3.1. Pd and Rh Induced Structural, Electronic, and Energetic Property Modifications of Au(111)
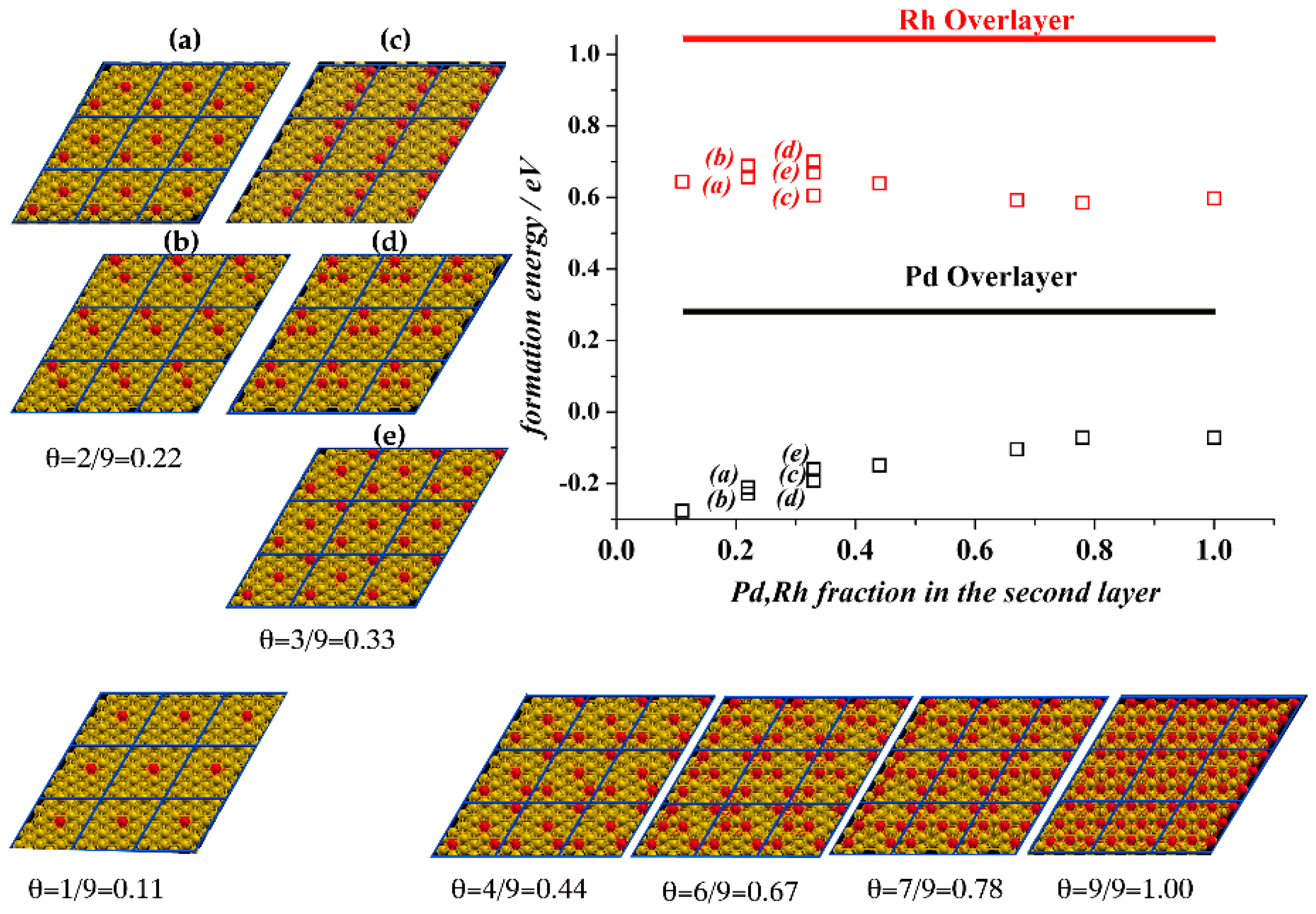
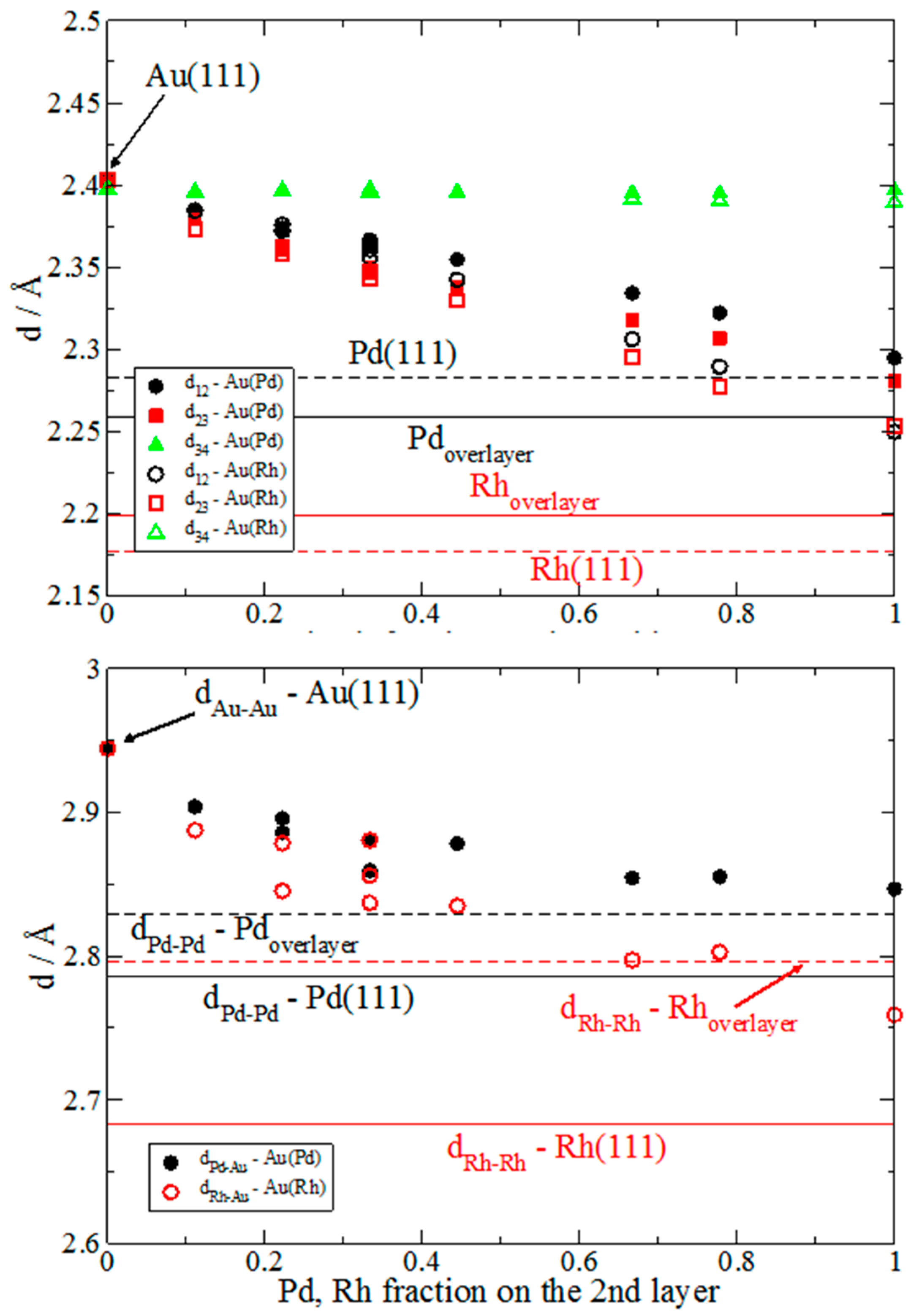
We first discuss the variation of the structural features of Au(111) because of the various concentrations of Rh and Pd dopants in the sublayers. The considered structures are drawn in
Figure 1 and the distances between the layers are plotted in
Figure 2 with the following labeling:
d12 is the distance between the top and the second layers;
d23 and
d34 are the separations between the second and the third layers, and the third and the fourth layers, respectively. Comparing these values in the ideal and doped Au(111) slab, it is first noting that the substitution of only one Au atom in the second layer (in the 3 × 3 cell) by Pd and Rh leads to bonding contractions. The Pd impurity has three first gold neighbors at 2.903 Å, whereas Rh-Au distances are 2.888 Å. By consequence the Au-Au distances at the surface layer decrease, as well as the separation between the first and the second layers (
d12 values in
Figure 2) shrinks. The third and fourth layers remain nearly unaffected with
d23 and
d34 distances practically equal to those in the undoped gold slab as follows from the results. In all of the cases the distances between Au and Rh atoms are smaller than Pd-Au distances, which is an indication for stronger Au-Rh interactions.
