Influence of Varying Functionalization on the Peroxidase Activity of Nickel(II)–Pyridine Macrocycle Catalysts: Mechanistic Insights from Density Functional Theory


Abstract
:
1. Introduction
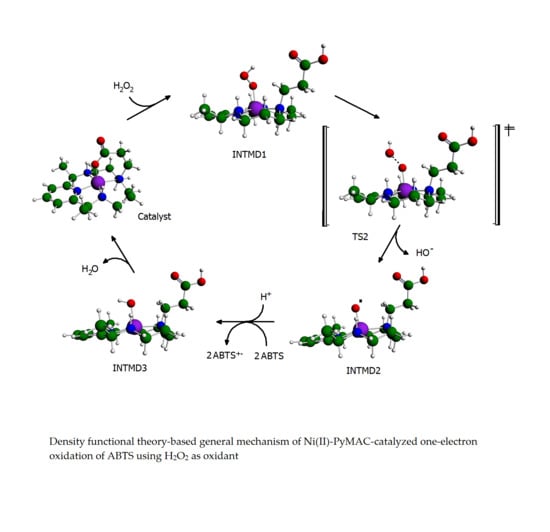
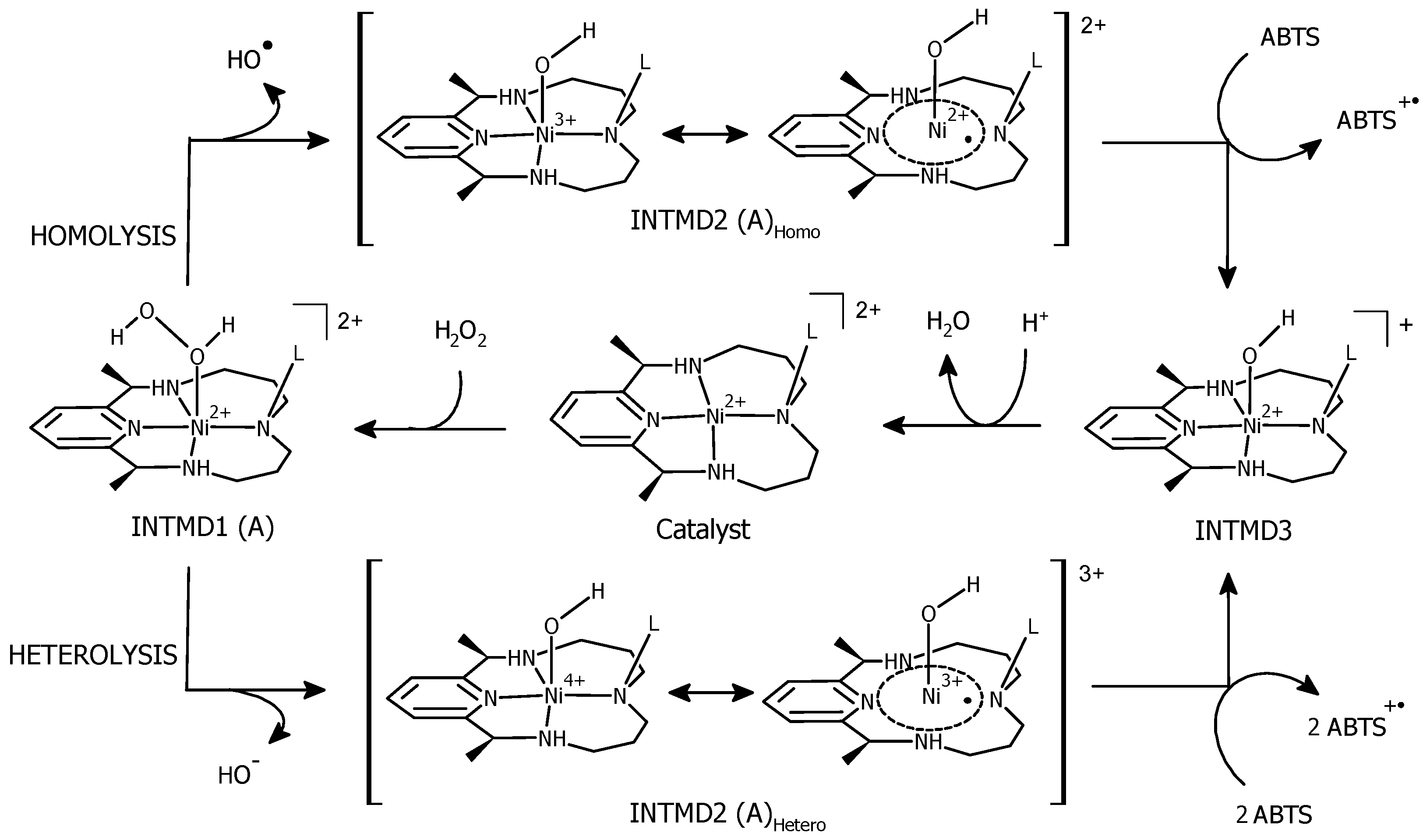
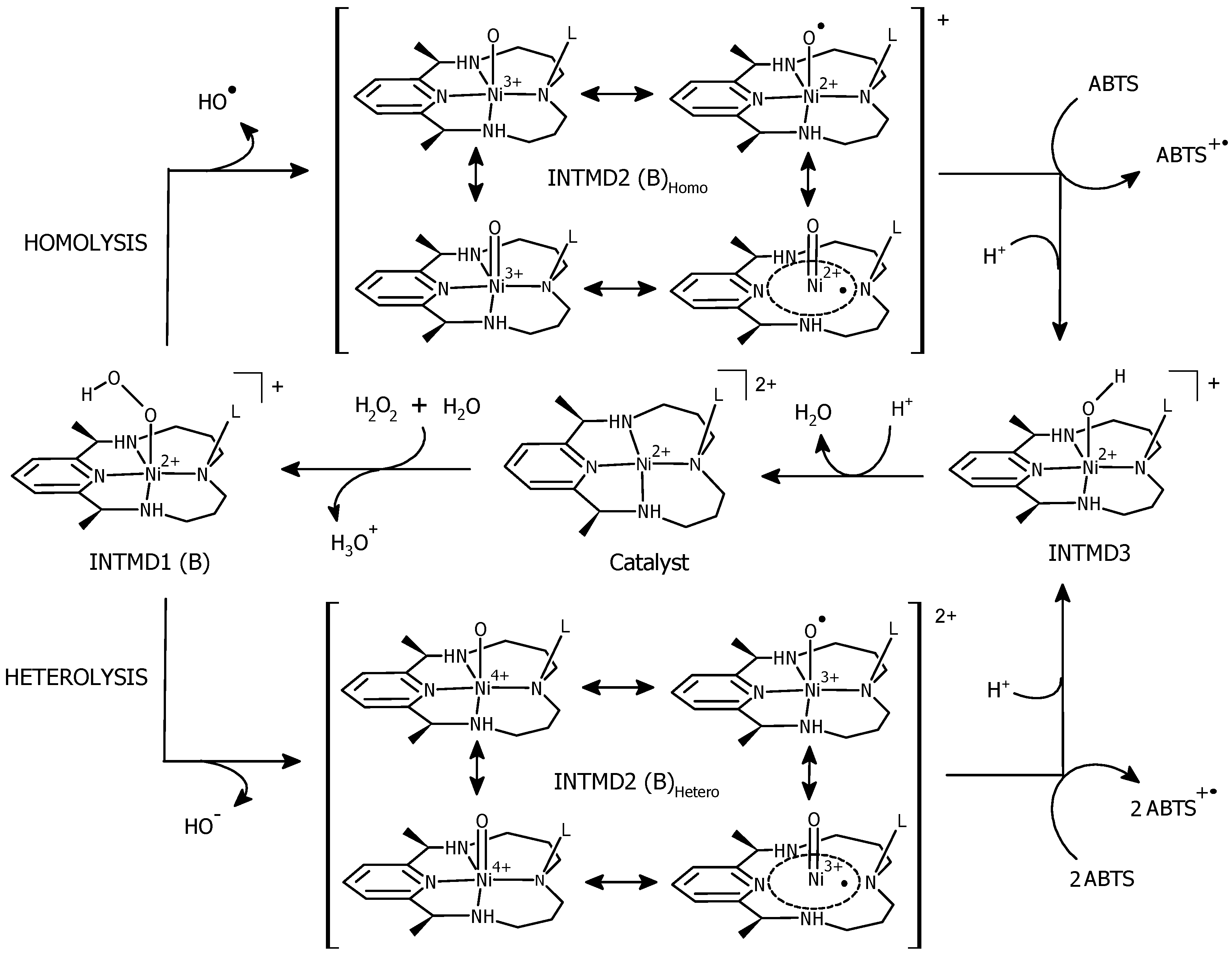
Proposed Reaction Pathways
2. Computational Methods
3. Results and Discussion
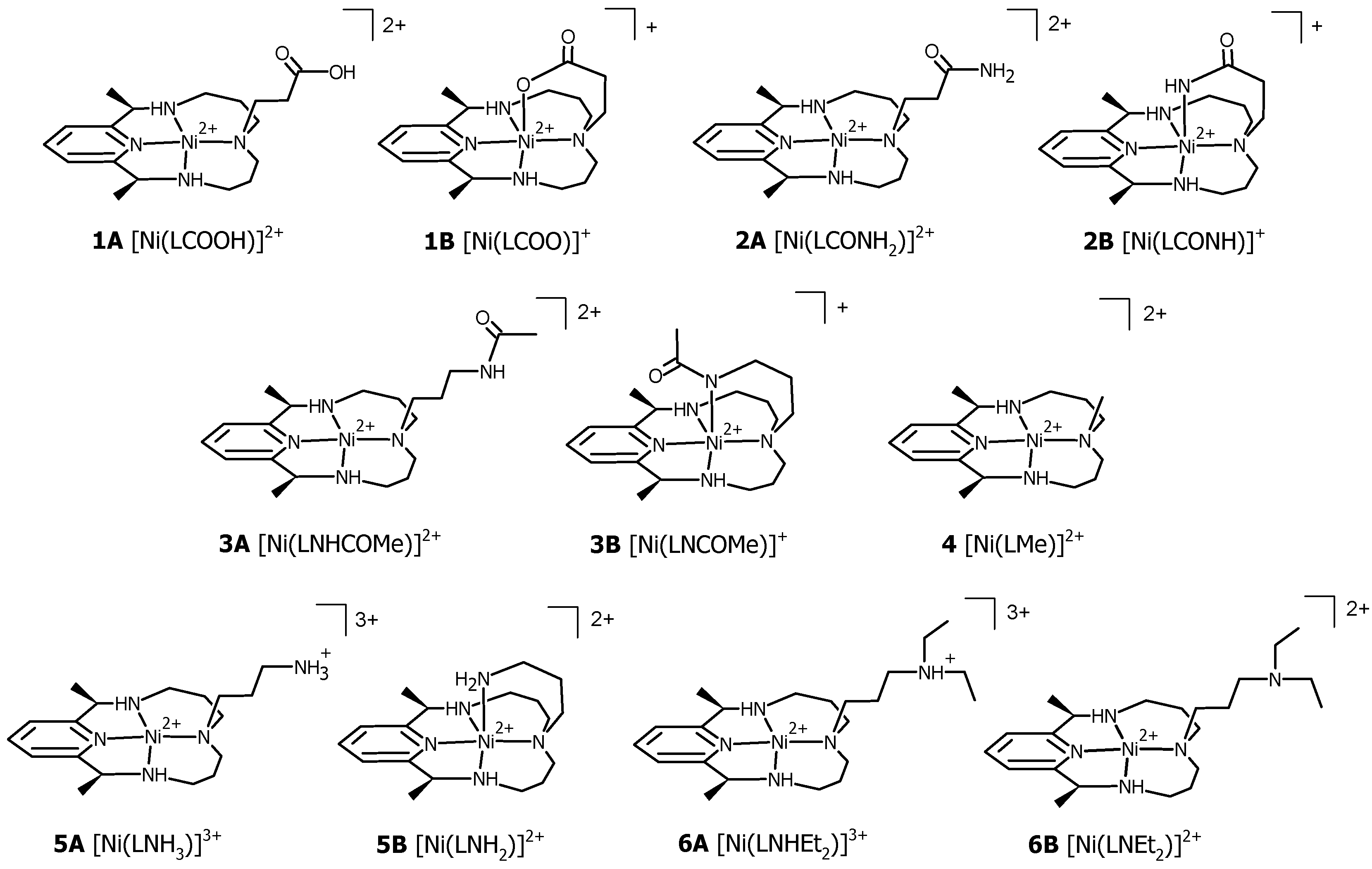
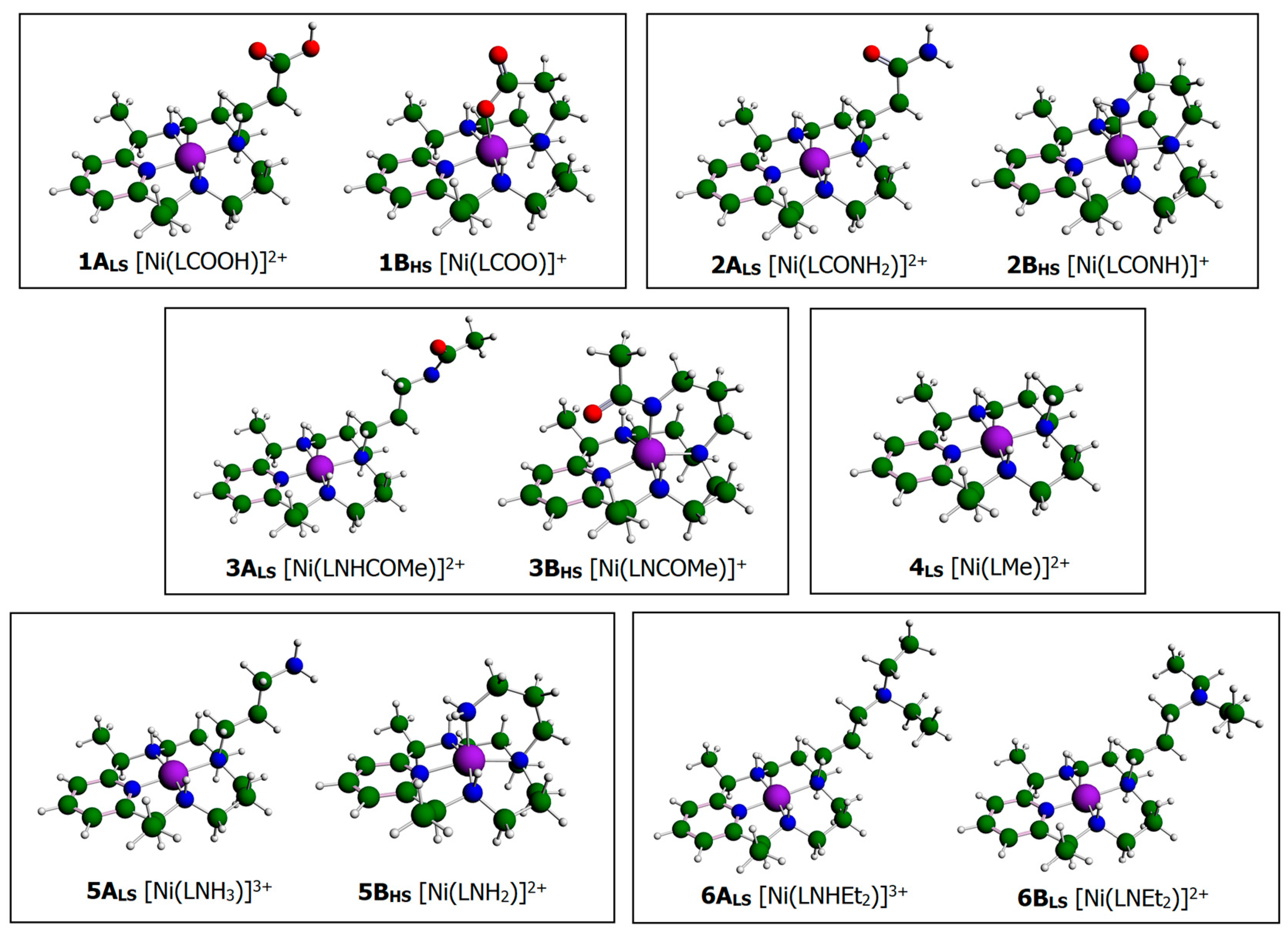
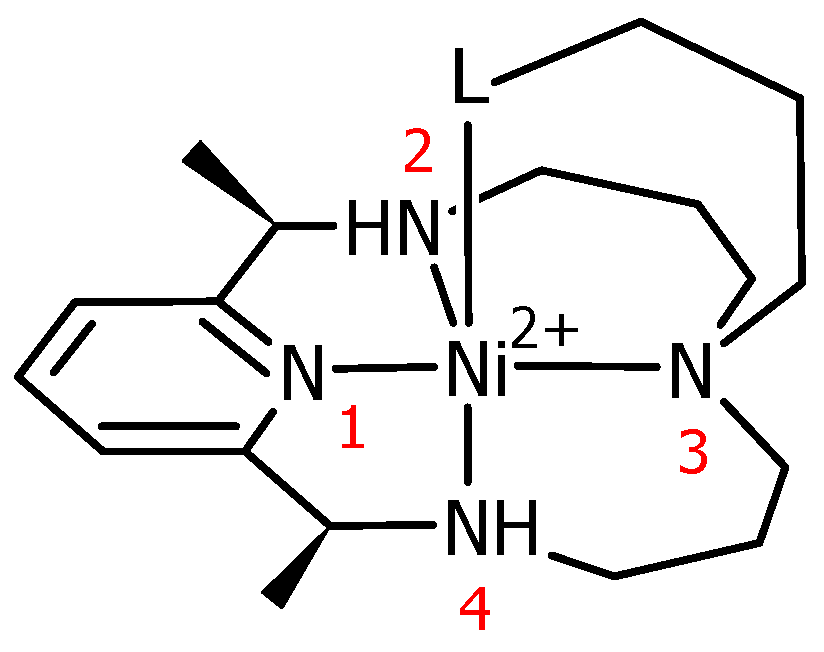
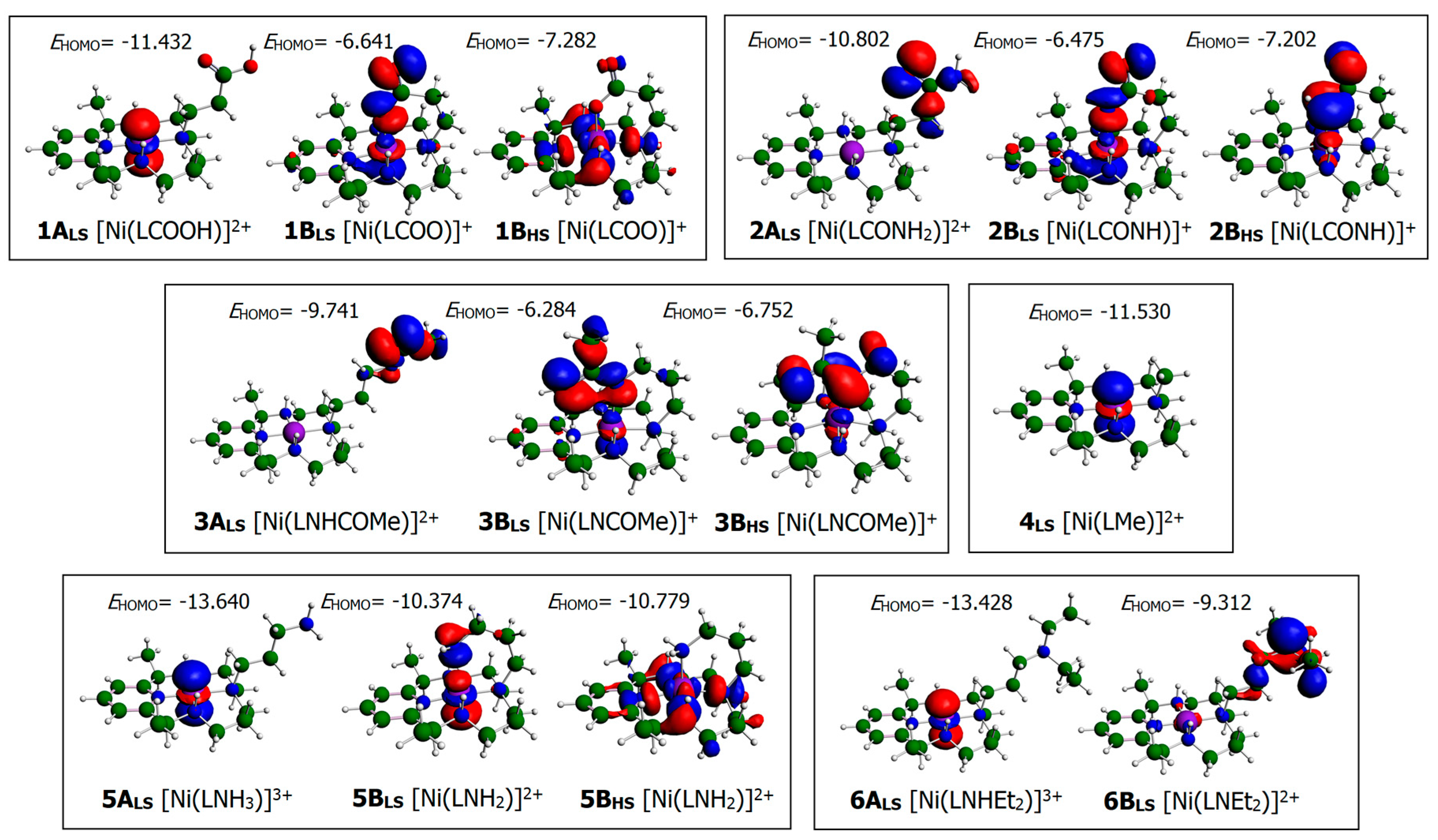
3.1. Structural Characterization of Ni(II)–PyMAC Complexes
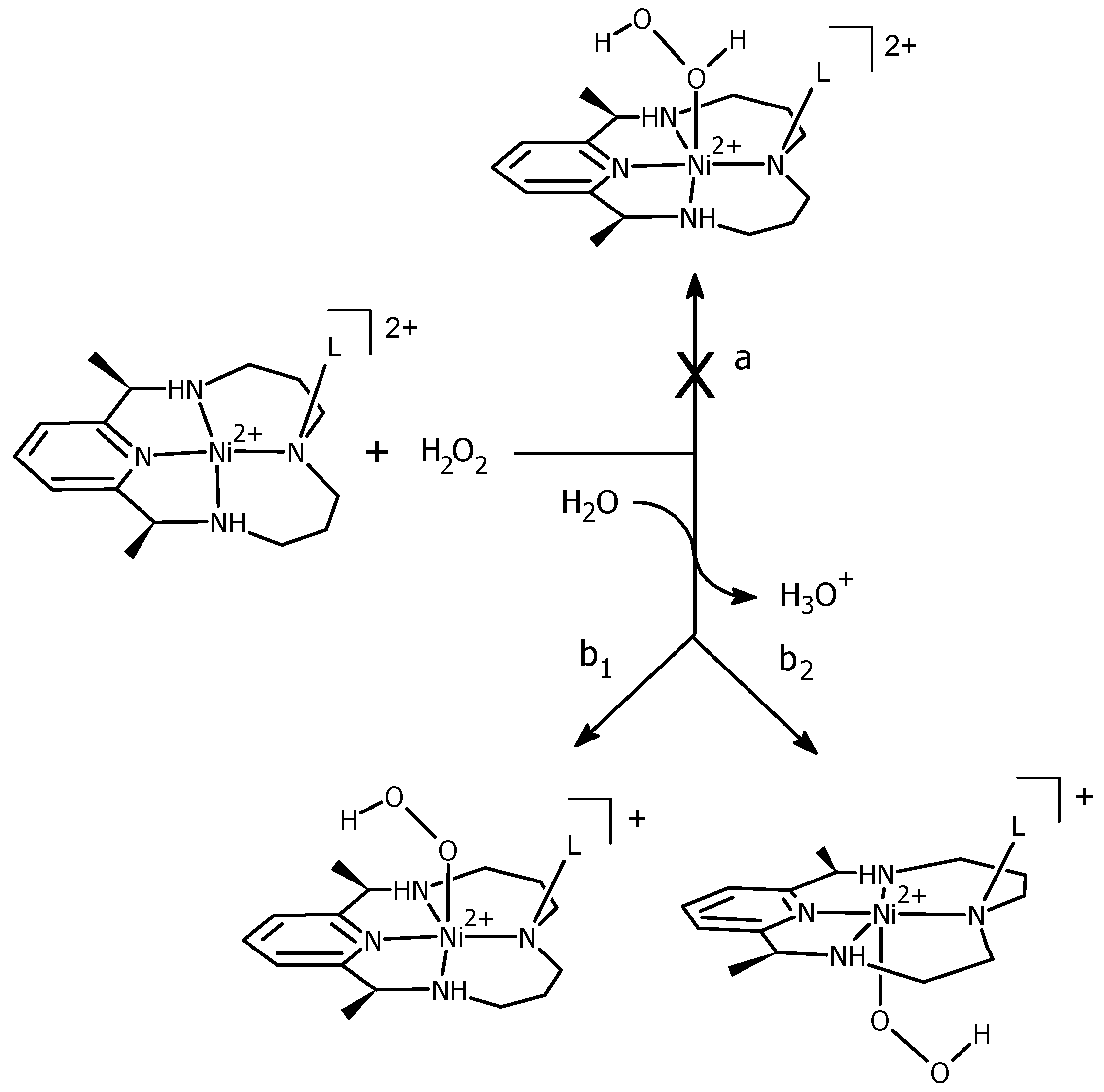
3.2. H2O2 Activation and Reactivity with Ni(II)–PyMACs
3.3. Structure and Energetics of Nickel-Hydroperoxo Species
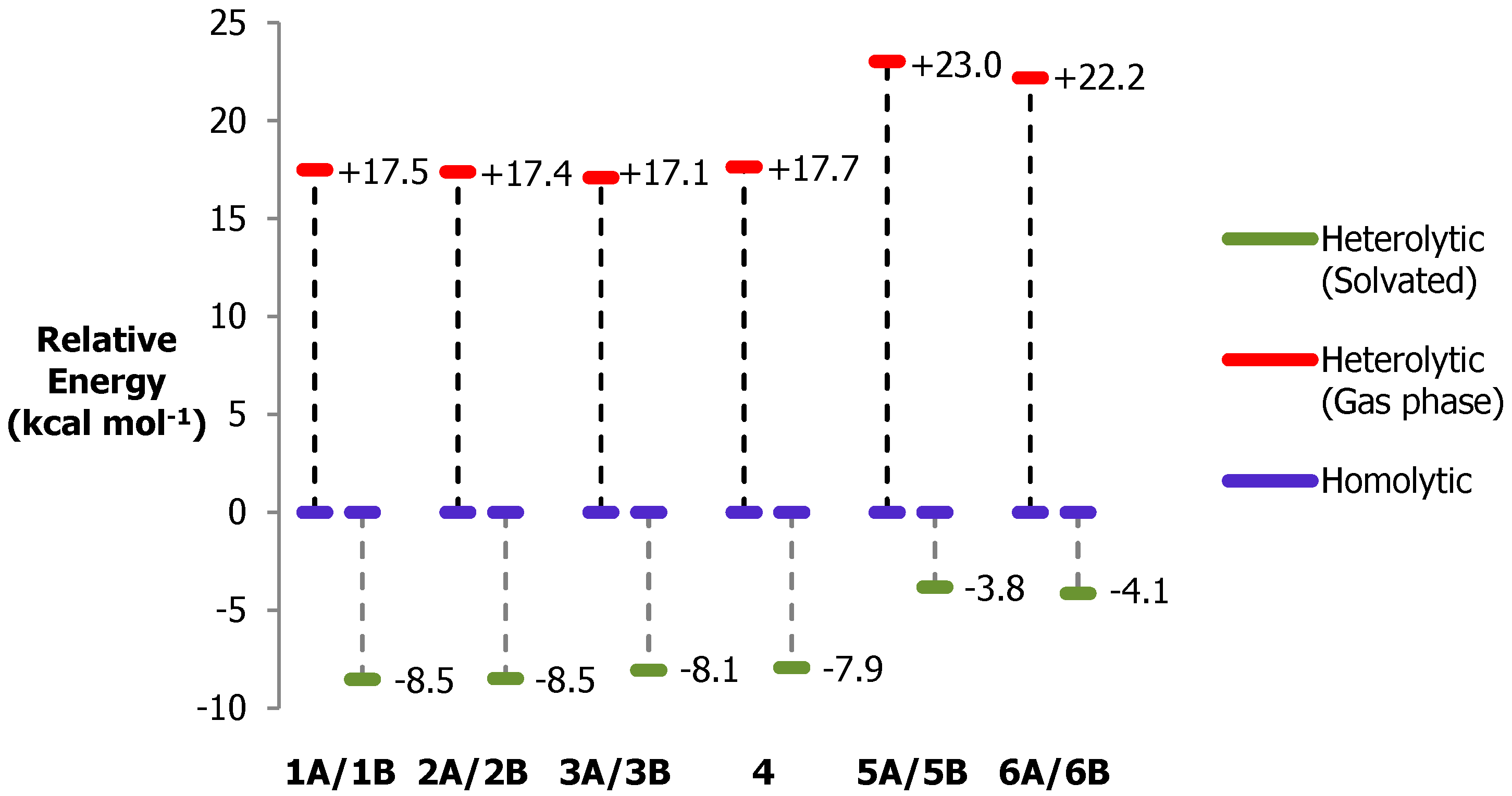
3.4. O–O Bond Cleavage: Heterolytic vs. Homolytic
3.5. Dissociation Energy of Nickel-Hydroxo O–O Bond
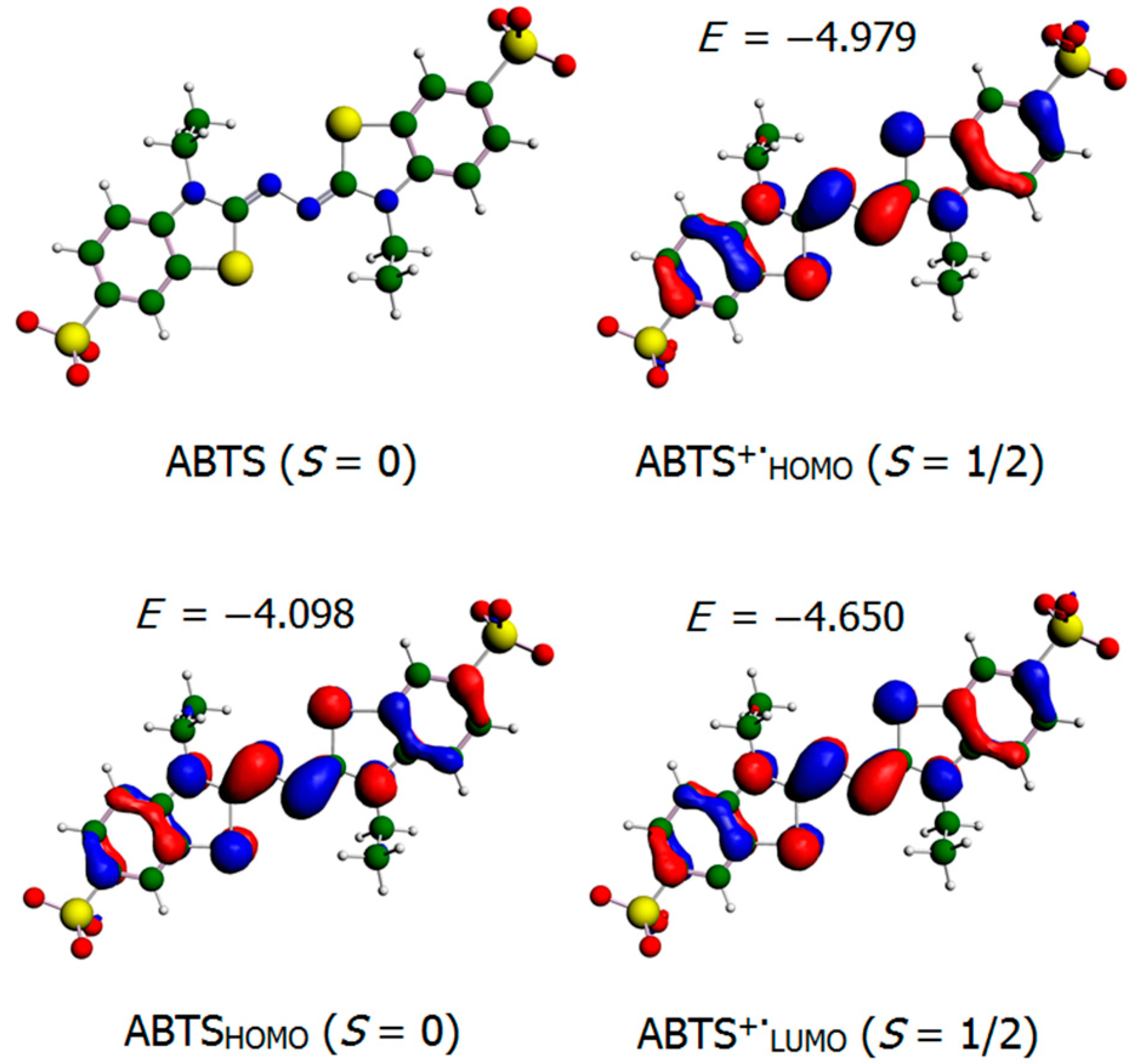
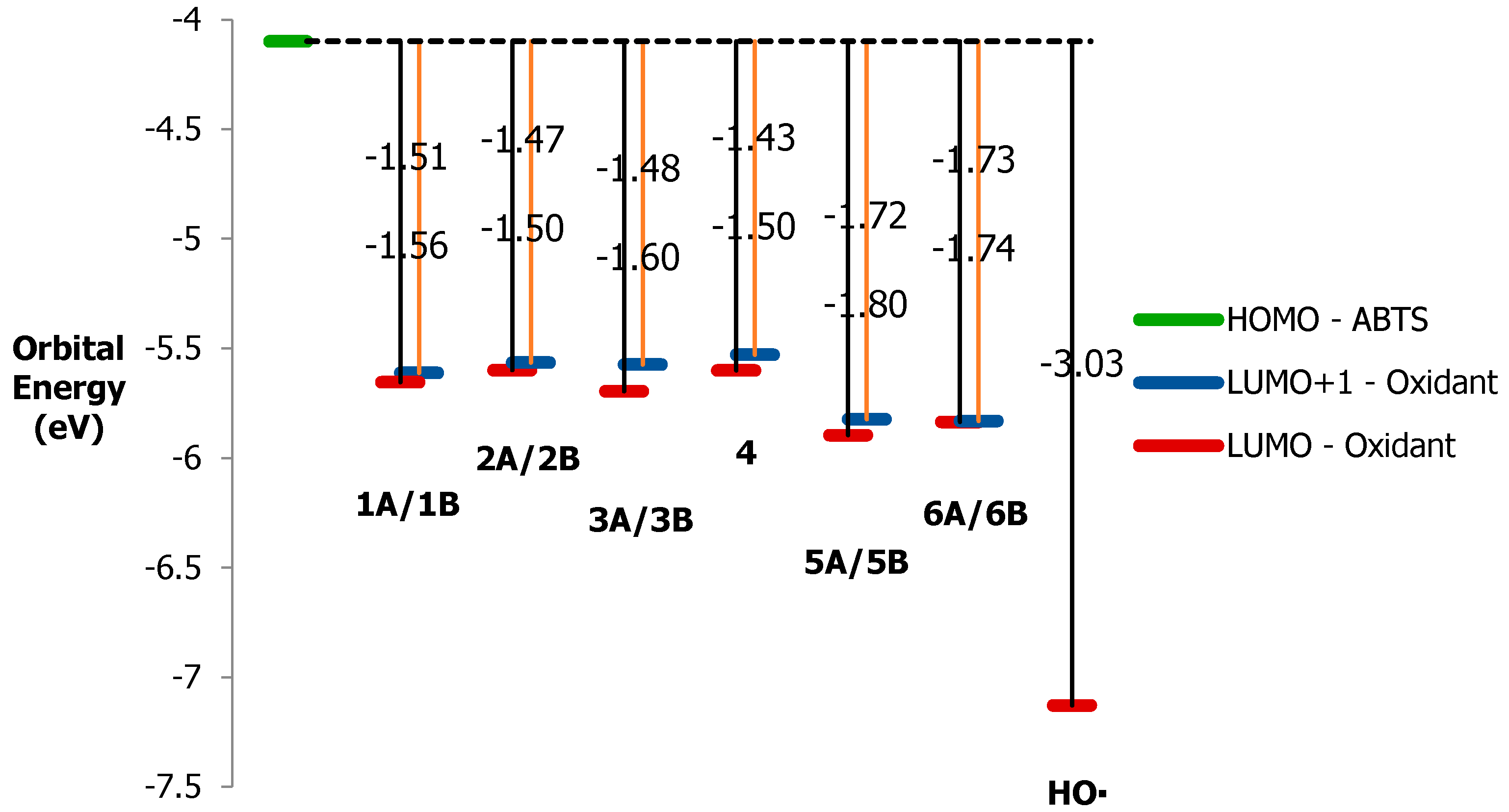
3.6. ABTS Oxidation by [(L)Ni3+–O·]2+ Active Intermediates
3.7. Formation of [(L)Ni2+–OH] and Regeneration of the Resting State
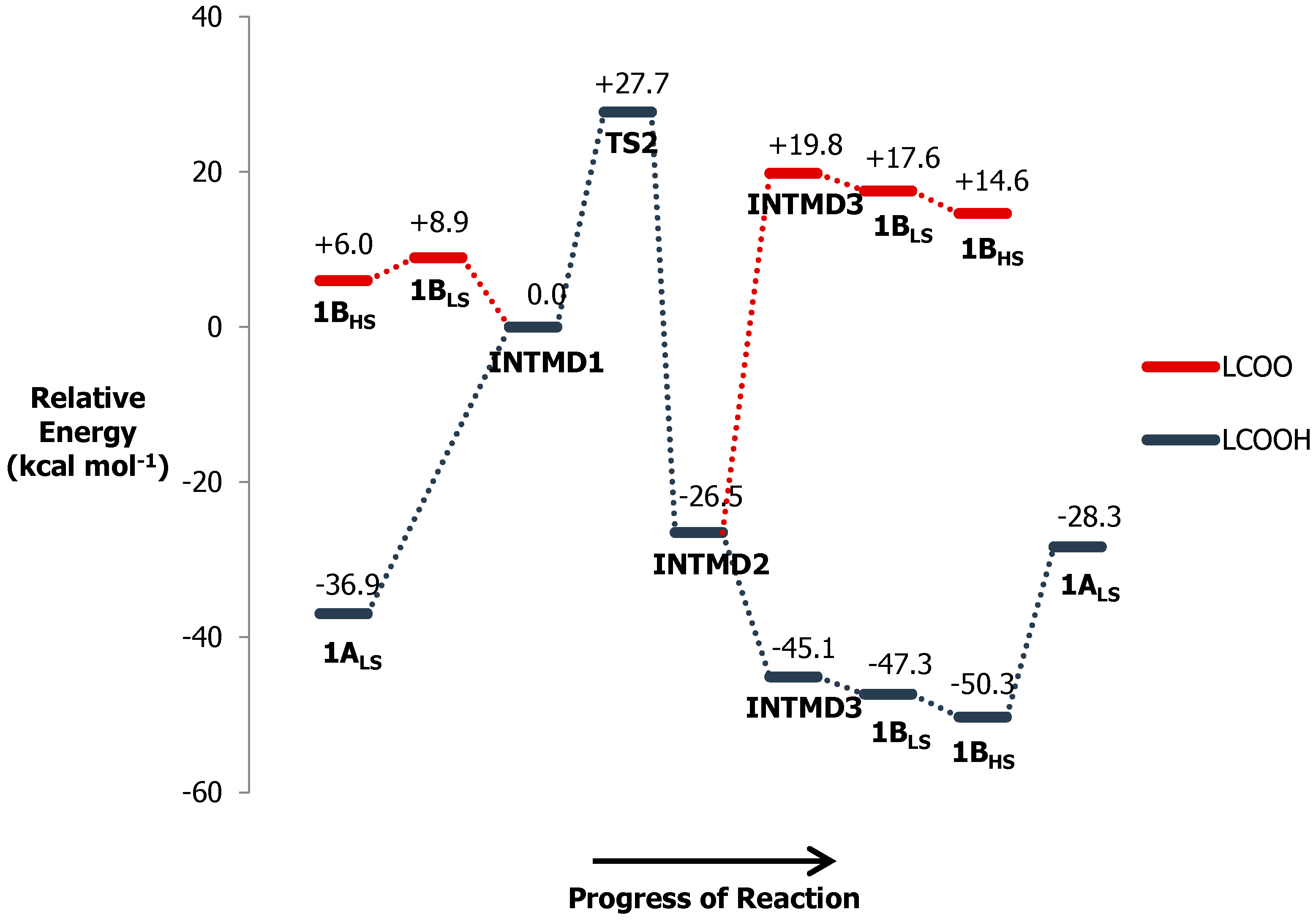
3.8. Reaction Energy Profile of Ni(II)–PyMAC Catalysis
4. Conclusions and Recommendation
Author Contributions
Funding
Conflicts of Interest
References
- Houmam, A. Electron transfer initiated reactions: Bond formation and bond dissociation. Chem. Rev. 2008, 108, 2180–2237. [Google Scholar] [CrossRef] [PubMed]
