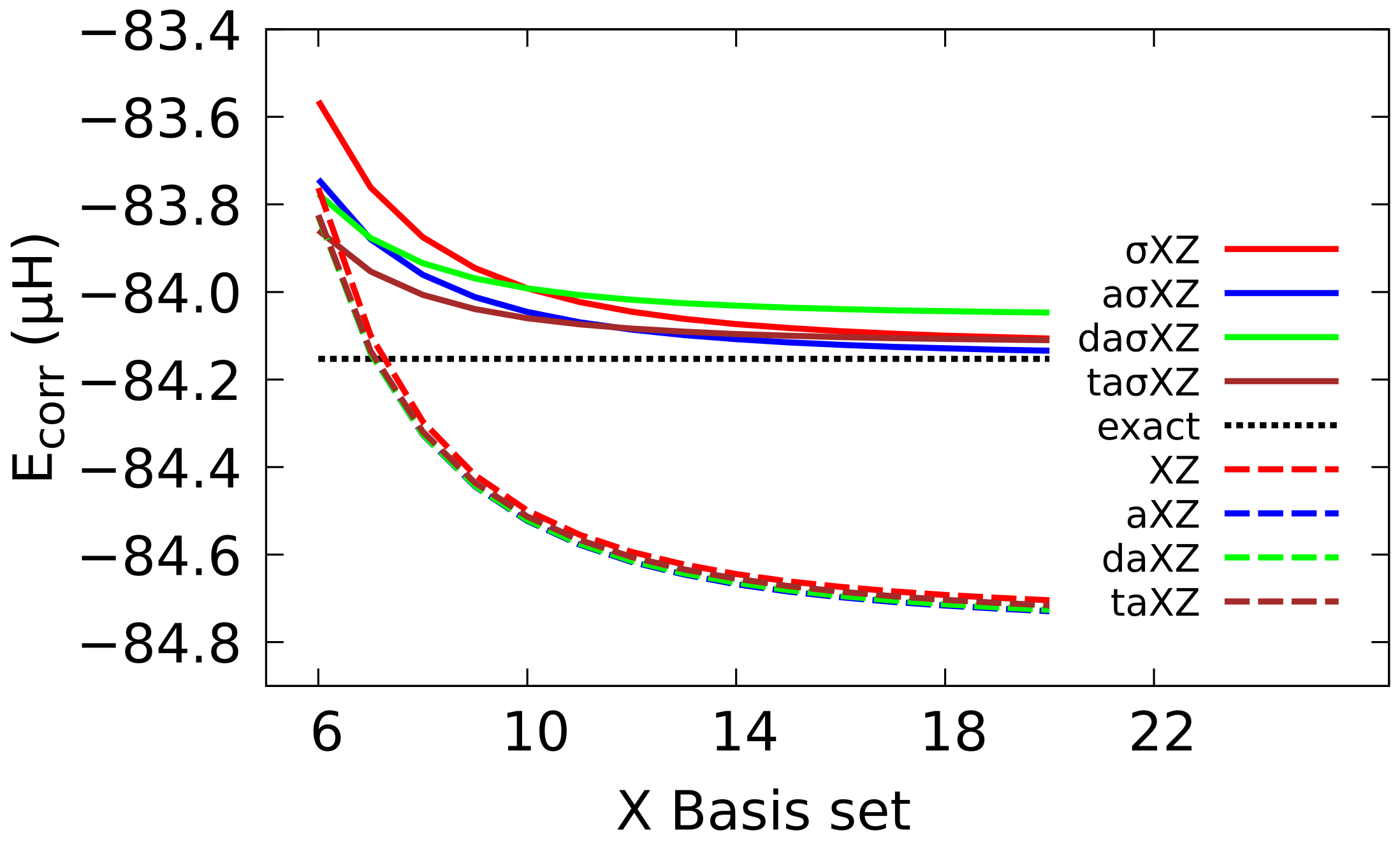
CBS extrapolation of the correlation energy of He at BED has also been examined, proving that the results with BS are clearly superior to those attained with Dunning BS. Especially remarkable is the result with the aXZ, which shows an agreement of five decimal figures with the best available result. Furthermore, it can be noticed that the CBS extrapolation with Dunning BS falls below the exact value, while the sigma converges to the right value.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}