The Impact of Stochasticity and Its Control on a Model of the Inflammatory Response

 ,
,
Abstract
:1. Introduction
2. Methods
2.1. Four-Variable Model
2.2. SDE Formulation and Integration
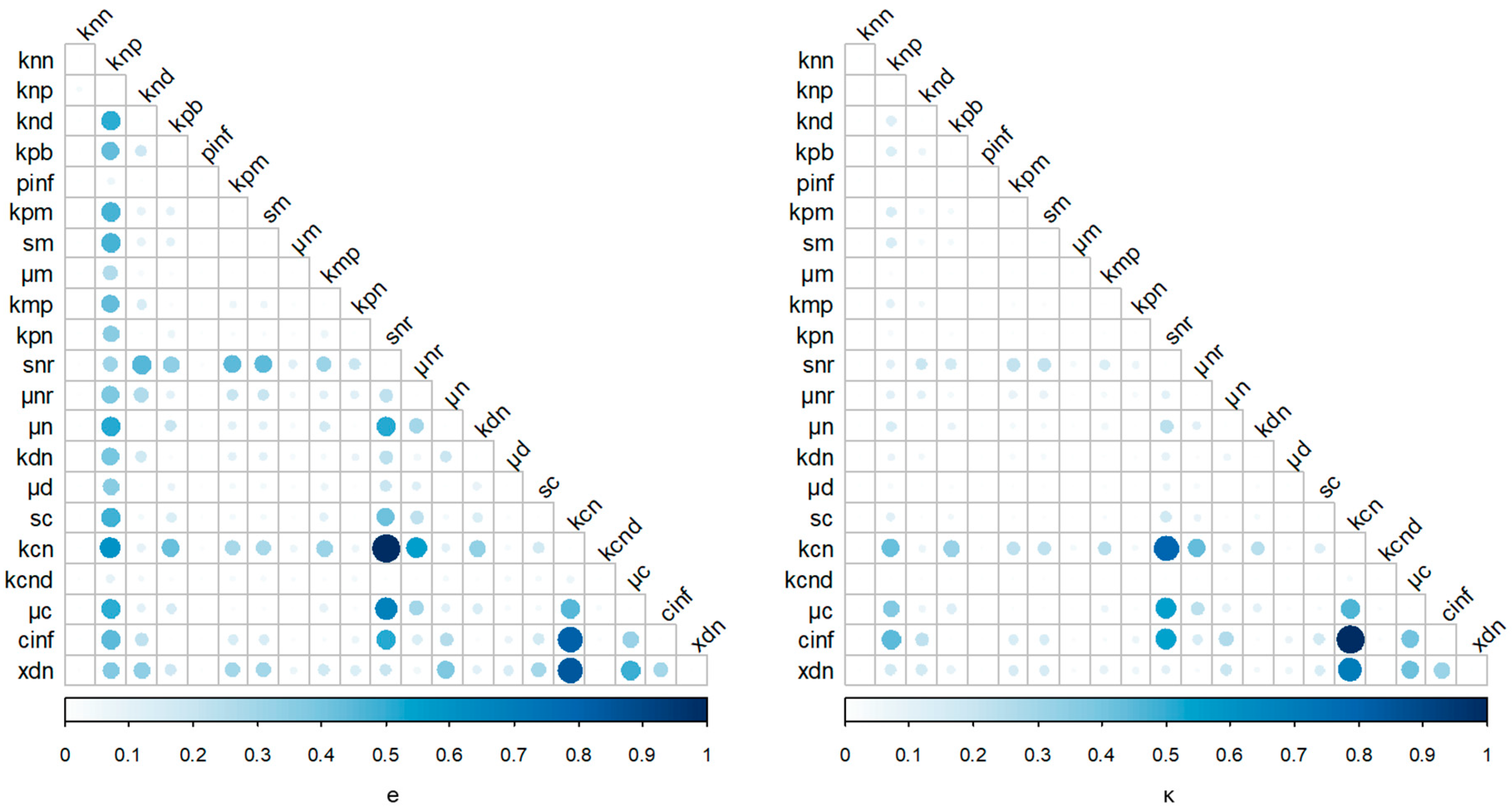
2.3. Stochastic Control Analysis
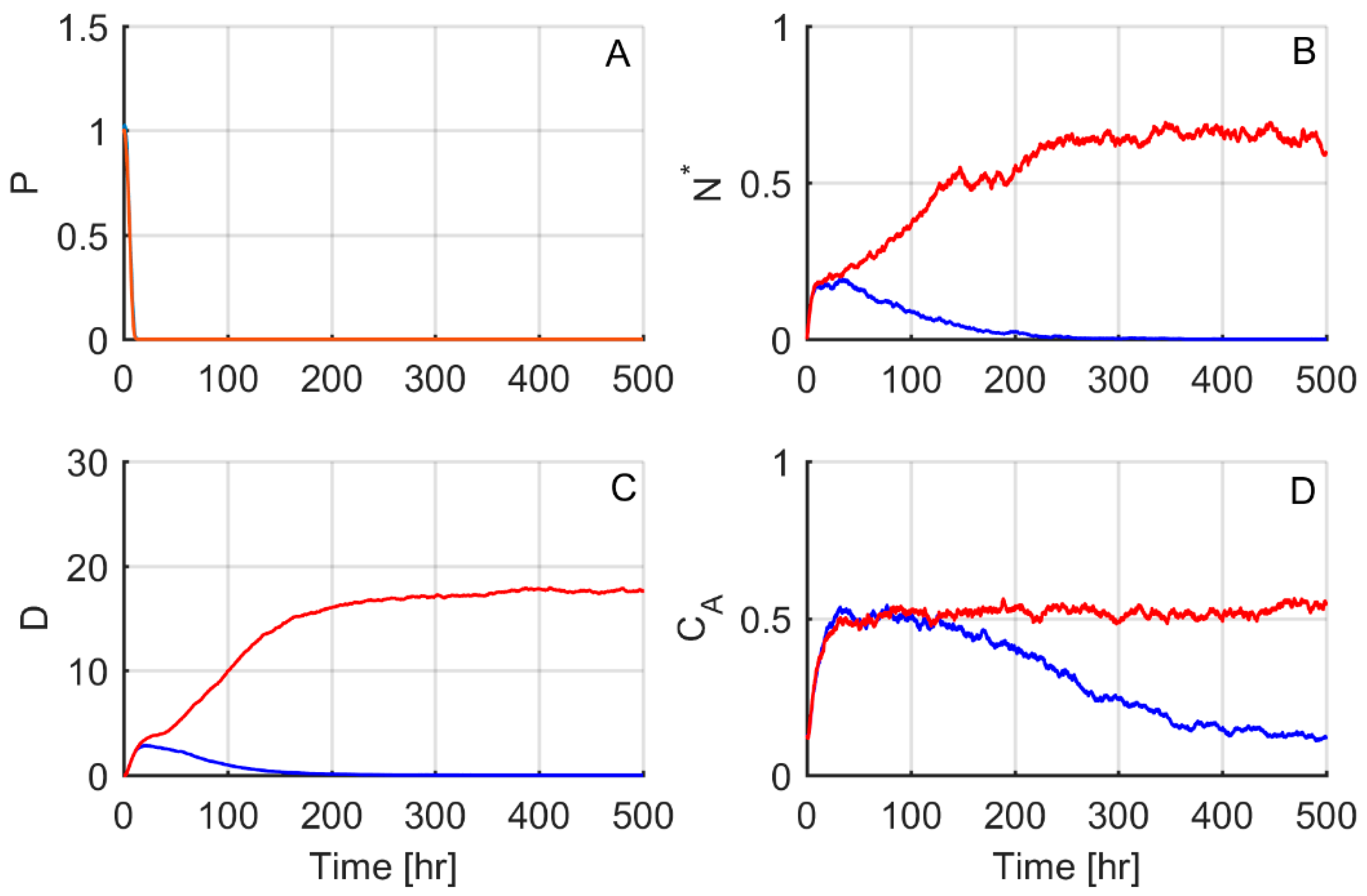
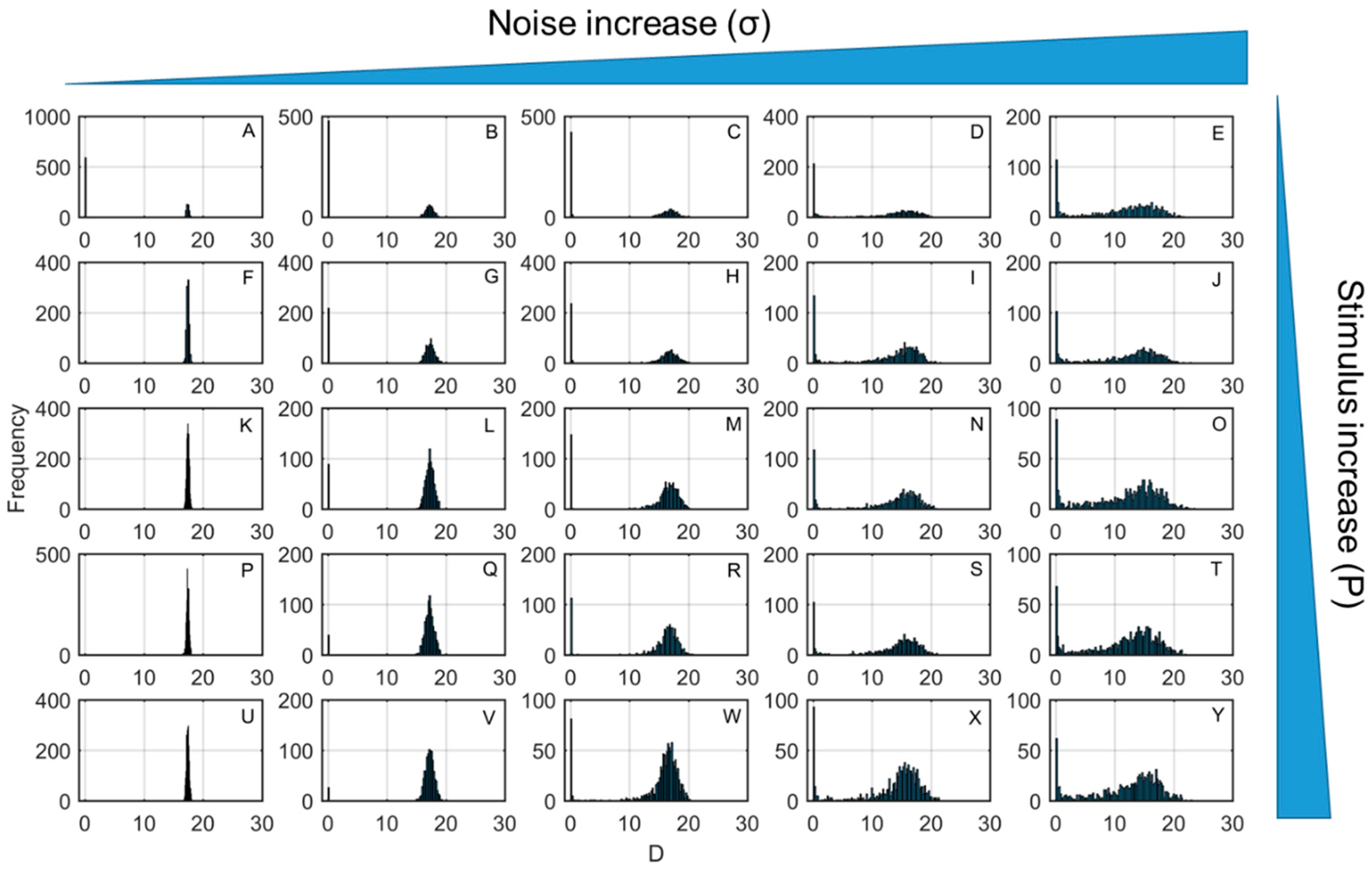
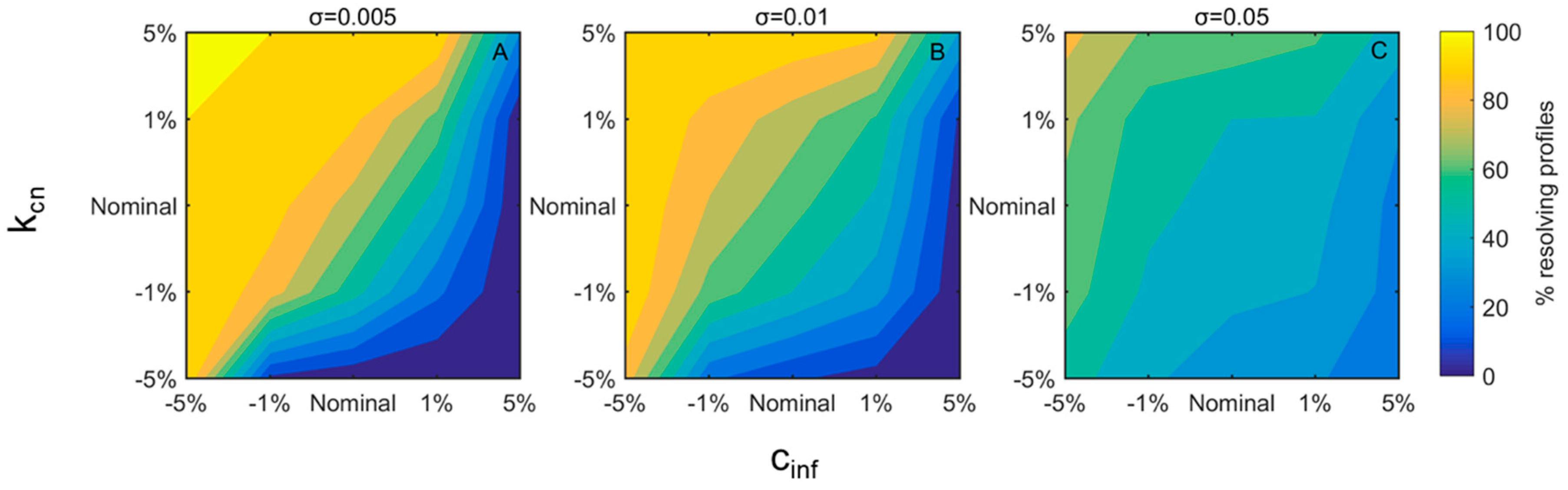
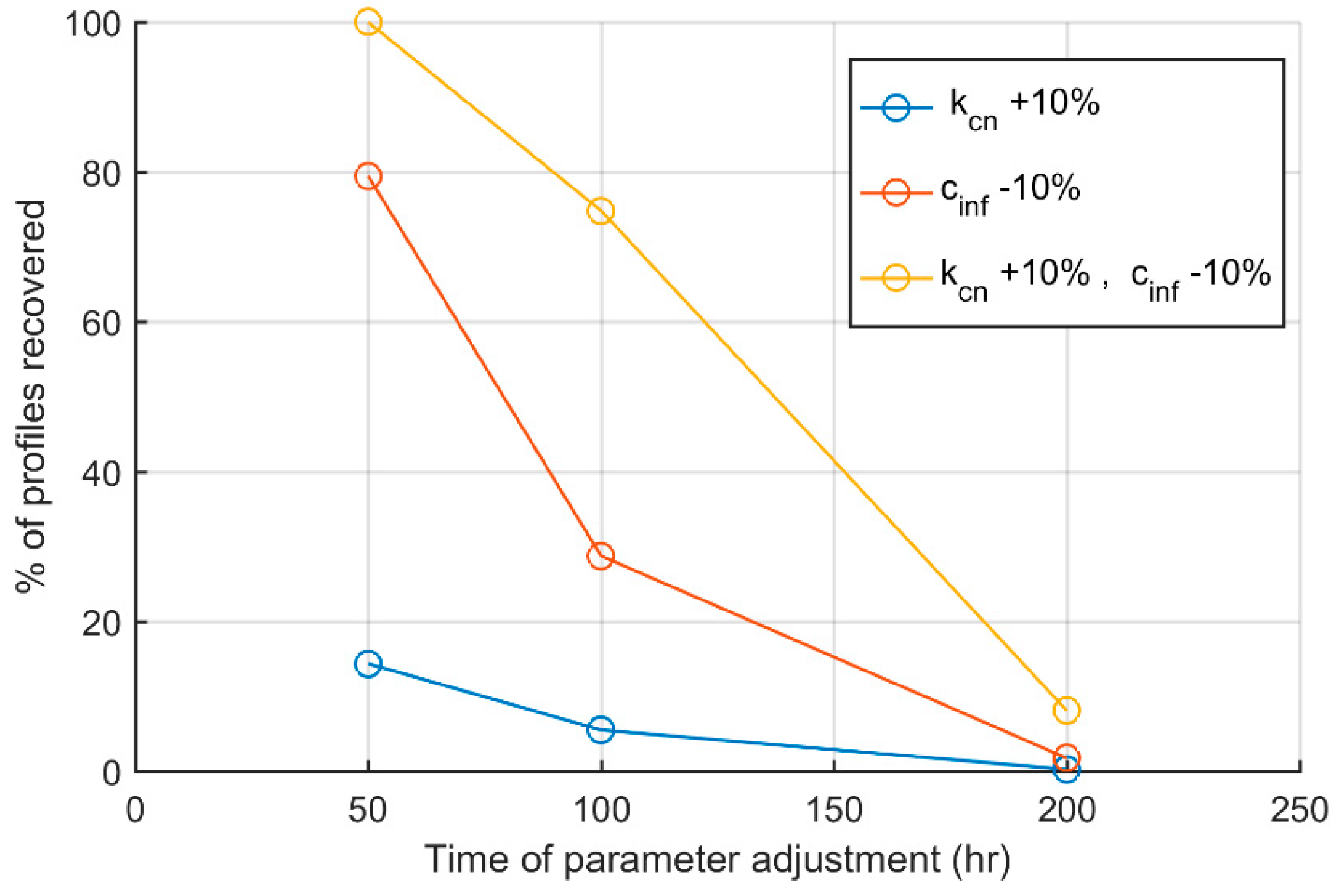
3. Results
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Nathan, C. Points of control in inflammation. Nature 2002, 420, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Reichlin, S. Neuroendocrine-immune interactions. N. Engl. J. Med. 1993, 329, 1246–1253. [Google Scholar]
- Tracey, K.J. The inflammatory reflex. Nature 2002, 420, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Bone, R.C. Immunologic dissonance: A continuing evolution in our understanding of the systemic inflammatory response syndrome (SIRS) and the multiple organ dysfunction syndrome (MODS). Ann. Intern. Med. 1996, 125, 680–687. [Google Scholar] [CrossRef]
- Seely, A.J.; Christou, N.V. Multiple organ dysfunction syndrome: Exploring the paradigm of complex nonlinear systems. Crit. Care Med. 2000, 28, 2193–2200. [Google Scholar] [CrossRef] [PubMed]
- Namas, R.; Zamora, R.; Namas, R.; An, G.; Doyle, J.; Dick, T.E.; Jacono, F.J.; Androulakis, I.P.; Chang, S.; Billiar, T.R.; et al. Sepsis: Something old, something new, and a systems view. J. Crit. Care 2012, 27, 314.e1–314.e11. [Google Scholar] [CrossRef] [PubMed]
- Vodovotz, Y.; Csete, M.; Bartels, J.; Chang, S.; An, G. Translational systems biology of inflammation. PLoS Comput. Biol. 2008, 4, e1000014. [Google Scholar] [CrossRef]
- Vodovotz, Y.; Constantine, G.; Rubin, J.; Csete, M.; Voit, E.O.; An, G. Mechanistic simulations of inflammation: Current state and future prospects. Math. Biosci. 2009, 217, 1–10. [Google Scholar] [CrossRef]
- Vodovotz, Y.; Clermont, G.; Chow, C.; An, G. Mathematical models of the acute inflammatory response. Curr. Opin. Crit. Care 2004, 10, 383–390. [Google Scholar] [CrossRef]
- Prince, J.M.; Levy, R.M.; Bartels, J.; Baratt, A.; Kane, J.M., III; Lagoa, C.; Rubin, J.; Day, J.; Wei, J.; Fink, M.P. In silico and in vivo approach to elucidate the inflammatory complexity of CD14-deficient mice. Mol. Med. 2006, 12, 88. [Google Scholar] [CrossRef]
