
First Principle Modelling of Materials and Processes in Dye-Sensitized Photoanodes for Solar Energy and Solar Fuels

Abstract
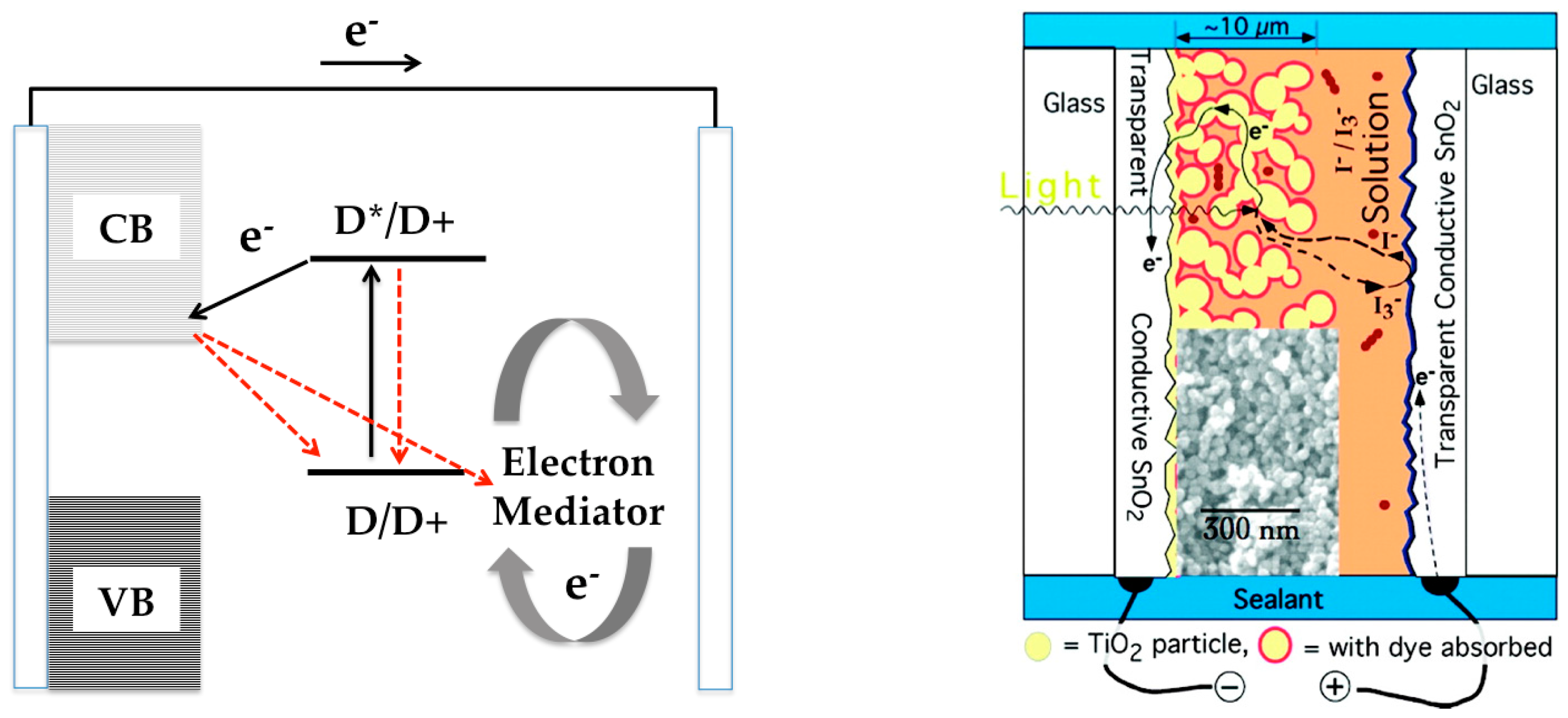
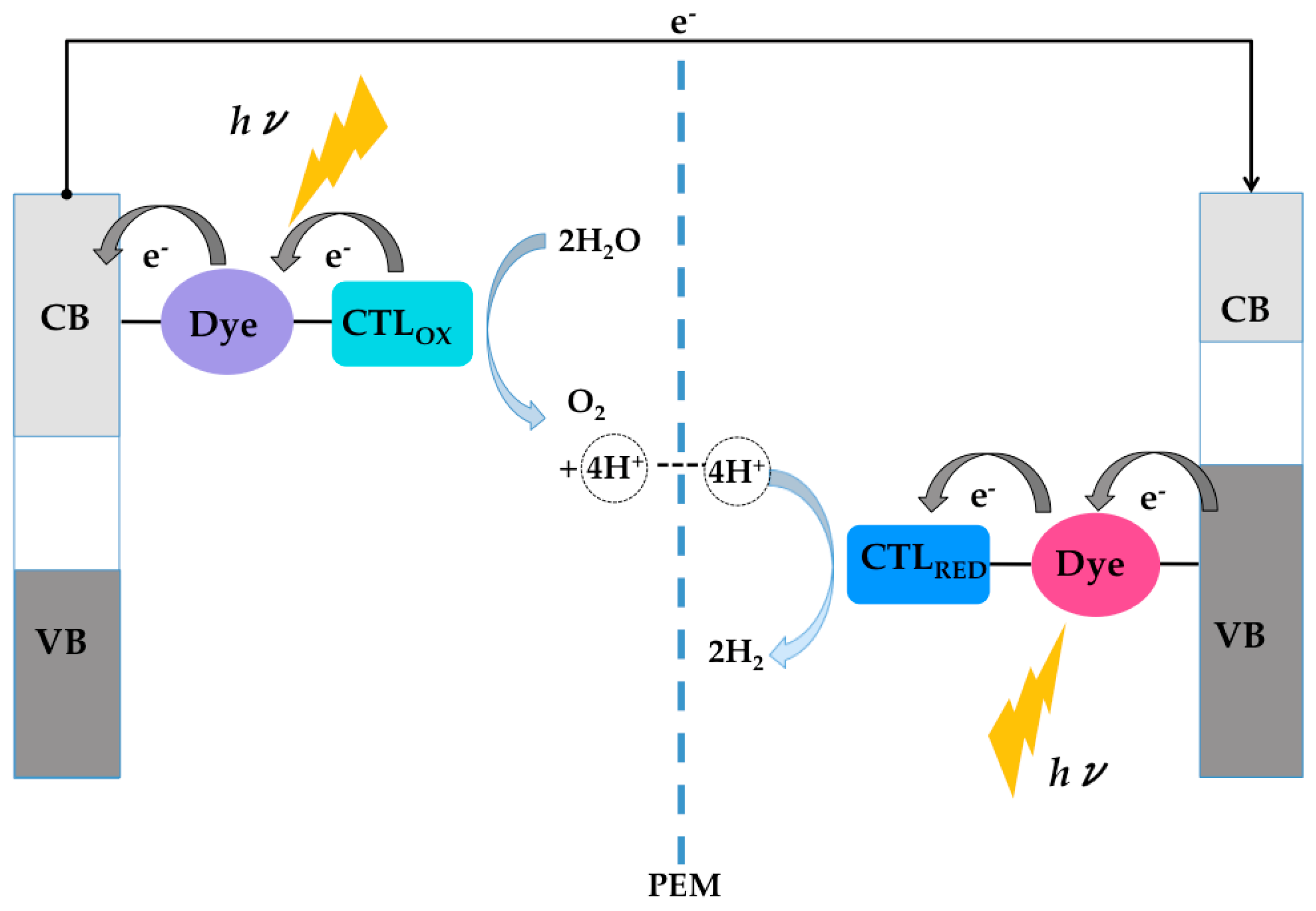
:1. Introduction
2. Dye’s Optical Properties
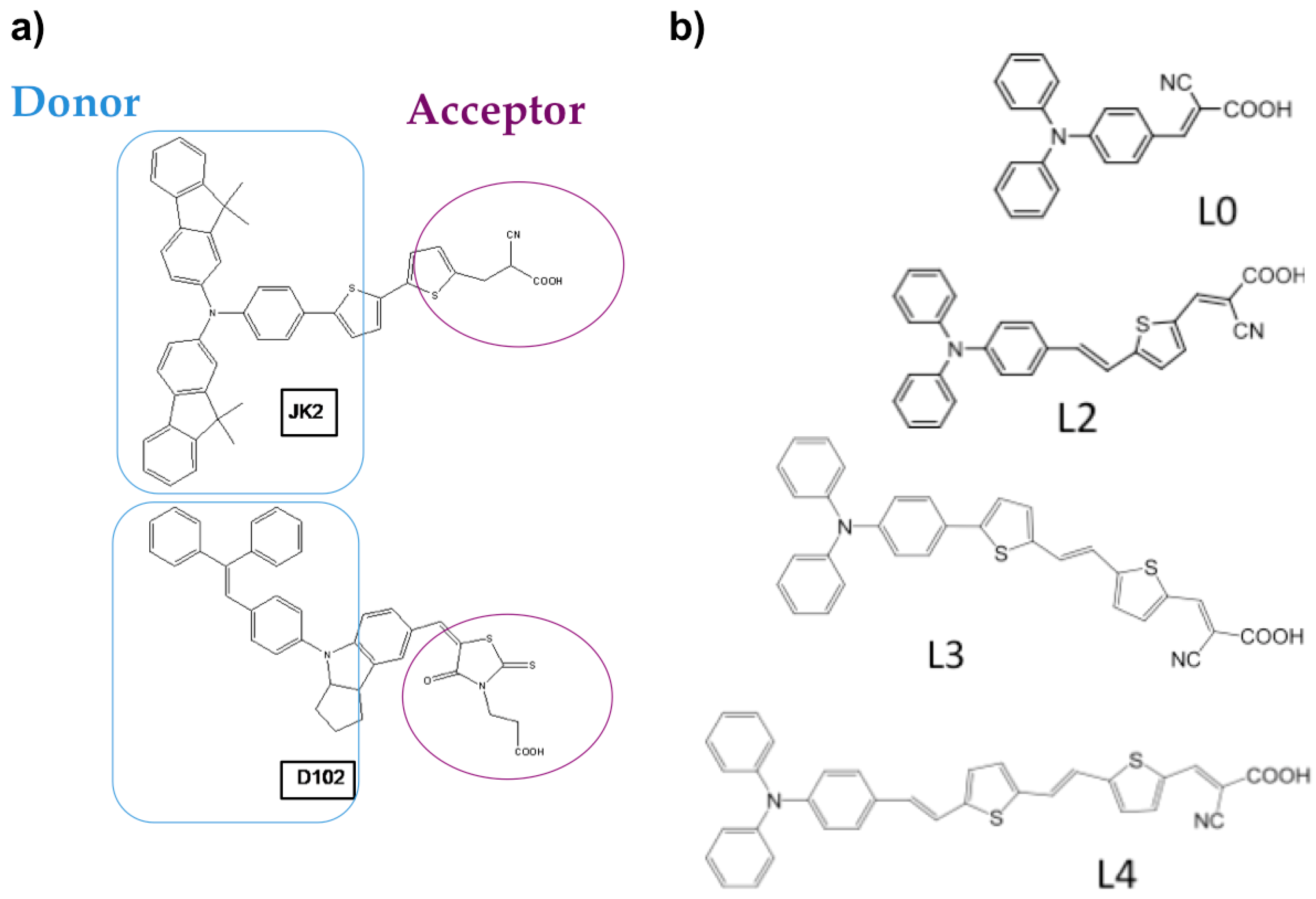
2.1. Organic Dyes
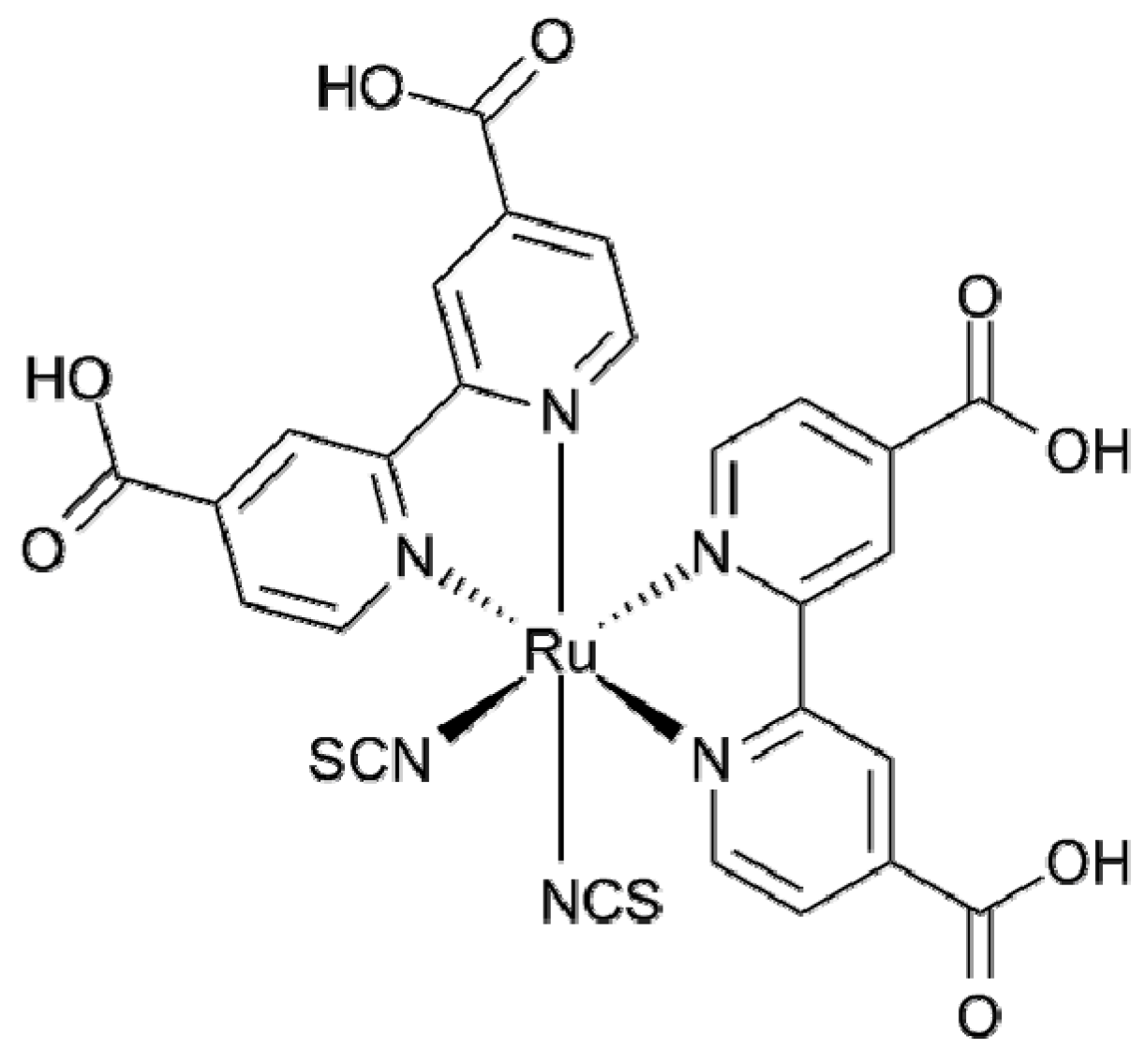
2.2. Transition Metal Complexes
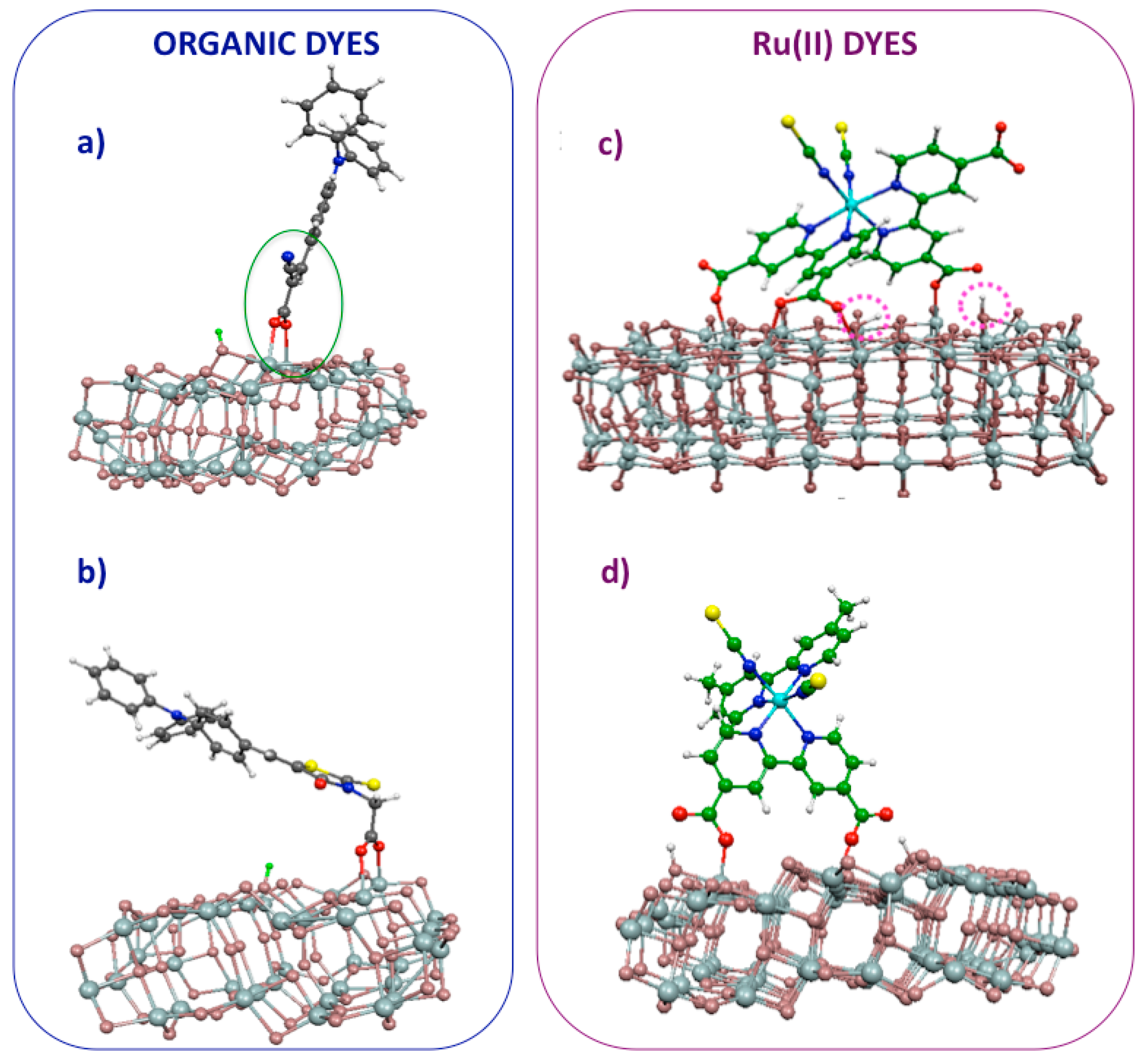
3. Dye/TiO2 Interfaces in DSCs
4. TiO2/Dye/Catalyst Multiple Interfaces in DSPECs
5. Conclusions
Conflicts of Interest
References
- Lewis, N.S.; Nocera, D.G. Powering the Planet: Chemical Challenges in Solar Energy Utilization. Proc. Natl. Acad. Sci. USA 2006, 103, 15729–15735. [Google Scholar] [CrossRef] [PubMed]
- Armaroli, N.; Balzani, V. Solar Electricity and Solar Fuels: Status and Perspectives in the Context of the Energy Transition. Chem. Eur. J. 2016, 22, 32–57. [Google Scholar] [CrossRef] [PubMed]
- Service, R.F. Is It Time to Shoot for the Sun? Science 2005, 309, 548–551. [Google Scholar] [CrossRef] [PubMed]
- Nocera, D.G. On the Future of Global Energy. Daedalus 2006, 135, 112–115. [Google Scholar] [CrossRef]
- Young, K.J.; Martini, L.A.; Milot, R.L.; Snoeberger, R.C., III; Batista, V.S.; Schmuttenmaer, C.A.; Crabtree, R.H.; Brudvig, G.W. Light-Driven Water Oxidation for Solar Fuels. Coord. Chem. Rev. 2012, 256, 2503–2520. [Google Scholar] [CrossRef] [PubMed]
- Hamann, T.W. Water Splitting: An Adaptive Junction. Nat. Mater. 2014, 13, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Concepcion, J.J.; House, R.L.; Papanikolas, J.M.; Meyer, T.J. Chemical Approaches to Artificial Photosynthesis. Proc. Natl. Acad. Sci. USA 2012, 109, 15560–15564. [Google Scholar] [CrossRef] [PubMed]
- Porter, G. Criteria for Solar Energy Conversion. In Light, Chemical Change and Life. A Source Book in Photochemistry; Coyle, J.D., Hill, R.R., Roberts, D.R., Eds.; Open University Press: Milton Keynes, UK, 1982; pp. 338–345. [Google Scholar]
- O’Regan, B.; Grätzel, M. A Low-Cost, High-Efficiency Solar Cell Based on Dye-Sensitized Colloidal TiO2 Films. Nature 1991, 353, 737–740. [Google Scholar] [CrossRef]
- Grätzel, M. Photoelectrochemical Cells. Nature 2001, 414, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.G.; Warren, E.L.; McKone, J.R.; Boettcher, S.W.; Mi, Q.; Santori, E.A.; Lewis, N.S. Solar Water Splitting Cells. Chem. Rev. 2010, 110, 6446–6473. [Google Scholar] [CrossRef] [PubMed]
- Alibabaei, L.; Brennaman, M.K.; Norris, M.R.; Kalanyan, B.; Song, W.; Losego, M.D.; Concepcion, J.J.; Binstead, R.A.; Parsons, G.N.; Meyer, T.J. Solar Water Splitting in a Molecular Photoelectrochemical Cell. Proc. Natl. Acad. Sci. USA 2013, 110, 20008–20013. [Google Scholar] [CrossRef] [PubMed]
- Boschloo, G.; Hagfeldt, A. Characteristics of the Iodide/Triiodide Redox Mediator in Dye-Sensitized Solar Cells. Acc. Chem. Res. 2009, 42, 1819–1826. [Google Scholar] [CrossRef] [PubMed]
- Hagfeldt, A.; Boschloo, G.; Sun, L.; Kloo, L.; Pettersson, H. Dye-Sensitized Solar Cells. Chem. Rev. 2010, 110, 6595–6663. [Google Scholar] [CrossRef] [PubMed]
- O’Regan, B.C.; Durrant, J.R. Kinetic and Energetic Paradigms for Dye-Sensitized Solar Cells: Moving from the Ideal to the Real. Acc. Chem. Res. 2009, 42, 1799–1808. [Google Scholar] [CrossRef] [PubMed]
- Pastore, M.; Etienne, T.; De Angelis, F. Structural and Electronic Properties of Dye-Sensitized TiO2 for Solar Cell Applications: From Single Molecules to Self-Assembled Monolayers. J. Mater. Chem. C 2016, 4, 4346–4373. [Google Scholar]
- Li, L.-L.; Diau, E.W.-G. Porphyrin-Sensitized Solar Cells. Chem. Soc. Rev. 2013, 42, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Clifford, J.N.; Martinez-Ferrero, E.; Viterisi, A.; Palomares, E. Sensitizer Molecular Structure-Device Efficiency Relationship in Dye Sensitized Solar Cells. Chem. Soc. Rev. 2011, 40, 1635–1646. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Guillen, E.; Kavan, L.; Gratzel, M.; Nazeeruddin, M.K. Metal Free Sensitizer and Catalyst for Dye Sensitized Solar Cells. Energy Environ. Sci. 2013, 6, 3439–3466. [Google Scholar] [CrossRef]
- Bozic-Weber, B.; Constable, E.C.; Housecroft, C.E. Light Harvesting with Earth Abundant D-Block Metals: Development of Sensitizers in Dye-Sensitized Solar Cells (Dscs). Coord. Chem. Rev. 2013, 257, 3089–3106. [Google Scholar] [CrossRef]
- Imahori, H.; Umeyama, T.; Ito, S. Large Π-Aromatic Molecules as Potential Sensitizers for Highly Efficient Dye-Sensitized Solar Cells. Acc. Chem. Res. 2009, 42, 1809–1818. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wu, J.; Desai, U.V.; Gao, D. High-Efficiency Solid-State Dye-Sensitized Solar Cells Based on TiO2-Coated Zno Nanowire Arrays. Nano Lett. 2012, 12, 2420–2424. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, Z.; Shi, J.; Yu, Y. One-Dimensional Titanium Dioxide Nanomaterials: Nanowires, Nanorods, and Nanobelts. Chem. Rev. 2014, 114, 9346–9384. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Grätzel, C.; Zakeeruddin, S.M.; Grätzel, M. Recent Developments in Redox Electrolytes for Dye-Sensitized Solar Cells. Energy Environ. Sci. 2012, 5, 9394–9405. [Google Scholar] [CrossRef]
- Wu, J.; Lan, Z.; Lin, J.; Huang, M.; Huang, Y.; Fan, L.; Luo, G. Electrolytes in Dye-Sensitized Solar Cells. Chem. Rev. 2015, 115, 2136–2173. [Google Scholar] [CrossRef] [PubMed]