- Legros, J.; Bolm, C. Iron-catalyzed asymmetric sulfide oxidation with aqueous hydrogen peroxide. Angew. Chem. Int. Ed. 2003, 42, 5487–5489. [Google Scholar] [CrossRef] [PubMed]
- Neumann, R.; Khenkin, A.M.; Juwiler, D.; Miller, H.; Gara, M. Catalytic oxidation with hydrogen peroxide catalyzed by ‘sandwich’ type transition metal substituted polyoxometalates. J. Mol. Catal. A Chem. 1997, 117, 169–183. [Google Scholar] [CrossRef]
- Prasad, R.V.; Thakkar, N.V. Study of cobalt complexes as catalysts in the decomposition of hydrogen peroxide. J. Mol. Catal. 1994, 92, 9–20. [Google Scholar] [CrossRef]
- Shivankar, V.S.; Thakkar, N.V. Decomposition of hydrogen peroxide in presence of mixed ligand cobalt (II) and nickel (II) complexes as catalysts. J. Sci. Ind. Res. (India) 2005, 64, 496–503. [Google Scholar]
- Haber, F.; Weiss, J. Uber die Katalyse des Hydroperoxydes. Die Nat. 1932, 20, 948–950. [Google Scholar] [CrossRef]
- Kremer, M.L. Oxidation reduction step in catalytic decomposition of hydrogen peroxide by ferric ions. Trans. Faraday Soc. 1963, 59, 2535. [Google Scholar] [CrossRef]
- Stephenson, N.A.; Bell, A.T. Mechanistic insights into iron porphyrin-catalyzed olefin epoxidation by hydrogen peroxide: Factors controlling activity and selectivity. J. Mol. Catal. A Chem. 2007, 275, 54–62. [Google Scholar] [CrossRef]