- Ben-David, I.; Price, S.E.; Bortz, D.M.; Greineder, C.F.; Cohen, S.E.; Bauer, A.L.; Jackson, T.L.; Younger, J.G. Dynamics of intrapulmonary bacterial growth in a murine model of repeated microaspiration. Am. J. Respir. Cell Mol. Boil. 2005, 33, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.C.; Clermont, G.; Kumar, R.; Lagoa, C.; Tawadrous, Z.; Gallo, D.; Betten, B.; Bartels, J.; Constantine, G.; Fink, M.P. The acute inflammatory response in diverse shock states. Shock 2005, 24, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Clermont, G.; Vodovotz, Y.; Chow, C.C. The dynamics of acute inflammation. J. Theor. Boil. 2004, 230, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Foteinou, P.; Calvano, S.; Lowry, S.; Androulakis, I. Modeling endotoxin-induced systemic inflammation using an indirect response approach. Math. Biosci. 2009, 217, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Vodovotz, Y.; Chow, C.C.; Bartels, J.; Lagoa, C.; Prince, J.M.; Levy, R.M.; Kumar, R.; Day, J.; Rubin, J.; Constantine, G. In silico models of acute inflammation in animals. Shock 2006, 26, 235–244. [Google Scholar] [CrossRef]
- Alt, W.; Lauffenburger, D.A. Transient behavior of a chemotaxis system modelling certain types of tissue inflammation. J. Math. Boil. 1987, 24, 691–722. [Google Scholar] [CrossRef]
- Reynolds, A.; Rubin, J.; Clermont, G.; Day, J.; Vodovotz, Y.; Ermentrout, G.B. A reduced mathematical model of the acute inflammatory response: I. Derivation of model and analysis of anti-inflammation. J. Theor. Boil. 2006, 242, 220–236. [Google Scholar] [CrossRef]
- Day, J.; Rubin, J.; Vodovotz, Y.; Chow, C.C.; Reynolds, A.; Clermont, G. A reduced mathematical model of the acute inflammatory response II. Capturing scenarios of repeated endotoxin administration. J. Theor. Boil. 2006, 242, 237–256. [Google Scholar] [CrossRef]
- An, G. Agent-based computer simulation and sirs: Building a bridge between basic science and clinical trials. Shock (Augusta, GA) 2001, 16, 266–273. [Google Scholar] [CrossRef]
- An, G. In silico experiments of existing and hypothetical cytokine-directed clinical trials using agent-based modeling. Crit. Care Med. 2004, 32, 2050–2060. [Google Scholar] [CrossRef]
- Cockrell, C.; An, G. Sepsis Reconsidered: Identifying novel metrics for behavioral landscape characterization with a high-performance computing implementation of an agent-based model. J. Theor. Boil. 2017, 430, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Scheff, J.D.; Mavroudis, P.D.; Calvano, S.E.; Lowry, S.F.; Androulakis, I.P. Modeling autonomic regulation of cardiac function and heart rate variability in human endotoxemia. Physiol. Genom. 2011, 43, 951–964. [Google Scholar] [CrossRef] [PubMed]