- Rüttinger, W.; Dismukes, G.C. Synthetic Water-Oxidation Catalysts for Artificial Photosynthetic Water Oxidation. Chem. Rev. 1997, 97, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Esswein, A.J.; Nocera, D.G. Hydrogen Production by Molecular Photocatalysis. Chem. Rev. 2007, 107, 4022–4047. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Eisenberg, R. Catalysts Made of Earth-Abundant Elements (Co, Ni, Fe) for Water Splitting: Recent Progress and Future Challenges. Energy Environ. Sci. 2012, 5, 6012–6021. [Google Scholar] [CrossRef]
- Yu, Z.; Li, F.; Sun, L. Recent Advances in Dye-Sensitized Photoelectrochemical Cells for Solar Hydrogen Production Based on Molecular Components. Energy Environ. Sci. 2015, 8, 760–775. [Google Scholar] [CrossRef]
- Hardin, B.E.; Snaith, H.J.; McGehee, M.D. The Renaissance of Dye-Sensitized Solar Cells. Nat. Photonics 2012, 6, 162. [Google Scholar] [CrossRef]
- Yen, Y.-S.; Chou, H.-H.; Chen, Y.-C.; Hsu, C.-Y.; Lin, J.T. Recent Developments in Molecule-Based Organic Materials for Dye-Sensitized Solar Cells. J. Mater. Chem. 2012, 22, 8734–8747. [Google Scholar] [CrossRef]
- Coutard, N.; Kaeffer, N.; Artero, V. Molecular Engineered Nanomaterials for Catalytic Hydrogen Evolution and Oxidation. Chem. Commun. 2016, 52, 13728–13748. [Google Scholar] [CrossRef] [PubMed]
- Brennaman, M.K.; Dillon, R.J.; Alibabaei, L.; Gish, M.K.; Dares, C.J.; Ashford, D.L.; House, R.L.; Meyer, G.J.; Papanikolas, J.M.; Meyer, T.J. Finding the Way to Solar Fuels with Dye-Sensitized Photoelectrosynthesis Cells. J. Am. Chem. Soc. 2016, 138, 13085–13102. [Google Scholar] [CrossRef] [PubMed]
- Bazzan, G.; Deneault, J.R.; Kang, T.-S.; Taylor, B.E.; Durstock, M.F. Nanoparticle/Dye Interface Optimization in Dye-Sensitized Solar Cells. Adv. Funct. Mater. 2011, 21, 3268–3274. [Google Scholar] [CrossRef]
- Cao, Y.; Cai, N.; Wang, Y.; Li, R.; Yuan, Y.; Wang, P. Modulating the Assembly of Organic Dye Molecules on Titania Nanocrystals Via Alkyl Chain Elongation for Efficient Mesoscopic Cobalt Solar Cells. Phys. Chem. Chem. Phys. 2012, 14, 8282–8286. [Google Scholar] [CrossRef] [PubMed]
- Johansson, V.; Ellis-Gibbings, L.; Clarke, T.; Gorlov, M.; Andersson, G.G.; Kloo, L. On the Correlation between Dye Coverage and Photoelectrochemical Performance in Dye-Sensitized Solar Cells. Phys. Chem. Chem. Phys. 2014, 16, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Pazoki, M.; Lohse, P.W.; Taghavinia, N.; Hagfeldt, A.; Boschloo, G. The Effect of Dye Coverage on the Performance of Dye-Sensitized Solar Cells with a Cobalt-Based Electrolyte. Phys. Chem. Chem. Phys. 2014, 16, 8503–8508. [Google Scholar] [CrossRef] [PubMed]
- Mahanta, S.; Matsuzaki, H.; Murakami, T.N.; Katoh, R.; Matsumoto, H.; Furube, A. Modulation of Electron Injection Dynamics of Ru-Based Dye/TiO2 System in the Presence of Three Different Organic Solvents: Role of Solvent Dipole Moment and Donor Number. ChemPhysChem 2015, 16, 1657–1662. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Leijtens, T.; Ronca, E.; Pastore, M.; Snaith, H.; De Angelis, F. Modeling the Effect of Ionic Additives on the Optical and Electronic Properties of a Dye-Sensitized TiO2 Heterointerface: Absorption, Charge Injection and Aggregation. J. Mater. Chem. A 2013, 1, 14675–14685. [Google Scholar] [CrossRef]
- Marotta, G.; Reddy, M.A.; Singh, S.P.; Islam, A.; Han, L.; De Angelis, F.; Pastore, M.; Chandrasekharam, M. Novel Carbazole-Phenothiazine Dyads for Dye-Sensitized Solar Cells: A Combined Experimental and Theoretical Study. ACS Appl. Mater. Interfaces 2013, 5, 9635–9647. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.S.V.; Zhang, J.; Marotta, G.; Reddy, M.A.; Singh, S.P.; Islam, A.; Han, L.; De Angelis, F.; Chandrasekharam, M.; Pastore, M. Effect of the Anchoring Group in the Performance of Carbazole-Phenothiazine Dyads for Dye-Sensitized Solar Cells. Dyes Pigment. 2015, 113, 536–545. [Google Scholar] [CrossRef]
- Vaissier, V.; Frost, J.M.; Barnes, P.R.F.; Nelson, J. Influence of Intermolecular Interactions on the Reorganization Energy of Charge Transfer between Surface-Attached Dye Molecules. J. Phys. Chem. C 2015, 119, 24337–24341. [Google Scholar] [CrossRef]
- Pastore, M.; Fantacci, S.; De Angelis, F. Modeling Excited States and Alignment of Energy Levels in Dye-Sensitized Solar Cells: Successes, Failures, and Challenges. J. Phys. Chem. C 2013, 117, 3685–3700. [Google Scholar] [CrossRef]
- Pastore, M.; Fantacci, S.; De Angelis, F. Ab Initio Determination of Ground and Excited State Oxidation Potentials of Organic Chromophores for Dye-Sensitized Solar Cells. J. Phys. Chem. C 2010, 114, 22742–22750. [Google Scholar] [CrossRef]
- Pastore, M.; Mosconi, E.; De Angelis, F. Computational Investigation of Dye-Iodine Interactions in Organic Dye-Sensitized Solar Cells. J. Phys. Chem. C 2012, 116, 5965–5973. [Google Scholar] [CrossRef]
- Labat, F.; Le Bahers, T.; Ciofini, I.; Adamo, C. First-Principles Modeling of Dye-Sensitized Solar Cells: Challenges and Perspectives. Acc. Chem. Res. 2012, 45, 1268–1277. [Google Scholar] [CrossRef] [PubMed]
- Labat, F.; Ciofini, I.; Hratchian, H.P.; Frisch, M.J.; Raghavachari, K.; Adamo, C. Insights into Working Principles of Ruthenium Polypyridyl Dye-Sensitized Solar Cells from First Principles Modeling. J. Phys. Chem. C 2011, 115, 4297–4306. [Google Scholar] [CrossRef]
- Martsinovich, N.; Troisi, A. Theoretical Studies of Dye-Sensitised Solar Cells: From Electronic Structure to Elementary Processes. Energy Environ. Sci. 2011, 4, 4473–4495. [Google Scholar] [CrossRef]
- Martsinovich, N.; Troisi, A. High-Throughput Computational Screening of Chromophores for Dye-Sensitized Solar Cells. J. Phys. Chem. C 2011, 115, 11781–11792. [Google Scholar] [CrossRef]