- Tanaka, M.; Matsuura, K.; Yoshioka, S.; Takahashi, S.; Ishimori, K.; Hori, H.; Morishima, I. Activation of hydrogen peroxide in horseradish peroxidase occurs within ~200 μs observed by a new freeze-quench device. Biophys. J. 2003, 84, 1998–2004. [Google Scholar] [CrossRef] [Green Version]
- Salem, I.A.; El-maazawi, M.; Zaki, A.B. Kinetics and mechanisms of decomposition reaction of hydrogen peroxide in presence of metal complexes. Int. J. Chem. Kinet. 2000, 32, 643–666. [Google Scholar] [CrossRef]
- Kuznetsov, M.L.; Teixeira, F.A.; Bokach, N.A.; Pombeiro, A.J.L.; Shul’pin, G.B. Radical decomposition of hydrogen peroxide catalyzed by aqua complexes [M(H2O)n]2+ (M=Be, Zn, Cd). J. Catal. 2014, 313, 135–148. [Google Scholar] [CrossRef]
- Aebi, H. Peroxidase. The properties and uses of a versatile enzyme and of some related catalysts. Angew. Chemie. 1965, 77, 1144. [Google Scholar] [CrossRef]
- Veitch, N.C.; Smith, A.T. Horseradish peroxidase. Adv. Inorg. Chem. 2000, 107–162. [Google Scholar] [CrossRef]
- English, A.M.; Tsaprailis, G. Catalytic structure–function relationships in heme peroxidases. Adv. Inorg. Chem. 1995, 79–125. [Google Scholar] [CrossRef]
- Pappa, H.S.; Cass, A.E.G. A step towards understanding the folding mechanism of horseradish peroxidase Tryptophan fluorescence and circular dichroism equilibrium studies. Eur. J. Biochem. 1993, 212, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Tams, J.W.; Welinder, K.G. Deglycosylation without anisole. Anal. Biochem. 1995, 228, 48–55. [Google Scholar] [CrossRef]