- Scheff, J.D.; Mavroudis, P.D.; Calvano, S.E.; Androulakis, I.P. Translational applications of evaluating physiologic variability in human endotoxemia. J. Clin. Monit. Comput. 2013, 27, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Scheff, J.D.; Mavroudis, P.D.; Foteinou, P.T.; Calvano, S.E.; Androulakis, I.P. Modeling physiologic variability in human endotoxemia. Crit. Rev. Biomed. Eng. 2012, 40, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Scheff, J.D.; Mavroudis, P.D.; Foteinou, P.T.; An, G.; Calvano, S.E.; Doyle, J.; Dick, T.E.; Lowry, S.F.; Vodovotz, Y.; Androulakis, I.P. A multiscale modeling approach to inflammation: A case study in human endotoxemia. J. Comput. Phys. 2013, 244, 279–289. [Google Scholar] [CrossRef]
- Scheff, J.D.; Mavroudis, P.; Calvano, S.E.; Lowry, S.F.; Androulakis, I.P. Towards in silico models of decomplexification in human endotoxemia. Comput. Aided Chem. Eng. 2011, 29, 1485–1489. [Google Scholar]
- Brown, E.N.; Meehan, P.M.; Dempster, A.P. A stochastic differential equation model of diurnal cortisol patterns. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E450–E461. [Google Scholar] [CrossRef]
- Lipniacki, T.; Paszek, P.; Brasier, A.R.; Luxon, B.A.; Kimmel, M. Stochastic regulation in early immune response. Biophys. J. 2006, 90, 725–742. [Google Scholar] [CrossRef]
- Dong, X.; Foteinou, P.T.; Calvano, S.E.; Lowry, S.F.; Androulakis, I.P. Agent-based modeling of endotoxin-induced acute inflammatory response in human blood leukocytes. PLoS ONE 2010, 5, e9249. [Google Scholar] [CrossRef]
- An, G. Introduction of an agent-based multi-scale modular architecture for dynamic knowledge representation of acute inflammation. Theor. Biol. Med. Model. 2008, 5, 11. [Google Scholar] [CrossRef]
- Mi, Q.; Riviere, B.; Clermont, G.; Steed, D.L.; Vodovotz, Y. Agent-based model of inflammation and wound healing: Insights into diabetic foot ulcer pathology and the role of transforming growth factor-beta1. Wound Repair Regen. 2007, 15, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Ziraldo, C.; Solovyev, A.; Allegretti, A.; Krishnan, S.; Henzel, M.K.; Sowa, G.A.; Brienza, D.; An, G.; Mi, Q.; Vodovotz, Y. A computational, tissue-realistic model of pressure ulcer formation in Individuals with spinal cord injury. PLoS Comput. Biol. 2015, 11, e1004309. [Google Scholar] [CrossRef]
- Foteinou, P.T.; Calvano, S.E.; Lowry, S.F.; Androulakis, I.P. A physiological model for autonomic heart rate regulation in human endotoxemia. Shock 2011, 35, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Foteinou, P.T.; Calvano, S.E.; Lowry, S.F.; Androulakis, I.P. Multiscale model for the assessment of autonomic dysfunction in human endotoxemia. Physiol. Genomics 2010, 42, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Scheff, J.D.; Calvano, S.E.; Lowry, S.F.; Androulakis, I.P. Modeling the influence of circadian rhythms on the acute inflammatory response. J. Theor. Biol. 2010, 264, 1068–1076. [Google Scholar] [CrossRef]