- Martsinovich, N.; Ambrosio, F.; Troisi, A. Adsorption and Electron Injection of the N3 Metal-Organic Dye on the TiO2 Rutile (110) Surface. Phys. Chem. Chem. Phys. 2012, 14, 16668–16676. [Google Scholar] [CrossRef] [PubMed]
- Fantacci, S.; De Angelis, F.; Selloni, A. Absorption Spectrum and Solvatochromism of the [Ru(4,4′-Cooh-2,2′-Bpy)2(Ncs)2] Molecular Dye by Time Dependent Density Functional Theory. J. Am. Chem. Soc. 2003, 125, 4381–4387. [Google Scholar] [CrossRef] [PubMed]
- Nazeeruddin, M.K.; De Angelis, F.; Fantacci, S.; Selloni, A.; Viscardi, G.; Liska, P.; Ito, S.; Takeru, B.; Grätzel, M. Combined Experimental and DFT-TDDFT Computational Study of Photoelectrochemical Cell Ruthenium Sensitizers. J. Am. Chem. Soc. 2005, 127, 16835–16847. [Google Scholar] [CrossRef] [PubMed]
- Fantacci, S.; De Angelis, F.; Nazeeruddin, M.K.; Grätzel, M. Electronic and Optical Properties of the Spiro-Meotad Hole Conductor in Its Neutral and Oxidized Forms: A DFT/TDDFT Investigation. J. Phys. Chem. C 2011, 115, 23126–23133. [Google Scholar] [CrossRef]
- Jacquemin, D.; Perpète, E.A.; Ciofini, I.; Adamo, C. Accurate Simulation of Optical Properties in Dyes. Acc. Chem. Res. 2009, 42, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Jacquemin, D.; Perpete, E.A.; Scuseria, G.E.; Ciofini, I.; Adamo, C. TD-DFT Performance for the Visible Absorption Spectra of Organic Dyes: Conventional Versus Long-Range Hybrids. J. Chem. Theor.Comp. 2008, 4, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Ertem, M.Z.; Gagliardi, L.; Cramer, C.J. Quantum Chemical Characterization of the Mechanism of an Iron-Based Water Oxidation Catalyst. Chem. Sci. 2012, 3, 1293–1299. [Google Scholar] [CrossRef]
- Mavros, M.G.; Tsuchimochi, T.; Kowalczyk, T.; McIsaac, A.; Wang, L.-P.; Voorhis, T.V. What Can Density Functional Theory Tell Us about Artificial Catalytic Water Splitting? Inorg. Chem. 2014, 53, 6386–6397. [Google Scholar] [CrossRef] [PubMed]
- Ping, Y.; Rocca, D.; Galli, G. Electronic Excitations in Light Absorbers for Photoelectrochemical Energy Conversion: First Principles Calculations Based on Many Body Perturbation Theory. Chem. Soc. Rev. 2013, 42, 2437–2469. [Google Scholar] [CrossRef] [PubMed]
- Pastore, M.; De Angelis, F.; Angeli, C. Optical Absorption Spectrum of the N3 Solar Cell Sensitizer by Second-Order Multireference Perturbation Theory. Theor. Chem. Acc. 2016, 135, 1–11. [Google Scholar] [CrossRef]
- Chen, P.; Yum, J.H.; De Angelis, F.; Mosconi, E.; Fantacci, S.; Moon, S.-J.; Baker, R.H.; Ko, J.; Nazeeruddin, M.K.; Grätzel, M. High Open-Circuit Voltage Solid-State Dye-Sensitized Solar Cells with Organic Dye. Nano Lett. 2009, 9, 2487–2492. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, F.; Fantacci, S.; Selloni, A.; Grätzel, M.; Nazeeruddin, M.K. Influence of the Sensitizer Adsorption Mode on the Open-Circuit Potential of Dye-Sensitized Solar Cells. Nano Lett. 2007, 7, 3189–3195. [Google Scholar] [CrossRef] [PubMed]
- Pastore, M.; Selloni, A.; Fantacci, S.; De Angelis, F. Electronic and Optical Properties of Dye-Sensitized TiO2 Interfaces. In First Principles Approaches to Spectroscopic Properties of Complex Materials; Springer: Berlin/Heidelberg, Germany, 2014; pp. 1–45. [Google Scholar]
- Maggio, E.; Martsinovich, N.; Troisi, A. Theoretical Study of Charge Recombination at the TiO2-Electrolyte Interface in Dye Sensitised Solar Cells. J. Chem. Phys. 2012, 137, 22A508. [Google Scholar] [CrossRef] [PubMed]
- Ambrosio, F.; Martsinovich, N.; Troisi, A. What Is the Best Anchoring Group for a Dye in a Dye-Sensitized Solar Cell? J. Phys. Chem. Lett. 2012, 3, 1531–1535. [Google Scholar] [CrossRef] [PubMed]
- Labat, F.; Ciofini, I.; Adamo, C. Revisiting the Importance of Dye Binding Mode in Dye-Sensitized Solar Cells: A Periodic Viewpoint. J. Mater. Chem. 2012, 22, 12205–12211. [Google Scholar] [CrossRef]
- Jakubikova, E.; Snoeberger Iii, R.C.; Batista, V.S.; Martin, R.L.; Batista, E.R. Interfacial Electron Transfer in TiO2 Surfaces Sensitized with Ru(Ii)−Polypyridine Complexes. J. Phys. Chem. A 2009, 113, 12532–12540. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Sproviero, E.M.; Snoeberger Iii, R.C.; Iguchi, N.; Blakemore, J.D.; Crabtree, R.H.; Brudvig, G.W.; Batista, V.S. Deposition of an Oxomanganese Water Oxidation Catalyst on TiO2 Nanoparticles: Computational Modeling, Assembly and Characterization. Energy Environ. Sci. 2009, 2, 230–238. [Google Scholar] [CrossRef]
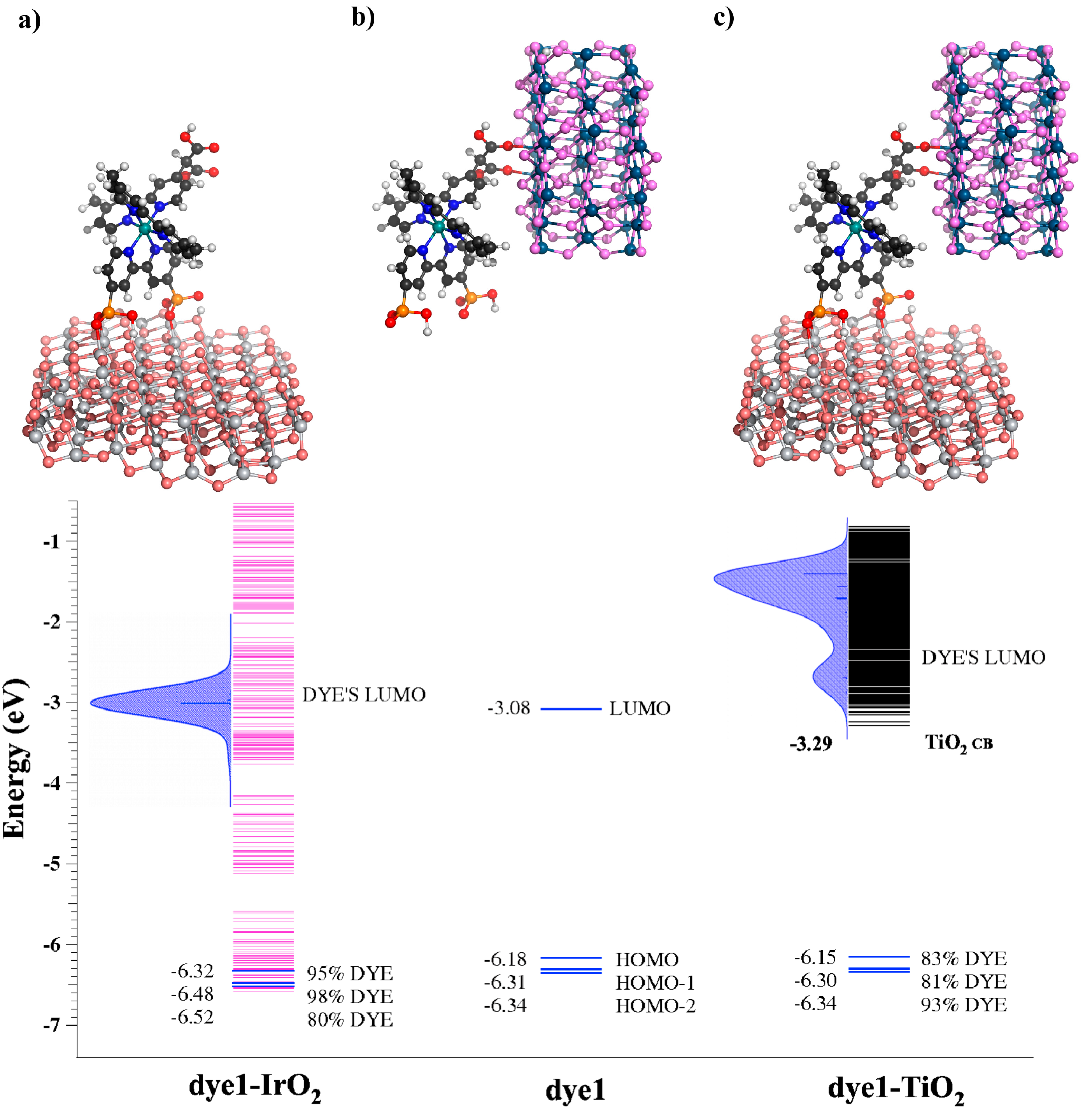
- Pastore, M.; De Angelis, F. First-Principles Modeling of a Dye-Sensitized TiO2/IrO2 Photoanode for Water Oxidation. J. Am. Chem. Soc. 2015, 137, 5798–5809. [Google Scholar] [CrossRef] [PubMed]
- Rego, L.G.C.; Batista, V.S. Quantum Dynamics Simulations of Interfacial Electron Transfer in Sensitized TiO2 Semiconductors. J. Am. Chem. Soc. 2003, 125, 7989–7997. [Google Scholar] [CrossRef] [PubMed]
- Negre, C.F.A.; Milot, R.L.; Martini, L.A.; Ding, W.; Crabtree, R.H.; Schmuttenmaer, C.A.; Batista, V.S. Efficiency of Interfacial Electron Transfer from Zn-Porphyrin Dyes into TiO2 Correlated to the Linker Single Molecule Conductance. J. Phys. Chem. C 2013, 117, 24462–24470. [Google Scholar] [CrossRef]
- Sproviero, E.M.; Shinopoulos, K.; Gascón, J.A.; McEvoy, J.P.; Brudvig, G.W.; Batista, V.S. Qm/Mm Computational Studies of Substrate Water Binding to the Oxygen-Evolving Centre of Photosystem II. Philos. Trans. R. Soc. B Biol. Sci. 2008, 363, 1149–1156. [Google Scholar] [CrossRef] [PubMed]