- Tams, J.W.; Welinder, K.G. Glycosylation and thermodynamic versus kinetic stability of horseradish peroxidase. Fed. Eur. Biochem. Soc. Lett. 1998, 421, 234–236. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Yang, Y.; Li, H.; Zhu, R.; Shao, Q.; Yang, S.; Xu, J. NiO nanoparticles modified with 5,10,15,20-tetrakis(4-carboxyl pheyl)-porphyrin: Promising peroxidase mimetics for H2O2 and glucose detection. Biosens. Bioelectron. 2014, 64, 147–153. [Google Scholar] [CrossRef]
- Kitamura, Y.; Mori, K.; Yamamoto, M.; Nozaki, A.; Saito, M.; Tsukamoto, I.; Mifune, M.; Saito, Y. Peroxidase-like catalytic activity of aqueous- and immobilized-Mn3+-octabromo-porphyrins on ion-exchange resin supplied as mimetic of horseradish peroxidase. Chem. Pharm. Bull. (Tokyo) 2008, 56, 1364–1366. [Google Scholar] [CrossRef] [Green Version]
- Shu, J.; Qiu, Z.; Wei, Q.; Zhuang, J.; Tang, D. Cobalt-porphyrin-platinum-functionalized reduced graphene oxide hybrid nanostructures: A novel peroxidase mimetic system for improved electrochemical immunoassay. Sci. Rep. 2015, 5, 15113. [Google Scholar] [CrossRef] [Green Version]
- Lousada, C.M.; Yang, M.; Nilsson, K.; Jonsson, M. Catalytic decomposition of hydrogen peroxide on transition metal and lanthanide oxides. J. Mol. Catal. A Chem. 2013, 379, 178–184. [Google Scholar] [CrossRef]