- Torres, A.; Bentley, T.; Bartels, J.; Sarkar, J.; Barclay, D.; Namas, R.; Constantine, G.; Zamora, R.; Puyana, J.C.; Vodovotz, Y. Mathematical modeling of posthemorrhage inflammation in mice: Studies using a novel, computer-controlled, closed-loop hemorrhage apparatus. Shock 2009, 32, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.; Namas, R.A.; Almahmoud, K.; Zaaqoq, A.; Sarkar, J.; Barclay, D.A.; Yin, J.; Ghuma, A.; Abboud, A.; Constantine, G.; et al. Trauma in silico: Individual-specific mathematical models and virtual clinical populations. Sci. Transl. Med. 2015, 7, 285ra261. [Google Scholar] [CrossRef] [PubMed]
- Arciero, J.C.; Ermentrout, G.B.; Upperman, J.S.; Vodovotz, Y.; Rubin, J.E. Using a mathematical model to analyze the role of probiotics and inflammation in necrotizing enterocolitis. PLoS ONE 2010, 5, e10066. [Google Scholar] [CrossRef] [PubMed]
- Foteinou, P.T.; Calvano, S.E.; Lowry, S.F.; Androulakis, I.P. In silico simulation of corticosteroids effect on an NFkB-dependent physicochemical model of systemic inflammation. PLoS ONE 2009, 4, e4706. [Google Scholar] [CrossRef]
- Vodovotz, Y. Translational systems biology of inflammation and healing. Wound Repair Regen. 2010, 18, 3–7. [Google Scholar] [CrossRef]
- Clermont, G.; Bartels, J.; Kumar, R.; Constantine, G.; Vodovotz, Y.; Chow, C. In silico design of clinical trials: A method coming of age. Crit. Care Med. 2004, 32, 2061–2070. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Sauro, H.M. Adjusting phenotypes by noise control. PLoS Comput. Biol. 2012, 8, e1002344. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, D.T. Exact stochastic simulation of coupled chemical reactions. J. Phys. Chem. 1977, 81, 2340–2361. [Google Scholar] [CrossRef]
- Higham, D.J. An algorithmic introduction to numerical simulation of stochastic differential equations. SIAM Rev. Soc. Ind. Appl. Math. 2001, 43, 525–546. [Google Scholar] [CrossRef]
- Saarinen, A.; Linne, M.L.; Yli-Harja, O. Modeling single neuron behavior using stochastic differential equations. Neurocomputing 2006, 69, 1091–1096. [Google Scholar] [CrossRef]
- Saarinen, A.; Linne, M.L.; Yli-Harja, O. Stochastic differential equation model for cerebellar granule cell excitability. PLoS Comput. Biol. 2008, 4, e1000004. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.C.; Wang, T.Y.; Tseng, H.H.; Huang, C.Y.; Kao, C.Y. A stochastic differential equation model for quantifying transcriptional regulatory network in Saccharomyces cerevisiae. Bioinformatics 2005, 21, 2883–2890. [Google Scholar] [CrossRef]