The computed formation energies, reported also in
Figure 1 as an inset, reveal negative Δ
Eform for Au(Pd) alloy at all the studied concentrations. This means that substituting gold by Pd is energetically favorable. This is in agreement with the phase diagram of the Au-Pd alloy [
32] reporting a total miscibility between Au and Pd.
All Rh-substituted structures in
Figure 1 are characterized by positive formation energies. This demonstrates that the formation of substituted Rh slabs is endothermic and would not occur spontaneously. Indeed, the Au(Rh) alloy is known for its immiscibility and it is expected to form Rh segregates within Au-solid (phase separation). The preference of Rh atoms to cluster between themselves rather than to coordinate Au atoms can be explained by the cohesive energies in
Table 1. Due to the high cohesive energy of Rh compared to that of Pd (−5.77 eV/Å for Rh and −3.72 eV/Å for Pd), the Δ
Eform of Au(Rh) alloy is positive, while Δ
Eform for Au(Pd) is negative. As follows from the inset in
Figure 1, where Δ
Eform are plotted versus the increase of the dopants concentration, Δ
Eform of Au(Rh) does not vary and is nearly constant (within 0.05 eV/atom) with the increase of the sublayer Rh coverage, at variance to the Au(Pd) as discussed above.
The other case of doping distribution in the Au matrix, considered here, are the structures with dopants overlayer. The overlayer formation energies, also reported in the inset in
Figure 1 are significantly higher than those computed for the sublayer doped alloys. The overlayer is destabilized by 1.05 and 0.29 eV/atom for Rh and Pd, respectively. This is rather expected, because Au has a lower surface energy than Pd and Rh, and in consequence it tends to segregate at the surface. We can also notice that the segregation energy of a monolayer of Pd from the second to the first layer of the slab, i.e., the energy difference between the system with the Pd-substituted first layer and system with the Pd-substituted second layer is 0.36 eV/atom. This value is similar to the calculated segregation energy of one Pd impurity in the gold matrix [
12]. The computed segregation energy of Rh is 0.45 eV/atom, which is in line with the expected trend from the cohesion energies [
34].
The bonding between a complete underlayer of Rh or Pd atoms and the gold slab is analyzed by means of the layers’ binding energy and electronic density difference. The binding energy between Au and Pd or Rh was calculated from:
The first term corresponds to the energy of the bimetallic system, being completely relaxed. The second term is the energy of the bimetallic system without the second layer, but with the coordinates of the Au layers fixed to the values obtained relaxing the whole bimetallic system. The third term is the energy of a single Pd or Rh layer with the coordinates fixed to those of the relaxed layer in the bimetallic slab.