- Wei, H.; Wang, E. Fe3O4 Magnetic nanoparticles as peroxidase mimetics and their applications in H2O2 and glucose detection. Anal. Chem. 2008, 80, 2250–2254. [Google Scholar] [CrossRef]
- McAteer, B.; Beattie, N.; Richens, D.T. Catalytic oxidation of cyclohexene by aqueous Iron(III)/H2O2 in mildly acidic solution: Epoxidation versus allylic oxidation. Inorg. Chem. Commun. 2013, 35, 284–289. [Google Scholar] [CrossRef]
- Organo, V.G.; Filatov, A.S.; Quartararo, J.S.; Friedman, Z.M.; Rybak-Akimova, E.V. Nickel(II) complexes of monofunctionalized pyridine-azamacrocycles: Synthesis, structures, pendant arm “on-off” coordination equilibria, and peroxidase-like activity. Inorg. Chem. 2009, 48, 8456–8468. [Google Scholar] [CrossRef]
- McKenzie, S.G.; Pallucio, T.D.; Patterson, J.D.; Rybak-Akimova, E.V. Synthesis, characterization, and oxidation catalysis studies of a monofunctionalized copper pyridine-aza macrocycle. Inorg. Chim. Acta 2018, 482, 732–737. [Google Scholar] [CrossRef]
- Chng, L.L.; Chang, C.J.; Nocera, D.G. Catalytic O−O activation chemistry mediated by iron hangman porphyrins with a wide range of proton-donating abilities. Am. Chem. Soc. 2003, 5, 1403–1406. [Google Scholar] [CrossRef]
- Aniagyei, A.; Tia, R.; Adei, E. A density functional theory study of the mechanisms of oxidation of ethylene by technetium oxo complexes. Comput. Theor. Chem. 2013, 1009, 70–80. [Google Scholar] [CrossRef]