- Monti, A.; de Ruiter, J.M.; de Groot, H.J.M.; Buda, F. A Dynamic View of Proton-Coupled Electron Transfer in Photocatalytic Water Splitting. J. Phys. Chem. C 2016, 120, 23074–23082. [Google Scholar] [CrossRef]
- Sulzer, D.; Iuchi, S.; Yasuda, K. A New Method to Evaluate Excited States Lifetimes Based on Green’s Function: Application to Dye-Sensitized Solar Cells. J. Chem. Theor. Comp. 2016, 12, 3074–3086. [Google Scholar] [CrossRef] [PubMed]
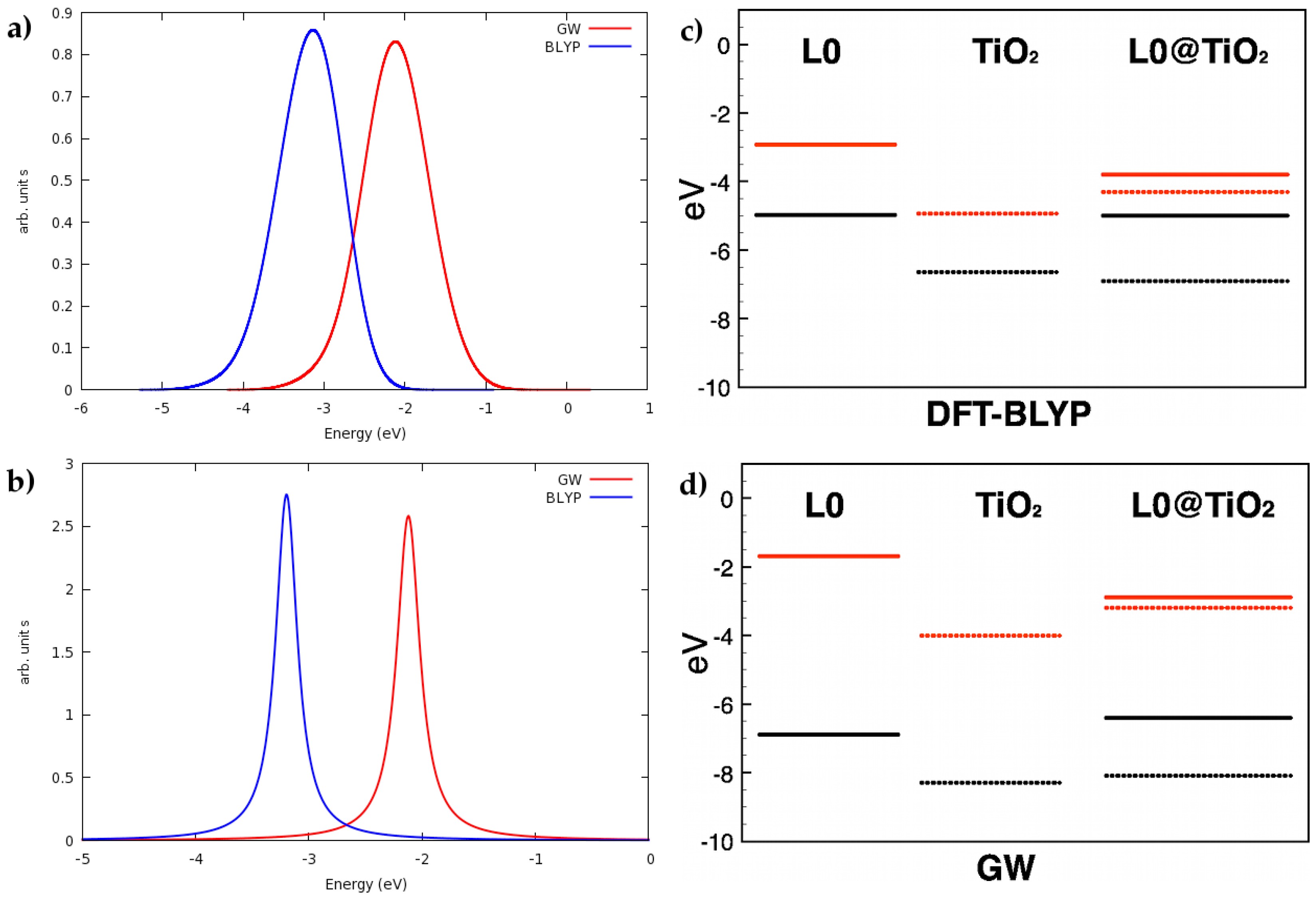
- Umari, P.; Giacomazzi, L.; De Angelis, F.; Pastore, M.; Baroni, S. Energy-Level Alignment in Organic Dye-Sensitized TiO2 from GW Calculations. J. Chem. Phys. 2013, 139, 014709. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Garcia, A.B.; Pavone, M. Structure and Energy Level Alignment at the Dye-Electrode Interface in P-Type Dsscs: New Hints on the Role of Anchoring Modes from Ab Initio Calculations. Phy. Chem. Chem. Phys. 2015, 17, 12238–12246. [Google Scholar] [CrossRef] [PubMed]
- Kontkanen, O.V.; Niskanen, M.; Hukka, T.I.; Rantala, T.T. Electronic Structure of P-Type Perylene Monoimide-Based Donor-Acceptor Dyes on the Nickel Oxide (100) Surface: A DFT Approach. Phys. Chem. Chem. Phys. 2016, 18, 14382–14389. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.J.; Laurent, A.D.; Boucher, F.; Odobel, F.; Jacquemin, D. Determining the Most Promising Anchors for Cuscn: Ab Initio Insights Towards P-Type Dsscs. J. Mater. Chem. A 2016, 4, 2217–2227. [Google Scholar] [CrossRef]
- Pastore, M.; Duchanois, T.; Liu, L.; Monari, A.; Assfeld, X.; Haacke, S.; Gros, P.C. Interfacial Charge Separation and Photovoltaic Efficiency in Fe(II)-Carbene Sensitized Solar Cells. Phys. Chem. Chem. Phys. 2016, 18, 28069–28081. [Google Scholar] [CrossRef] [PubMed]
- Salvatori, P.; Amat, A.; Pastore, M.; Vitillaro, G.; Sudhakar, K.; Giribabu, L.; Soujanya, Y.; De Angelis, F. Corrole Dyes for Dye-Sensitized Solar Cells: The Crucial Role of the Dye/Semiconductor Energy Level Alignment. Comp. Theor. Chem. 2014, 1030, 59–66. [Google Scholar] [CrossRef]
- Stier, W.; Prezhdo, O.V. Nonadiabatic Molecular Dynamics Simulation of Light-Induced Electron Transfer from an Anchored Molecular Electron Donor to a Semiconductor Acceptor. J. Phys. Chem. B 2002, 106, 8047–8054. [Google Scholar] [CrossRef]
- Kondov, I.; Čížek, M.; Benesch, C.; Wang, H.; Thoss, M. Quantum Dynamics of Photoinduced Electron-Transfer Reactions in Dye-Semiconductor Systems: First-Principles Description and Application to Coumarin 343−TiO2. J. Phys. Chem. C 2007, 111, 11970–11981. [Google Scholar] [CrossRef]
- Meng, S.; Ren, J.; Kaxiras, E. Natural Dyes Adsorbed on TiO2 Nanowire for Photovoltaic Applications: Enhanced Light Absorption and Ultrafast Electron Injection. Nano Lett. 2008, 8, 3266–3272. [Google Scholar] [CrossRef] [PubMed]
- Monti, A.; Negre, C.F.A.; Batista, V.S.; Rego, L.G.C.; de Groot, H.J.M.; Buda, F. Crucial Role of Nuclear Dynamics for Electron Injection in a Dye-Semiconductor Complex. J. Phys. Chem. Lett. 2015, 6, 2393–2398. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Trivedi, D.J.; Akimov, A.V.; Aradi, B.; Frauenheim, T.; Prezhdo, O.V. Nonadiabatic Molecular Dynamics for Thousand Atom Systems: A Tight-Binding Approach toward Pyxaid. J. Chem. Theor. Comp. 2016, 12, 1436–1448. [Google Scholar] [CrossRef] [PubMed]
- Abuabara, S.G.; Rego, L.G.C.; Batista, V.S. Influence of Thermal Fluctuations on Interfacial Electron Transfer in Functionalized TiO2 Semiconductors. J. Am. Chem. Soc. 2005, 127, 18234–18242. [Google Scholar] [CrossRef] [PubMed]
- Duncan, W.R.; Stier, W.M.; Prezhdo, O.V. Ab Initio Nonadiabatic Molecular Dynamics of the Ultrafast Electron Injection across the Alizarin-TiO2 Interface. J. Am. Chem. Soc. 2005, 127, 7941–7951. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, H.; Persson, P.; Thoss, M. Photoinduced Electron Transfer Processes in Dye-Semiconductor Systems with Different Spacer Groups. J. Chem. Phys. 2012, 137, 22A529. [Google Scholar] [CrossRef] [PubMed]
- Marques, M.A.L.; López, X.; Varsano, D.; Castro, A.; Rubio, A. Time-Dependent Density-Functional Approach for Biological Chromophores: The Case of the Green Fluorescent Protein. Phys. Rev. Lett. 2003, 90, 258101–258104. [Google Scholar] [CrossRef] [PubMed]
- Meng, S.; Kaxiras, E. Electron and Hole Dynamics in Dye-Sensitized Solar Cells: Influencing Factors and Systematic Trends. Nano Lett. 2010, 10, 1238–1247. [Google Scholar] [CrossRef] [PubMed]
- Angeli, C.; Pastore, M.; Cimiraglia, C. New Perspectives in Multireference Perturbation Theory: The N-Electron Valence State Approach. Theor. Chem. Acc. 2007, 117, 743–754. [Google Scholar] [CrossRef]
- Angeli, C.; Cimiraglia, R.; Evangelisti, S.; Leininger, T.; Malrieu, J.-P. Introduction of N-Electron Valence States for Multireference Perturbation Theory. J. Chem. Phys. 2001, 114, 10252–10264. [Google Scholar] [CrossRef]
- Angeli, C.; Cimiraglia, R.; Malrieu, J.-P. N-Electron Valence State Perturbation Theory: A Fast Implementation of the Strongly Contracted Variant. Chem. Phys. Lett. 2001, 350, 297–305. [Google Scholar] [CrossRef]
- Neese, F. The Orca Program System. WIREs Comp. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Hedin, L. New Method for Calculating the One-Particle Green’s Function with Application to the Electron-Gas Problem. Phys. Rev. 1965, 139, A796–A823. [Google Scholar] [CrossRef]
- Hybertsen, M.S.; Louie, S.G. Electron Correlation in Semiconductors and Insulators: Band Gaps and Quasiparticle Energies. Phys. Rev. B 1986, 34, 5390–5413. [Google Scholar] [CrossRef]