- Higham, D.J. Stochastic ordinary differential equations in applied and computational mathematics. Univ. Strathclyde Math. Stat. Res. Rep. 2010, 7, 1–32. [Google Scholar] [CrossRef]
- Lamba, H. An adaptive timestepping algorithm for stochastic differential equations. J. Comput. Appl. Math. 2003, 161, 417–430. [Google Scholar] [CrossRef]
- Rao, C.V.; Wolf, D.M.; Arkin, A.P. Control, exploitation and tolerance of intracellular noise. Nature 2002, 420, 231–237. [Google Scholar] [CrossRef]
- Arkin, A.; Ross, J.; McAdams, H.H. Stochastic kinetic analysis of developmental pathway bifurcation in phage lambda-infected Escherichia coli cells. Genetics 1998, 149, 1633–1648. [Google Scholar] [PubMed]
- Samoilov, M.; Plyasunov, S.; Arkin, A.P. Stochastic amplification and signaling in enzymatic futile cycles through noise-induced bistability with oscillations. Proc. Natl. Acad. Sci. USA 2005, 102, 2310–2315. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Shi, P.Z.; Xing, J. Stochastic bifurcation, slow fluctuations, and bistability as an origin of biochemical complexity. Phys. Chem. Chem. Phys. 2009, 11, 4861–4870. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Horng, T. Transcriptional control of the inflammatory response. Nat. Rev. Immunol. 2009, 9, 692–703. [Google Scholar] [CrossRef] [PubMed]
- Majno, G.; Joris, I. Cells, Tissues, and Disease: Principles of General Pathology; Oxford University Press: Oxford, UK, 2004. [Google Scholar]
- Serhan, C.N.; Savill, J. Resolution of inflammation: The beginning programs the end. Nat. Immunol. 2005, 6, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Andersson, U.; Tracey, K.J. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu. Rev. Immunol. 2011, 29, 139–162. [Google Scholar] [CrossRef]
- Beutler, B. Microbe sensing, positive feedback loops, and the pathogenesis of inflammatory diseases. Immunol. Rev. 2009, 227, 248–263. [Google Scholar] [CrossRef]
- Krzyzanski, W.; Jusko, W.J. Integrated functions for four basic models of indirect pharmacodynamic response. J. Pharm. Sci. 1998, 87, 67–72. [Google Scholar] [CrossRef]
- Scheff, J.D.; Almon, R.R.; Dubois, D.C.; Jusko, W.J.; Androulakis, I.P. Assessment of pharmacologic area under the curve when baselines are variable. Pharm. Res. 2011, 28, 1081–1089. [Google Scholar] [CrossRef] [PubMed]
- Eldar, A.; Elowitz, M.B. Functional roles for noise in genetic circuits. Nature 2010, 467, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Balazsi, G.; van Oudenaarden, A.; Collins, J.J. Cellular decision making and biological noise: From microbes to mammals. Cell 2011, 144, 910–925. [Google Scholar] [CrossRef] [PubMed]