Table 2 collects the binding energies of Au-Au, Au-Pd, and Au-Rh layers calculated from a pure gold slab, and from a complete underlayer of Pd or Rh, respectively.
The bonding energy between the gold layers is higher by 1 eV compared to the binding energies of gold-palladium and gold-rhodium layers. As a possible reason, the increased electronic density between the new metal layer and the gold layers was pointed out [
19]. It is worth to note that despite the computed positive formation for the Au(Rh) alloy with an underlayer of Rh (vide supra), the Au-Rh binding between Au-Rh layers is stronger than that between Au-Pd ones. Several reasons for this discrepancy can be pointed out. An increased attraction between Au and X layers is most probably arising from an increased electron density between the two adjacent Au and X layers leading to density depletion between the remaining Au-Au layers, thus rendering the whole slab energetically less stable. Another possible reason could be the effect of the significantly larger cohesive energy of Rh (−5.8 eV/atom) compared to the cohesive energies of Au and Pd, being almost equal to −3.8 eV/atom (see the computed and experimental [
31,
33] values in
Table 1).
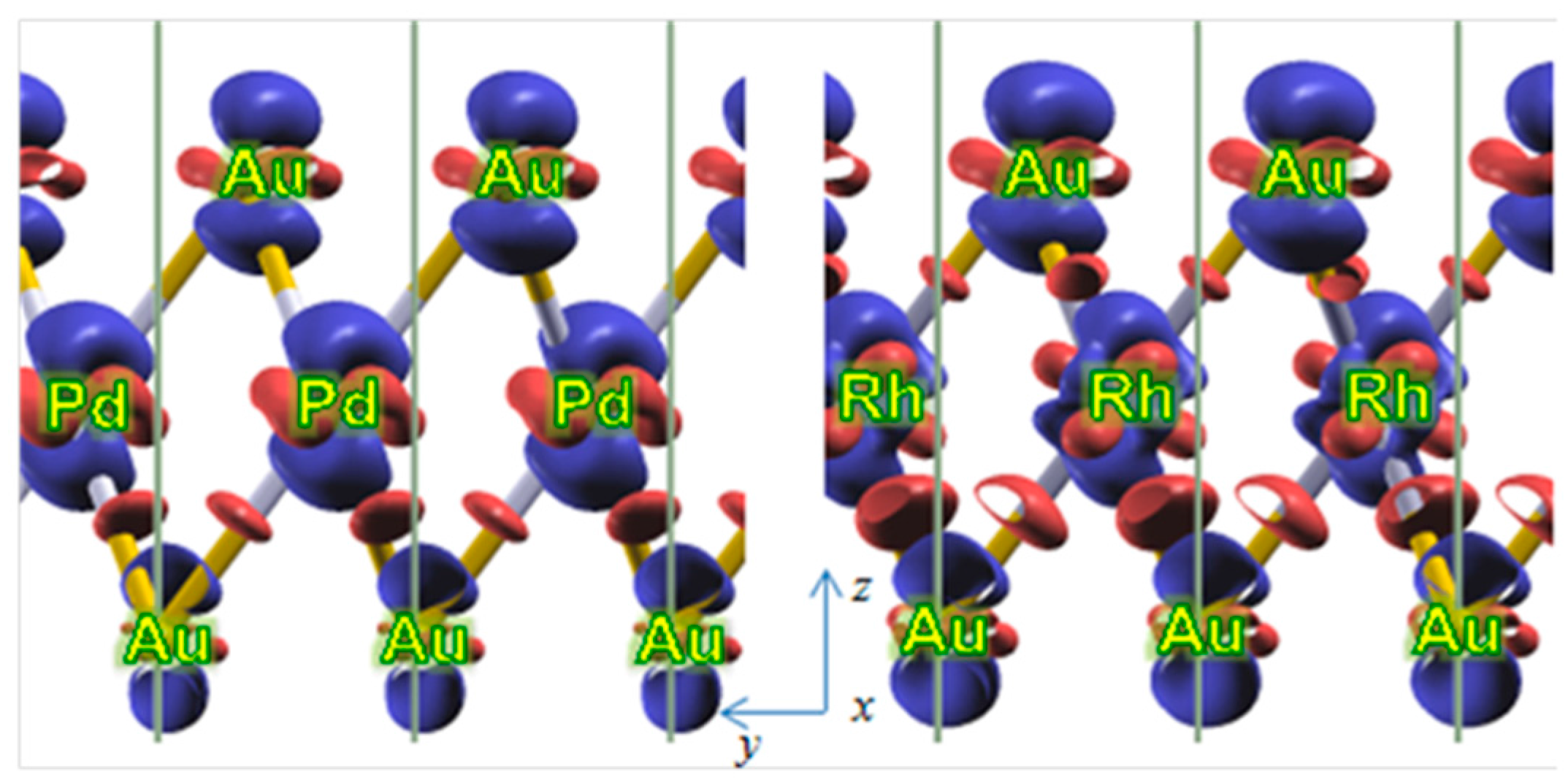
In order to understand whether significant electron density redistribution occurs, to which one can attribute the increased X-Au interactions, we have computed and analyzed the density charge differences, from which the electron density redistribution in the real space can be inferred. The charge differences computed as:
are shown in
Figure 3 for both Pd (left) and Rh (right) underlayers in Au(X) alloys. In Equation (5) ρ
Au/X/Au(111), ρ
Au/vacancy/Au(111), and ρ
X(2ndL) are the spatial electron charge density distributions of the relaxed bimetallic system, the Au(111) system without the second layer of Au with the coordinates fixed to the relaxed bimetallic system, and the isolated second layer of Pd or Rh with the coordinates of Pd or Rh in the bimetallic system.
This analysis reveals a charge depletion along
z-direction at both Au and X sites and a charge accumulation in the
xy-plane at the atomic sites as well as along the X-Au (inner, third Au- layer) bonds. Therefore, a charge distribution from
z- toward