- Pastore, M.; Mosconi, E.; De Angelis, F.; Grätzel, M. A Computational Investigation of Organic Dyes for Dye-Sensitized Solar Cells: Benchmark, Strategies, and Open Issues. J. Phys. Chem. C 2010, 114, 7205–7212. [Google Scholar] [CrossRef]
- Delgado, A.; Corni, S.; Goldoni, G. Low-Lying Electronic Excitations and Optical Absorption Spectra of the Black Dye Sensitizer: A First-Principles Study. Theor. Chem. Acc. 2012, 131, 1–14. [Google Scholar] [CrossRef]
- Faber, C.; Duchemin, I.; Deutsch, T.; Blase, X. Many-Body Green’s Function Study of Coumarins for Dye-Sensitized Solar Cells. Phys. Rev. B 2012, 86, 155315. [Google Scholar] [CrossRef]
- Umari, P.; Mosconi, E.; De Angelis, F. Relativistic GW Calculations on CH3NH3PbI3 and CH3NH3SnI3 Perovskites for Solar Cell Applications. Sci. Rep. 2014, 4, 4467. [Google Scholar] [CrossRef] [PubMed]
- Marom, N.; Moussa, J.E.; Ren, X.; Tkatchenko, A.; Chelikowsky, J.R. Electronic Structure of Dye-Sensitized TiO2 Clusters from Many-Body Perturbation Theory. Phys. Rev. B 2011, 84, 245115. [Google Scholar] [CrossRef]
- Sproviero, E.M.; Gascón, J.A.; McEvoy, J.P.; Brudvig, G.W.; Batista, V.S. Quantum Mechanics/Molecular Mechanics Study of the Catalytic Cycle of Water Splitting in Photosystem II. J. Am. Chem. Soc. 2008, 130, 3428–3442. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Hall, M.B. Mechanism of Water Splitting and Oxygen−Oxygen Bond Formation by a Mononuclear Ruthenium Complex. J. Am. Chem. Soc. 2009, 132, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Sobolewski, A.L.; Domcke, W. Computational Model of Photocatalytic Water Splitting. J. Phys. Chem. A 2008, 112, 7311–7313. [Google Scholar] [CrossRef] [PubMed]
- Rivalta, I.; Brudvig, G.W.; Batista, V.S. Computational Studies of the Oxygen-Evolving Complex of Photosystem II and Biomimetic Oxomanganese Complexes for Renewable Energy Applications. In Applications of Molecular Modeling to Challenges in Clean Energy; American Chemical Society: Washington, DC, USA, 2013; Volume 1133, pp. 203–215. [Google Scholar]
- Pastore, M.; Mosconi, E.; Fantacci, S.; De Angelis, F. Computational Investigations on Organic Sensitizers for Dye- Sensitized Solar Cells. Curr. Org. Synth. 2012, 9, 215–232. [Google Scholar] [CrossRef]
- Tognetti, V.; Joubert, L. Unraveling Charge Transfer Processes with the Quantum Theory of Atoms-in-Molecules. Theor. Chem. Acc. 2016, 135, 124. [Google Scholar] [CrossRef]
- Ronca, E.; Angeli, C.; Belpassi, L.; De Angelis, F.; Tarantelli, F.; Pastore, M. Density Relaxation in Time-Dependent Density Functional Theory: Combining Relaxed Density Natural Orbitals and Multireference Perturbation Theories for an Improved Description of Excited States. J. Chem. Theor. Comp. 2014, 10, 4014–4024. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Zhang, Q.; Shiota, Y.; Nakagawa, T.; Kuwabara, K.; Yoshizawa, K.; Adachi, C. Computational Prediction for Singlet- and Triplet-Transition Energies of Charge-Transfer Compounds. J. Chem. Theor. Comp. 2013, 9, 3872–3877. [Google Scholar] [CrossRef] [PubMed]
- Yanai, T.; Tew, D.P.; Handy, N.C. A New Hybrid Exchange-Correlation Functional Using the Coulomb-Attenuating Method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Baer, R.; Neuhauser, D. Density Functional Theory with Correct Long-Range Asymptotic Behavior. Phys. Rev. Lett. 2005, 94, 043002. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.-D.; Head-Gordon, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom–Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Vydrov, O.A.; Scuseria, G.E. Assessment of a Long-Range Corrected Hybrid Functional. J. Chem. Phys. 2006, 125, 234109. [Google Scholar] [CrossRef] [PubMed]
- Rohrdanz, M.A.; Martins, K.M.; Herbert, J.M. A Long-Range-Corrected Density Functional That Performs Well for Both Ground-State Properties and Time-Dependent Density Functional Theory Excitation Energies, Including Charge-Transfer Excited States. J. Chem. Phys. 2009, 130, 054112. [Google Scholar] [CrossRef] [PubMed]
- Baer, R.; Livshits, E.; Salzner, U. Tuned Range-Separated Hybrids in Density Functional Theory. Annu. Rev. Phys. Chem. 2010, 61, 85–109. [Google Scholar] [CrossRef] [PubMed]
- Moore, B.; Charaf-Eddin, A.; Planchat, A.; Adamo, C.; Autschbach, J.; Jacquemin, D. Electronic Band Shapes Calculated with Optimally Tuned Range-Separated Hybrid Functionals. J. Chem. Theor. Comp. 2014, 10, 4599–4608. [Google Scholar] [CrossRef] [PubMed]
- Kronik, L.; Stein, T.; Refaely-Abramson, S.; Baer, R. Excitation Gaps of Finite-Sized Systems from Optimally Tuned Range-Separated Hybrid Functionals. J. Chem. Theor. Comp. 2012, 8, 1515–1531. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhong, C.; Brédas, J.-L. Reliable Prediction with Tuned Range-Separated Functionals of the Singlet–Triplet Gap in Organic Emitters for Thermally Activated Delayed Fluorescence. J. Chem. Theor. Comp. 2015, 11, 3851–3858. [Google Scholar] [CrossRef] [PubMed]
- Tortorella, S.; Talamo, M.M.; Cardone, A.; Pastore, M.; De Angelis, F. Benchmarking DFT and Semi-Empirical Methods for a Reliable and Cost-Efficient Computational Screening of Benzofulvene Derivatives as Donor Materials for Small-Molecule Organic Solar Cells. J. Phys. Condens. Matter 2016, 28, 074005. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, J.K.; Kang, S.O.; Ko, J.; Yum, J.H.; Fantacci, S.; De Angelis, F.; Di Censo, D.; Nazeeruddin, M.K.; Grätzel, M. Molecular Engineering of Organic Sensitizers for Solar Cell Applications. J. Am. Chem. Soc. 2006, 128, 16701–16707. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Mende, L.; Bach, U.; Humphry-Baker, R.; Horiuchi, T.; Miura, H.; Ito, S.; Uchida, S.; Grätzel, M. Organic Dye for Highly Efficient Solid-State Dye-Sensitized Solar Cells. Adv. Mater. 2005, 17, 813–815. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- Azzaroli, N.; Lobello, M.G.; Lapini, A.; Iagatti, A.; Bussotti, L.; Di Donato, M.; Calogero, G.; Pastore, M.; De Angelis, F.; Foggi, P. Monitoring the Intramolecular Charge Transfer Process in the Z907 Solar Cell Sensitizer: A Transient Vis and Ir Spectroscopy and Ab Initio Investigation. Phys. Chem. Chem. Phys. 2015, 17, 21594–21604. [Google Scholar] [CrossRef] [PubMed]