- Qian, H. Cooperativity in cellular biochemical processes: Noise-enhanced sensitivity, fluctuating enzyme, bistability with nonlinear feedback, and other mechanisms for sigmoidal responses. Annu. Rev. Biophys. 2012, 41, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Blake, W.J.; Kærn, M.; Cantor, C.R.; Collins, J.J. Noise in eukaryotic gene expression. Nature 2003, 422, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Paulsson, J.; Berg, O.G.; Ehrenberg, M. Stochastic focusing: Fluctuation-enhanced sensitivity of intracellular regulation. Proc. Natl. Acad. Sci. USA 2000, 97, 7148–7153. [Google Scholar] [CrossRef] [PubMed]
- Kærn, M.; Elston, T.C.; Blake, W.J.; Collins, J.J. Stochasticity in gene expression: From theories to phenotypes. Nat. Rev. Genet. 2005, 6, 451–464. [Google Scholar] [CrossRef]
- Kussell, E.; Leibler, S. Phenotypic diversity, population growth, and information in fluctuating environments. Science 2005, 309, 2075–2078. [Google Scholar] [CrossRef]
- Read, E.L.; Tovo-Dwyer, A.A.; Chakraborty, A.K. Stochastic effects are important in intrahost HIV evolution even when viral loads are high. Proc. Natl Acad. Sci. USA 2012, 109, 19727–19732. [Google Scholar] [CrossRef]
- Luzyanina, T.; Bocharov, G. Stochastic modeling of the impact of random forcing on persistent hepatitis B virus infection. Math. Comput. Simul. 2014, 96, 54–65. [Google Scholar] [CrossRef]
- Wiesenfeld, K.; Moss, F. Stochastic resonance and the benefits of noise: From ice ages to crayfish and SQUIDs. Nature 1995, 373, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Hänggi, P. Stochastic resonance in biology how noise can enhance detection of weak signals and help improve biological information processing. ChemPhysChem 2002, 3, 285–290. [Google Scholar] [CrossRef]
- Longtin, A.; Bulsara, A.; Moss, F. Time-interval sequences in bistable systems and the noise-induced transmission of information by sensory neurons. Phys. Rev. Lett. 1991, 67, 656–659. [Google Scholar] [CrossRef] [PubMed]
- Douglass, J.K.; Wilkens, L.; Pantazelou, E.; Moss, F. Noise enhancement of information transfer in crayfish mechanoreceptors by stochastic resonance. Nature 1993, 365, 337–340. [Google Scholar] [CrossRef] [PubMed]
- Lowry, S.F. The stressed host response to infection: The disruptive signals and rhythms of systemic inflammation. Surg. Clin. North Am. 2009, 89, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Lowry, S.F.; Calvano, S.E. Challenges for modeling and interpreting the complex biology of severe injury and inflammation. J. Leukoc. Biol. 2008, 83, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Mi, Q.; Constantine, G.; Ziraldo, C.; Solovyev, A.; Torres, A.; Namas, R.; Bentley, T.; Billiar, T.R.; Zamora, R.; Puyana, J.C.; et al. A dynamic view of trauma/hemorrhage-induced inflammation in mice: Principal drivers and networks. PLoS ONE 2011. [Google Scholar] [CrossRef]