xy-plane occurred because of the Au-substitution by Pd and Rh. The picture is similar for Pd and Rh layers, although a slightly higher charge accumulation along Rh-Au bonds is observed. There is not a charge accumulation between the surface gold layer and the Pd layer at variance to the Rh-Au (Au surface layer). The withdrawal of electron density toward gold atoms is explained by the higher Au electronegativity than that of Pd and Rh. The larger charge accumulation found for Rh-Au bonds favors the Au-Rh interlayer binding compared to the Pd-Au one. This agrees as well with the shorter Rh-Au than Pd-Au distances reported in
Figure 2. The charge density redistribution is observed only for the adjacent Au-X layers and not for the Au-Au layers (see
Figure 3). It follows that the dopants cause only locally a density drift from
z- to
xy- plane. This is the most likely reason for the gold-dopant layer stabilization and inter-atomic, and inter-layer geometrical modifications (vide supra) in the doped gold slab compared to the pure Au(111). The local in space electron density reorganization affects predominantly the adjacent dopant-gold layers and not the remaining gold layers in the gold slab.
3.2. Hydrogen Ab- and Adsorption
To study the reactivity of the doped Au(111) slab, hydrogen ab- and adsorption reactions were considered for the Au-X systems with a complete X monolayer underneath the Au-surface layer. For comparison, the hydrogen interaction with undoped Au(111) is also considered. Many previous investigations devoted to hydrogen ab- and adsorption on ideal monometallic surfaces, including Au(111), provided already sound information [
35,
36,
37,
38,
39]. Concerning the most favored adsorption sites, there is a general consensus that H adsorbs preferably on both hexagonal closed packed (hcp) and face-centered cubic (fcc) three-fold hollow sites on a large variety of metal surfaces. In this study we will also consider hcp and fcc sites because the absorption process should be different for H atoms diffusing to subsurfaces from both slab sites. To characterize different sites, first the adsorption energies of H atoms on top of the monometallic Rh, Pd, and Au surfaces were computed and are collected in
Table 3. Our results are in very good agreement with those reported previously [
36,
38,
39].