- Improta, R.; Barone, V. Absorption and Fluorescence Spectra of Uracil in the Gas Phase and in Aqueous Solution: A TD-DFT Quantum Mechanical Study. J. Am. Chem. Soc. 2004, 126, 14320–14321. [Google Scholar] [CrossRef] [PubMed]
- Jacquemin, D.; Preat, J.; Wathelet, V.; Perpète, E.A. Substitution and Chemical Environment Effects on the Absorption Spectrum of Indigo. J. Chem. Phys. 2006, 124, 074104. [Google Scholar] [CrossRef] [PubMed]
- Jacquemin, D.; Perpète, E.A.; Ciofini, I.; Adamo, C. On the TD-DFT UV/Vis Spectra Accuracy: The Azoalkanes. Theor. Chem. Acc. 2008, 120, 405–410. [Google Scholar] [CrossRef]
- Umari, P.; Stenuit, G.; Baroni, S. GW Quasiparticle Spectra from Occupied States Only. Phys. Rev. B 2010, 81, 115104–115109. [Google Scholar] [CrossRef]
- Umari, P.; Stenuit, G.; Baroni, S. Optimal Representation of the Polarization Propagator for Large-Scale GW Calculations. Phys. Rev. B 2009, 79, 201104. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. Quantum Espresso: A Modular and Open-Source Software Project for Quantum Simulations of Materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Bruneval, F. GW Approximation of the Many-Body Problem and Changes in the Particle Number. Phys. Rev. Lett. 2009, 103, 176403. [Google Scholar] [CrossRef] [PubMed]
- Körzdörfer, T.; Marom, N. Strategy for Finding a Reliable Starting Point for G0W0 Demonstrated for Molecules. Phys. Rev. B 2011, 86, 041110. [Google Scholar] [CrossRef]
- Bruneval, F.; Marques, M.A.L. Benchmarking the Starting Points of the GW Approximation for Molecules. J. Chem. Theor. Comp. 2013, 9, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Jacquemin, D.; Duchemin, I.; Blase, X. Benchmarking the Bethe–Salpeter Formalism on a Standard Organic Molecular Set. J. Chem. Theor. Comp. 2015, 11, 3290–3304. [Google Scholar] [CrossRef] [PubMed]
- Marinado, T.; Hagberg, D.; Hedlund, M.; Edvinsson, T.; Johansson, E.; Boschloo, G.; Rensmo, H.; Brinck, T.; Sun, L.; Hagfeldt, A. Rhodanine Dyes for Dye-Sensitized Solar Cells: Spectroscopy, Energy Levels and Photovoltaic Performance. Phys. Chem. Chem. Phys. 2009, 11, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Nazeeruddin, M.K.; Kay, A.; Rodicio, I.; Humphry-Baker, R.; Mueller, E.; Liska, P.; Vlachopoulos, N.; Graetzel, M. Conversion of Light to Electricity by Cis-X2bis(2,2′-Bipyridyl-4,4′-Dicarboxylate)Ruthenium(II) Charge-Transfer Sensitizers (X = Cl-, Br-, I-, CN-, and SCN-) on Nanocrystalline Titanium Dioxide Electrode. J. Am. Chem. Soc. 1993, 115, 6382–6390. [Google Scholar] [CrossRef]
- Nazeeruddin, M.K.; Péchy, P.; Grätzel, M. Efficient Panchromatic Sensitization of Nanocrystalline TiO2 Films by a Black Dye Based on Atrithiocyanato–Ruthenium Complex. Chem. Commun. 1997, 18, 1705–1706. [Google Scholar] [CrossRef]
- Nazeeruddin, M.K.; Péchy, P.; Renouard, T.; Zakeeruddin, S.M.; Humphry-Baker, R.; Comte, P.; Liska, P.; Cevey, L.; Costa, E.; Shklover, V.; et al. Engineering of Efficient Panchromatic Sensitizers for Nanocrystalline TiO2-Based Solar Cells. J. Am. Chem. Soc. 2001, 123, 1613–1624. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Islam, A.; Chen, H.; Malapaka, C.; Chiranjeevi, B.; Zhang, S.; Yang, X.; Yanagida, M. High-Efficiency Dye-Sensitized Solar Cell with a Novel Co-Adsorbent. Energy Environ. Sci. 2012, 5, 6057–6060. [Google Scholar] [CrossRef]
- Becke, A.D. A New Mixing of Hartree–Fock and Local Density-Functional Theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- De Angelis, F.; Fantacci, S.; Selloni, A. Time Dependent Density Functional Theory Study of the Absorption Spectrum of the [Ru(4,4′-COO−-2,2′-bpy)2(X)2]4− (X = NCS, Cl) Dyes in Water Solution. Chem. Phys. Lett. 2005, 415, 115–120. [Google Scholar] [CrossRef]
- De Angelis, F.; Fantacci, S.; Selloni, A. Time-Dependent Density Functional Theory Study of the Absorption Spectrum of [Ru(4,4′-COOH-2,2′-bpy)2(NCS)2] in Water Solution: Influence of the pH. Chem. Phys. Lett. 2004, 389, 204–208. [Google Scholar] [CrossRef]
- Monat, J.E.; Rodriguez, J.H.; McCusker, J.K. Ground- and Excited-State Electronic Structures of the Solar Cell Sensitizer Bis(4,4′-Dicarboxylato-2,2′-Bipyridine)Bis(Isothiocyanato)Ruthenium(II). J. Phys. Chem. A 2002, 106, 7399–7406. [Google Scholar] [CrossRef]
- Jäger, M.; Freitag, L.; González, L. Using Computational Chemistry to Design Ru Photosensitizers with Directional Charge Transfer. Coord. Chem. Rev. 2015, 304–305, 146–165. [Google Scholar] [CrossRef]
- Daniel, C. Photochemistry and Photophysics of Transition Metal Complexes: Quantum Chemistry. Coord. Chem. Rev. 2015, 282–283, 19–32. [Google Scholar] [CrossRef]
- Piau, R.E.; Guillon, T.; Lebon, E.; Perrot, N.; Alary, F.; Boggio-Pasqua, M.; Heully, J.-L.; Juris, A.; Sutra, P.; Igau, A. Photophysical and Electrochemical Properties of Polypyridine Imine Ruthenium(II) Complexes: A Comparative Experimental and Theoretical Study. New J. Chem. 2012, 36, 2484–2492. [Google Scholar] [CrossRef]