- Mavroudis, P.D.; Corbett, S.A.; Calvano, S.E.; Androulakis, I.P. Circadian characteristics of permissive and suppressive effects of cortisol and their role in homeostasis and the acute inflammatory response. Math. Biosci. 2015, 260, 54–64. [Google Scholar] [CrossRef]
- Foteinou, P.T.; Calvano, S.E.; Lowry, S.F.; Androulakis, I.P. Translational potential of systems-based models of inflammation. Clin. Transl. Sci. 2009, 2, 85–89. [Google Scholar] [CrossRef]
- Day, J.; Rubin, J.; Clermont, G. Using nonlinear model predictive control to find optimal therapeutic strategies to modulate inflammation. Math. Biosci. Eng. 2010, 7, 739–763. [Google Scholar] [CrossRef]
- Petersen, B.K.; Uang, J.; Grathwohl, W.S.; Cockrell, C.; Santiago, C.; An, G.; Faissol, D.M. Precision medicine as a control problem: Using simulation and deep reinforcement learning to discover adaptive, personalized multi-cytokine therapy for sepsis. arXiv, 2018; arXiv:1802.10440. [Google Scholar]
- Lestas, I.; Vinnicombe, G.; Paulsson, J. Fundamental limits on the suppression of molecular fluctuations. Nature 2010, 467, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Manninen, T.; Linne, M.L.; Ruohonen, K. Developing Ito stochastic differential equation models for neuronal signal transduction pathways. Comput. Biol. Chem. 2006, 30, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Steuer, R. Effects of stochasticity in models of the cell cycle: From quantized cycle times to noise-induced oscillations. J. Theor. Biol. 2004, 228, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.A.; O’Brien, J.M., Jr.; Dungan, K.; Phillips, G.; Marsh, C.B.; Lemeshow, S.; Connors, A.F., Jr.; Preiser, J.C. Glucose variability and mortality in patients with sepsis. Crit. Care Med. 2008, 36, 2316–2321. [Google Scholar] [CrossRef] [PubMed]
- Spapen, H.D. The glycemia threat in sepsis: Too high, too low, or too... variable! Crit. Care Med. 2008, 36, 2459–2460. [Google Scholar] [CrossRef] [PubMed]
- Annane, D.; Trabold, F.; Sharshar, T.; Jarrin, I.; Blanc, A.S.; Raphael, J.C.; Gajdos, P. Inappropriate sympathetic activation at onset of septic shock: A spectral analysis approach. Am. J. Respir. Crit. Care Med. 1999, 160, 458–465. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value | Units | Description |
|---|---|---|---|
| knn | 0.01 | N*-units/h | Activation of resting phagocytes by previously activated phagocytes and their cytokines |
| kpg | 0.3 | 1/h | The growth rate of pathogen |