- Hou, L.-J.; Wu, B.-W.; Han, Y.-X.; Kong, C.; Chen, D.-P.; Gao, L.-G. Density functional theoretical study on the reaction mechanism of HNCS with SiHF radical. Comput. Theor. Chem. 2015, 1051, 57–61. [Google Scholar] [CrossRef]
- Lundberg, M.; Borowski, T. Oxoferryl species in mononuclear non-heme iron enzymes: Biosynthesis, properties and reactivity from a theoretical perspective. Coord. Chem. Rev. 2013, 257, 277–289. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Maeda, S.; Ohno, K. Decomposition of alkyl hydroperoxide by a copper(I) complex: Insights from density functional theory. Tetrahedron Lett. 2008, 49, 6841–6845. [Google Scholar] [CrossRef]
- Mothana, B.; Boyd, R.J. A density functional theory study of the mechanism of the Paal-Knorr pyrrole synthesis. J. Mol. Struct. THEOCHEM. 2007, 811, 97–107. [Google Scholar] [CrossRef]
- Vafaeezadeh, M.; Fattahi, A. DFT investigations for “Fischer” esterification mechanism over silica-propyl-SO3H catalyst: Is the reaction reversible? Comput. Theor. Chem. 2015, 1071, 27–32. [Google Scholar] [CrossRef]
- Hynninen, P.H.; Kaartinen, V.; Kolehmainen, E. Horseradish peroxidase-catalyzed oxidation of chlorophyll a with hydrogen peroxide Characterization of the products and mechanism of the reaction. Biochim. Biophys. Acta Bioenerg. 2010, 1797, 531–542. [Google Scholar] [CrossRef] [Green Version]