- Charlot, M.-F.; Aukauloo, A. Highlighting the Role of the Medium in DFT Analysis of the Photophysical Properties of Ruthenium(II) Polypyridine-Type Complexes. J. Phys. Chem. A 2007, 111, 11661–11672. [Google Scholar] [CrossRef] [PubMed]
- Stoyanov, S.R.; Villegas, J.M.; Paul Rillema, D. The Charge Transfer Band Solvent-Dependence of [Ru(bpy)2(CNx)Cl]+, CNx = 2,6-Dimethylphenylisocyanide: A Polarizable Continuum Model/Time-Dependent Density Functional Theory Study. Inorg. Chem. Commun. 2004, 7, 838–841. [Google Scholar] [CrossRef]
- Fantacci, S.; De Angelis, F.; Sgamellotti, A.; Re, N. A Tddft Study of the Ruthenium(II) Polyazaaromatic Complex [Ru(dppz)(phen)2]2+ in Solution. Chem. Phys. Lett. 2004, 396, 43–48. [Google Scholar] [CrossRef]
- Vlček, A., Jr.; Záliš, S. Modeling of Charge-Transfer Transitions and Excited States in D6 Transition Metal Complexes by DFT Techniques. Coord. Chem. Rev. 2007, 251, 258–287. [Google Scholar] [CrossRef]
- Escudero, D.; González, L. Raspt2/Rasscf vs. Range-Separated/Hybrid DFT Methods: Assessing the Excited States of a Ru(II)Bipyridyl Complex. J. Chem. Theor. Comp. 2011, 8, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Chantzis, A.; Very, T.; Monari, A.; Assfeld, X. Improved Treatment of Surrounding Effects: UV/Vis Absorption Properties of a Solvated Ru(II) Complex. J. Chem. Theor. Comp. 2012, 8, 1536–1541. [Google Scholar] [CrossRef] [PubMed]
- Dalton, a Molecular Electronic Structure Program, Release 2.0 (2005). Available online: http://daltonprogram.org/ (accessed on 29 December 2016).
- Pierloot, K.; Vancoillie, S. Relative Energy of the High-(5T2g) and Low-(1A1g) Spin States of [Fe(H2O)6]2+, [Fe(NH3)6]2+, and [Fe(bpy)3]2+: CASPT2 Versus Density Functional Theory. J. Chem. Phys. 2006, 125, 124303. [Google Scholar] [CrossRef] [PubMed]
- Gindensperger, E.; Koppel, H.; Daniel, C. Mechanism of Visible-Light Photoisomerization of a Rhenium(I) Carbonyl-Diimine Complex. Chem. Comm. 2010, 46, 8225–8227. [Google Scholar] [CrossRef] [PubMed]
- Escudero, D.; Thiel, W. Assessing the Density Functional Theory-Based Multireference Configuration Interaction (DFT/MRCI) Method for Transition Metal Complexes. J. Chem. Phys. 2014, 140, 194105. [Google Scholar] [CrossRef] [PubMed]
- Domingo, A.; Carvajal, M.; de Graaf, C.; Sivalingam, K.; Neese, F.; Angeli, C. Metal-to-Metal Charge-Transfer Transitions: Reliable Excitation Energies from Ab Initio Calculations. Theor. Chem. Acc. 2012, 131, 1–13. [Google Scholar] [CrossRef]
- De Angelis, F.; Fantacci, S.; Selloni, A.; Nazeeruddin, M.K.; Graetzel, M. Time-Dependent Density Functional Theory Investigations on the Excited States of Ru(II)-Dye-Sensitized TiO2 Nanoparticles: The Role of Sensitizer Protonation. J. Am. Chem. Soc. 2007, 129, 14156–14157. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, F.; Fantacci, S.; Selloni, A.; Nazeeruddin, M.K.; Grätzel, M. First-Principles Modeling of the Adsorption Geometry and Electronic Structure of Ru(II) Dyes on Extended TiO2 Substrates for Dye-Sensitized Solar Cell Applications. J. Phys. Chem. C 2010, 114, 6054–6061. [Google Scholar] [CrossRef]
- Wiberg, J.; Marinado, T.; Hagberg, D.P.; Sun, L.; Hagfeldt, A.; Albinsson, B. Effect of Anchoring Group on Electron Injection and Recombination Dynamics in Organic Dye-Sensitized Solar Cells. J. Phys. Chem. C 2009, 113, 3881–3886. [Google Scholar] [CrossRef]
- Pastore, M.; De Angelis, F. Aggregation of Organic Dyes on TiO2 in Dye-Sensitized Solar Cells Models: An Ab Initio Investigation. ACS Nano 2010, 4, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Pastore, M.; De Angelis, F. Computational Modelling of TiO2 Surfaces Sensitized by Organic Dyes with Different Anchoring Groups: Adsorption Modes, Electronic Structure and Implication for Electron Injection/Recombination. Phys. Chem. Chem. Phys. 2012, 14, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Grätzel, M. Conversion of Sunlight to Electric Power by Nanocrystalline Dye-Sensitized Solar Cells. J. Photochem. Photobiol. A 2004, 164, 3–14. [Google Scholar] [CrossRef]
- Odobel, F.; Blart, E.; Lagrée, M.; Villieras, M.; Boujtita, H.; El Murr, N.; Caramori, S.; Bignozzi, C.A. Porphyrin Dyes for TiO2 Sensitization. J. Mater. Chem. 2003, 13, 502–510. [Google Scholar] [CrossRef]
- Abbotto, A.; Manfredi, N.; Marinzi, C.; De Angelis, F.; Mosconi, E.; Yum, J.; Xianxi, Z.; Nazeeruddin, M.K.; Grätzel, M. Di-Branched Di-Anchoring Organic Dyes for Dye-Sensitized Solar Cells. Energy Environ. Sci. 2009, 2, 1094. [Google Scholar] [CrossRef]
- Anselmi, C.; Mosconi, E.; Pastore, M.; Ronca, E.; De Angelis, F. Adsorption of Organic Dyes on TiO2 Surfaces in Dye-Sensitized Solar Cells: Interplay of Theory and Experiment. Phy. Chem. Chem. Phys. 2012, 14, 15963–15974. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, F.; VandeVondele, J.; Hutter, J.; Wirz, R.; Urakawa, A.; Baiker, A. Protonation-Dependent Binding of Ruthenium Bipyridyl Complexes to the Anatase(101) Surface. J. Phys. Chem. C 2010, 114, 8398–8404. [Google Scholar] [CrossRef] [Green Version]
- De Angelis, F.; Vitillaro, G.; Kavan, L.; Nazeeruddin, M.K.; Grätzel, M. Modeling Ruthenium-Dye-Sensitized TiO2 Surfaces Exposing the (001) or (101) Faces: A First-Principles Investigation. J. Phys. Chem. C 2012, 116, 18124–18131. [Google Scholar] [CrossRef]
- Pastore, M.; De Angelis, F. First-Principles Computational Modeling of Fluorescence Resonance Energy Transfer in Co-Sensitized Dye Solar Cells. J. Phys. Chem. Lett. 2012, 3, 2146–2153. [Google Scholar] [CrossRef] [PubMed]