| sm | 0.005 | M-units/h | Source of non-specific local response (M) |
| kpn | 1.8 | N*-units/h | Rate at which activated phagocytes (N*) consume pathogen |
| μn | 0.05 | 1/h | Decay rate of activated phagocytes |
| sc | 0.0125 | CA-units/h | Source of the anti-inflammatory mediator |
| μc | 0.1 | 1/h | Decay rate of the anti-inflammatory mediator |
| knp | 0.1 | P-units/h | Activation of resting phagocytes by previously activated phagocytes and their cytokines |
| pinf | 20 | cc | Maximum pathogen population |
| μm | 0.002 | 1/h | Decay rate for the non-specific local response |
| snr | 0.08 | NR-units/h | Source of resting phagocytes (NR) |
| kdn | 0.35 | D-units/h | Maximum rate of damage produced by activated phagocytes (and/or associated cytokines and free radicals) |
| kcn | 0.04 | CA-units/h | Maximum production rate of the anti-inflammatory mediator |
| cinf | 0.28 | CA-units | Controls the strength of the anti-inflammatory mediator (CA) |
| knd | 0.02 | D-units/h | Activation of resting phagocytes by tissue damage (D) |
| kpm | 0.6 | M-units/h | Rate at which the non-specific local response (M) eliminates pathogen |
| kmp | 0.01 | P-units/h | Rate at which the non-specific local response is exhausted by pathogen (P) |
| μnr | 0.12 | 1/h | Decay rate of resting phagocytes |
| μd | 0.02 | 1/h | Decay rate of damage; a combination of repair, resolution, and regeneration of tissue |
| kcnd | 48 | N*-units/D-units | Relative effectiveness of activated phagocytes and damaged tissue in inducing production of the anti-inflammatory mediator |
| xdn | 0.06 | N*-units | Determines level of activated phagocytes needed to bring damage production up to half its maximum |
| Variable | Initial Condition |
|---|---|
| Pathogen (P) | 1 |
| Tissue Damage (D) | 0 |
| Activated Phagocytes (N*) | 0 |
| Anti-inflammatory Mediators (CA) | 0.125 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mavroudis, P.D.; Scheff, J.D.; Doyle, J.C.; Vodovotz, Y.; Androulakis, I.P. The Impact of Stochasticity and Its Control on a Model of the Inflammatory Response. Computation 2019, 7, 3. https://0-doi-org.brum.beds.ac.uk/10.3390/computation7010003
Mavroudis PD, Scheff JD, Doyle JC, Vodovotz Y, Androulakis IP. The Impact of Stochasticity and Its Control on a Model of the Inflammatory Response. Computation. 2019; 7(1):3. https://0-doi-org.brum.beds.ac.uk/10.3390/computation7010003
Chicago/Turabian StyleMavroudis, Panteleimon D., Jeremy D. Scheff, John C. Doyle, Yoram Vodovotz, and Ioannis P. Androulakis. 2019. "The Impact of Stochasticity and Its Control on a Model of the Inflammatory Response" Computation 7, no. 1: 3. https://0-doi-org.brum.beds.ac.uk/10.3390/computation7010003