- Keilin, D.; Hartree, E.F. Purification of horse-radish peroxidase and comparison of its properties with those of catalase and methaemoglobin. Biochem. J. 1951, 49, 88–106. [Google Scholar] [CrossRef] [Green Version]
- Gajhede, M.; Schuller, D.J.; Henriksen, A.; Smith, A.T.; Poulos, T.L. Crystal structure of horseradish peroxidase C at 2.15 Å resolution. Nat. Struct. Biol. 1997, 4, 1032–1038. [Google Scholar] [CrossRef]
- Poulos, T.; Kraut, J. A hypothetical model of the cytochrome c peroxidase. Cytochrome c electron transfer complex. J. Biol. Chem. 1980, 255, 10322–10330. [Google Scholar]
- Henriksen, A.; Smith, A.T.; Gajhede, M. The structures of the horseradish peroxidase c-ferulic acid complex and the ternary complex with cyanide suggest how peroxidases oxidize small phenolic substrates. J. Biol. Chem. 1999, 274, 35005–35011. [Google Scholar] [CrossRef] [Green Version]
- Stephenson, N.A.; Bell, A.T. Effects of porphyrin composition on the activity and selectivity of the iron(III) porphyrin catalysts for the epoxidation of cyclooctene by hydrogen peroxide. J. Mol. Catal. A Chem. 2007, 272, 108–117. [Google Scholar] [CrossRef]
- Song, W.J.; Ryu, Y.O.; Song, R.; Nam, W. Oxoiron(IV) porphyrin π-cation radical complexes with a chameleon behavior in cytochrome P450 model reactions. J. Biol. Inorg. Chem. 2005, 10, 294–304. [Google Scholar] [CrossRef]