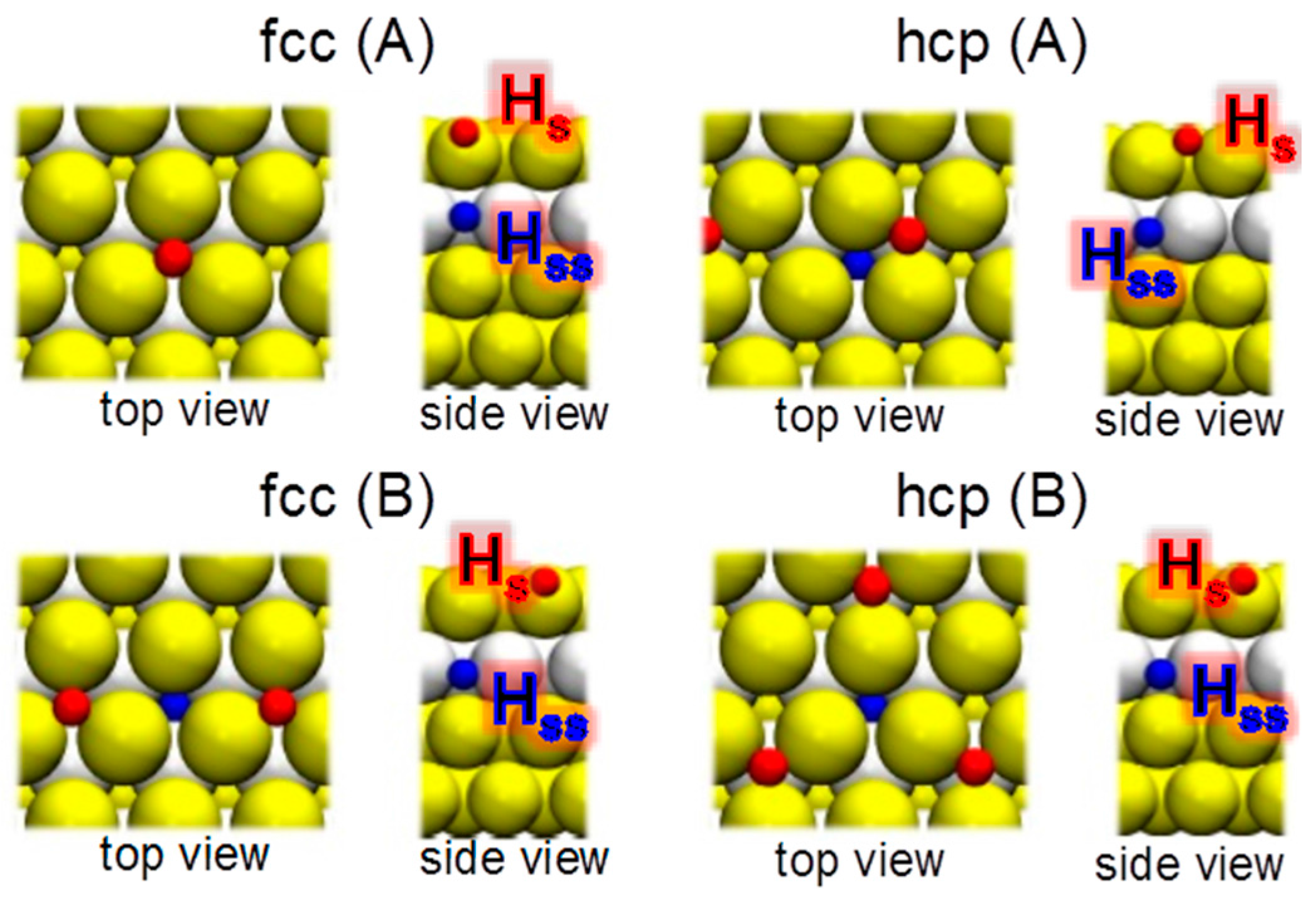
The hydrogen atoms adsorbed on the surface gold atoms (z
H > z
Au_top_layer) are labeled as H
s and those bound to the atoms of the underlying second layer (H-absorption; z
H < z
Au_top_layer) are labeled as H
ss. The computed binding energies for the ad- and absorption of one H atom are presented in
Table 3 and
Table 4, respectively. The adsorption energies on the bimetallic systems (without pre-absorbed H
ss underneath the surface) are the same as on the pure gold surface, within the DFT error. There is only a small decrease in the energy (and an increase in the bond length) in the Au(Rh) slab. When the H-coverage is increased, the H binding energies decrease. At low hydrogen coverage, the H-interaction with the second layer atoms (H-absorption) is higher than
Eads that indicates that H-penetration into the interstitial space is favored compared to the surface adsorption. The latter result is expected because H in the second layer interacts with three Pd or Rh dopant atoms and only one Au atom. It is, thus, not surprising that the interaction with the surface gold atoms remains weaker. Nevertheless those surface atoms are bound to the dopants in the second layer. This is in line with the above-discussed result that there is not an important charge drift in Au