- Ronca, E.; Pastore, M.; Belpassi, L.; Tarantelli, F.; De Angelis, F. Influence of the Dye Molecular Structure on the TiO2 Conduction Band in Dye-Sensitized Solar Cells: Disentangling Charge Transfer and Electrostatic Effects. Energy Environ. Sci. 2013, 6, 183–193. [Google Scholar] [CrossRef]
- Kang, W.; Hybertsen, M.S. Quasiparticle and Optical Properties of Rutile and Anatase TiO2. Phys. Rev. B 2010, 82, 085203. [Google Scholar] [CrossRef]
- Chiodo, L.; García-Lastra, J.M.; Iacomino, A.; Ossicini, S.; Zhao, J.; Petek, H.; Rubio, A. Self-Energy and Excitonic Effects in the Electronic and Optical Properties of TiO2 Crystalline Phases. Phys. Rev. B 2010, 82, 045207. [Google Scholar] [CrossRef]
- Landmann, M.; Rauls, E.; Schmidt, W.G. The Electronic Structure and Optical Response of Rutile, Anatase and Brookite TiO2. J. Phys. Condens. Matter 2012, 24, 195503. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.R.; Kim, K.J. Structural and Optical Properties of Rutile and Anatase TiO2 Thin Films: Effects of Co Doping. Thin Solid Films 2005, 484, 34–38. [Google Scholar] [CrossRef]
- Neaton, J.B.; Hybertsen, M.S.; Louie, S.G. Renormalization of Molecular Electronic Levels at Metal-Molecule Interfaces. Phys. Rev. Lett. 2006, 97, 216405. [Google Scholar] [CrossRef] [PubMed]
- Persson, P.; Lundqvist, M.J.; Ernstorfer, R.; Goddard, W.A.; Willig, F. Quantum Chemical Calculations of the Influence of Anchor-Cum-Spacer Groups on Femtosecond Electron Transfer Times in Dye-Sensitized Semiconductor Nanocrystals. J. Chem. Theory Comput. 2006, 2, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Muscat, J.P.; Newns, D.M. Chemisorption on Metals. Prog. Surf. Sci. 1978, 9, 1–43. [Google Scholar] [CrossRef]
- Lundqvist, M.J.; Nilsing, M.; Persson, P.; Lunel, S. DFT Study of Bare and Dye-Sensitized TiO2 Clusters and Nanocrystals. Int. J. Quantum Chem. 2006, 106, 3214–3234. [Google Scholar] [CrossRef]
- De Angelis, F.; Fantacci, S.; Mosconi, E.; Nazeeruddin, M.K.; Grätzel, M. Absorption Spectra and Excited State Energy Levels of the N719 Dye on TiO2 in Dye-Sensitized Solar Cell Models. J. Phys. Chem. C 2011, 115, 8825–8831. [Google Scholar] [CrossRef]
- Caramori, S.; Ronconi, F.; Argazzi, R.; Carli, S.; Boaretto, R.; Busatto, E.; Bignozzi, C.A. Solar Energy Conversion in Photoelectrochemical Systems. In Applied Photochemistry: When Light Meets Molecules; Bergamini, G., Silvi, S., Eds.; Springer: Cham, Switzerland, 2016; pp. 67–143. [Google Scholar]
- Youngblood, W.J.; Lee, S.-H.A.; Kobayashi, Y.; Hernandez-Pagan, E.A.; Hoertz, P.G.; Moore, T.A.; Moore, A.L.; Gust, D.; Mallouk, T.E. Photoassisted Overall Water Splitting in a Visible Light-Absorbing Dye-Sensitized Photoelectrochemical Cell. J. Am. Chem. Soc. 2009, 131, 926–927. [Google Scholar] [CrossRef] [PubMed]
- Hoertz, P.G.; Kim, Y.-I.; Youngblood, W.J.; Mallouk, T.E. Bidentate Dicarboxylate Capping Groups and Photosensitizers Control the Size of IrO2 Nanoparticle Catalysts for Water Oxidation. J. Phys. Chem. B 2007, 111, 6845–6856. [Google Scholar] [CrossRef] [PubMed]
- She, C.; Guo, J.; Irle, S.; Morokuma, K.; Mohler, D.L.; Zabri, H.; Odobel, F.; Youm, K.-T.; Liu, F.; Hupp, J.T.; et al. Comparison of Interfacial Electron Transfer through Carboxylate and Phosphonate Anchoring Groups†. J. Phys. Chem. A 2007, 111, 6832–6842. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dye | B3LYP | MPW1K | CAM-B3LYP | Exp. (EtOH) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1H | 0H | 1H | 0H | 1H | 0H | |||||
| Vac | Solv | Solv | Vac | Solv | Solv | Vac | Solv | Solv | ||
| JK2 | 1.99 | 1.82 | 2.26 | 2.60 | 2.45 | 2.81 | 2.78 | 2.62 | 2.94 | 2.84 [119] |
| D102 | 2.61 | 2.29 | 2.37 | 3.07 | 2.78 | 2.89 | 3.11 | 2.86 | 2.90 | 2.53 [120] |
| Dye | IP (eV) | EA (eV) |
|---|---|---|
| L0 | 6.87 | 1.66 |
| L2 | 6.48 | 2.31 |
| L3 | 6.37 | 2.28 |
| L4 | 6.21 | 2.21 |
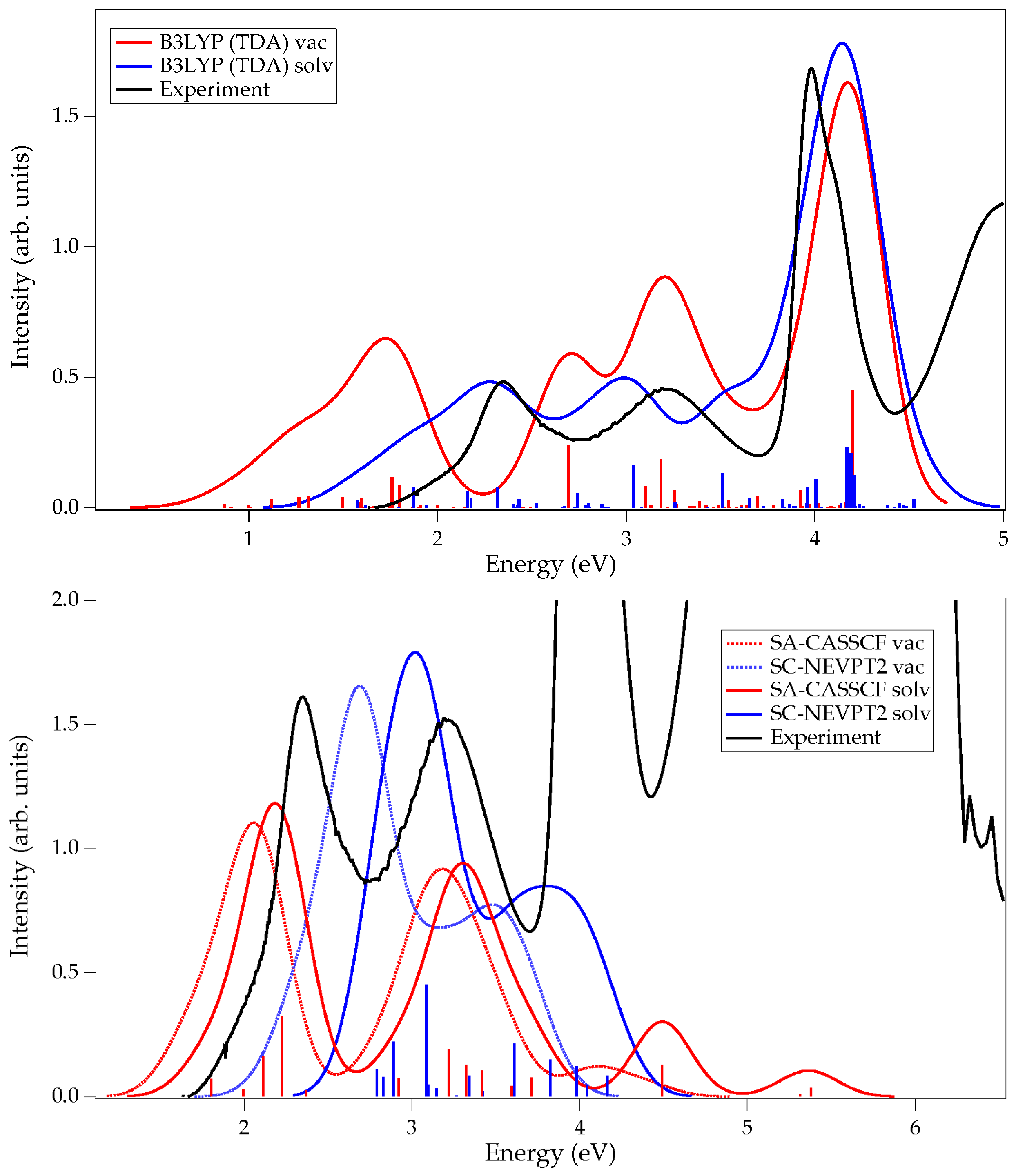
| Emax (I) | Emax (II) | E (III) | ∆E (I-II) | ∆E (II-III) | ||
|---|---|---|---|---|---|---|
| Experimental (H2O, pH = 1) | 2.35 | 3.19 | 3.99 | 0.84 | 0.80 | |
| Calculated | 15 states | |||||
| SA-CASSCF solv | 2.18 | 3.30 | - | 1.12 | - | |
| SC-NEVPT2 solv | 3.24 | 4.13 | - | 0.89 | - | |
| TDA-B3LYP | ||||||
| Gas phase | 1.72 | 2.70–3.20 | 4.17 | - | - | |
| Water | 2.27 a/2.39 b | 2.99 a/2.95 b | 4.14 a/3.98 b | 0.72 a/0.56 b | 1.15 a/1.03 b | |
| System | Γ (eV) | DOS (States/eV) | kinj (s−1) |
|---|---|---|---|
| Dye* + TiO2 → Dye+ + TiO2 (e−) | |||
| dye1-TiO2 | 0.1128 | 106 | 1.6 × 1014 |
| Dye* + IrO2 → Dye+ + IrO2 (e−) | |||
| dye1-IrO2 | 2.5 × 10−4 | 37 | 3.3 × 1011 |
| IrO2 + Dye (h+) → IrO2 (h+) + Dye | |||
| dye1-IrO2 | 4.5 × 10−4 | 48 | 1.0 × 1012 |
| TiO2-dye1-IrO2 | 2.7 × 10−4 | 45 | 0.5 × 1011 |
| TiO2 (e−) + Dye (h+) → TiO2 + Dye | |||
| dye1-TiO2 | 6.3 × 10−11 | 1.5 × 10−9 | 1.0 × 104 |
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pastore, M. First Principle Modelling of Materials and Processes in Dye-Sensitized Photoanodes for Solar Energy and Solar Fuels. Computation 2017, 5, 5. https://0-doi-org.brum.beds.ac.uk/10.3390/computation5010005
Pastore M. First Principle Modelling of Materials and Processes in Dye-Sensitized Photoanodes for Solar Energy and Solar Fuels. Computation. 2017; 5(1):5. https://0-doi-org.brum.beds.ac.uk/10.3390/computation5010005
Chicago/Turabian StylePastore, Mariachiara. 2017. "First Principle Modelling of Materials and Processes in Dye-Sensitized Photoanodes for Solar Energy and Solar Fuels" Computation 5, no. 1: 5. https://0-doi-org.brum.beds.ac.uk/10.3390/computation5010005