surf-X bond region, contrary to the charge accumulation in the X-Au (3rd layer) space (see
Figure 3).
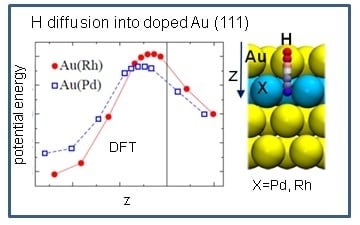
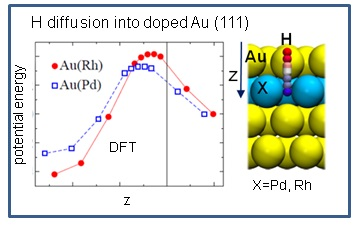
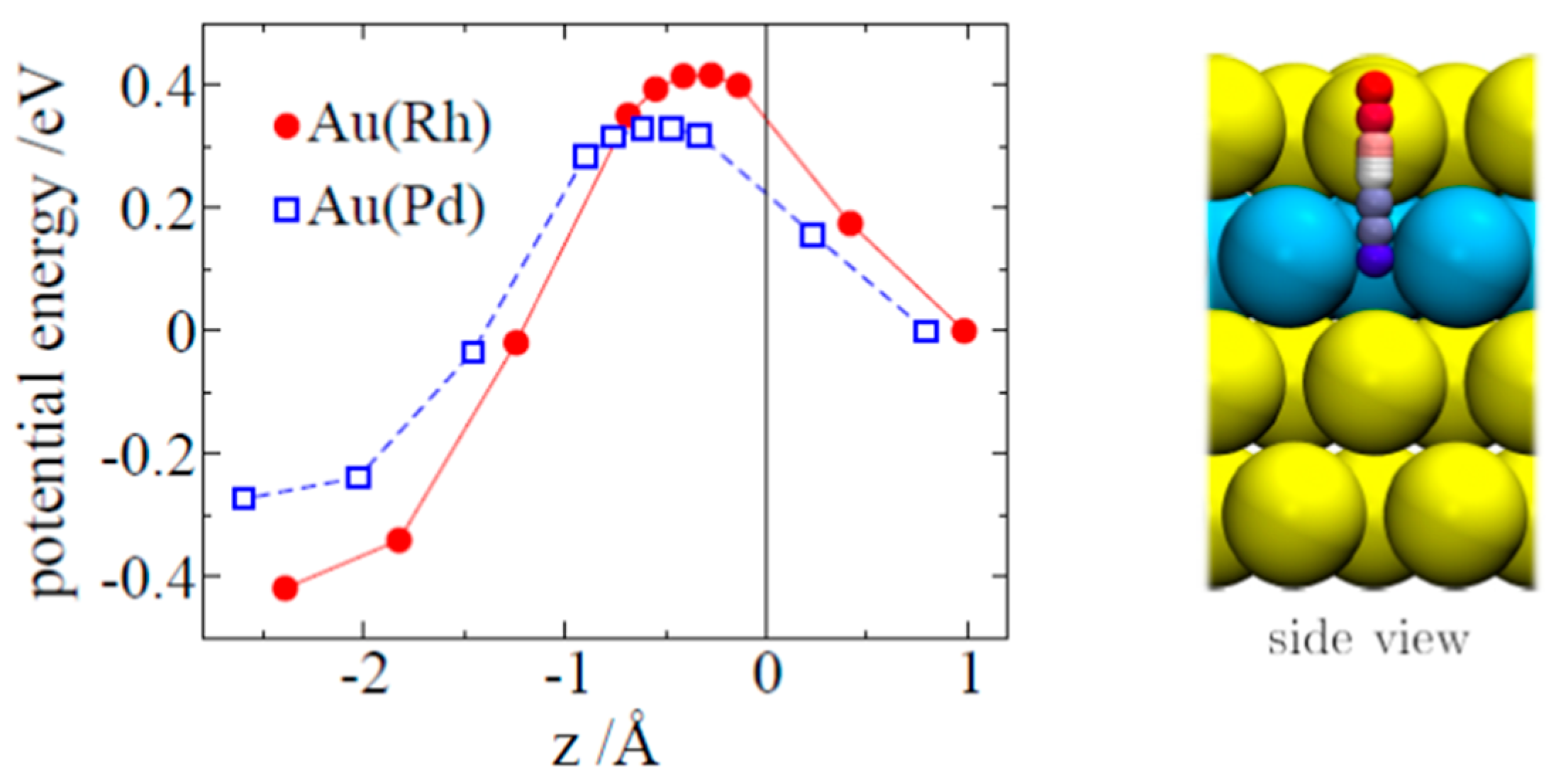
In addition to the minima structures, the effect of dopants on the hydrogen absorption energy was studied. The saddle points on the PES were localized by using the nudge elastic band technique as implemented in the VASP code.
Figure 4 shows the potential energy of hydrogen penetrating the alloy as a function of the z-coordinates of the hydrogen. The reaction and activation energies are collected in
Table 5. The activation energy for hydrogen absorption is 0.42 eV for Au(Rh) and 0.33 eV for Au(Pd), which is of the same order of magnitude as that on Pd(111) (see
Table 5). This is perfectly in line with the computed stronger Rh-Au bond energies (
Table 2), indicating that a strongly bound Rh to Au would rend the Au-Rh dimer less reactive.
We also studied the desorption process and found a significantly higher activation barrier (>0.6 eV), which demonstrates that the absorbed hydrogen would be hardly evacuated from the underneath layers. Further on, by comparing the adsorption energies on clean bimetallic surfaces (θ(H
ss) = 0.00) and on pre-H-covered surfaces (θ(H
ss) = 0.25, or 1.00), it appears that the effect of the underneath hydrogen atoms is weak. The 0.25 ML hydrogen precovered underlayer structures are shown in
Figure 5. Nevertheless, an increase in the bond strength (or a decrease in the adsorption energy) is observed for a complete layer of hydrogen atoms under the surface. This suggests that by changing the degree of hydrogen coverage one can modify the gold surface reactivity toward interaction with hydrogen and, thus modifying the reactivity of the bimetallic gold-covered electrodes.
Finally, the change in the work functions at high coverage was calculated and the results are reported in
Table 6. As expected the adsorption of hydrogen lowers the work function of the surface, whereas absorption increases it. When hydrogen atoms are located in/on the alloy surface the work function is lower and similar to that of the work function of the system with adsorbed hydrogen.


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}