- Almarsson, O.; Bruice, T.C. A homolytic mechanism of O-O bond scission prevails in the reactions of alkyl hydroperoxides with an octacationic tetraphenylporphinato-iron(III) complex in aqueous solution. J. Am. Chem. Soc. 1995, 117, 4533–4544. [Google Scholar] [CrossRef]
- Traylor, T.G.; Kim, C.; Richards, J.L.; Xu, F.; Perrin, C.L. Reactions of iron(III) porphyrins with oxidants. Structure-reactivity studies. J. Am. Chem. Soc. 1995, 117, 3468–3474. [Google Scholar] [CrossRef]
- Nam, W.; Han, H.J.; Oh, S.-Y.; Lee, Y.J.; Choi, M.-H.; Han, S.-Y.; Kim, C.; Woo, S.K.; Shin, W. New insights into the mechanisms of O−O bond cleavage of hydrogen peroxide and tert -alkyl hydroperoxides by iron(III) porphyrin complexes. J. Am. Chem. Soc. 2000, 122, 8677–8684. [Google Scholar] [CrossRef]
- Nagataki, T.; Ishii, K.; Tachi, Y.; Itoh, S. Ligand effects on NiII-catalysed alkane-hydroxylation with m-CPBA. Dalt. Trans. 2007, 1120. [Google Scholar] [CrossRef]
- Pfaff, F.F.; Heims, F.; Kundu, S.; Mebs, S.; Ray, K. Spectroscopic capture and reactivity of S = 1/2 nickel(III)–oxygen intermediates in the reaction of a Ni(II)-salt with mCPBA. Chem. Commun. 2012, 48, 3730. [Google Scholar] [CrossRef]
- Nagataki, T.; Tachi, Y.; Itoh, S. NiII(TPA) as an efficient catalyst for alkane hydroxylation with m-CPBA. Chem. Commun. 2006, 4016. [Google Scholar] [CrossRef]
- Schröder, D.; Schwarz, H. C-H and C-C bond activation by bare transition-metal oxide cations in the gas phase. Angew. Chemie Int. Ed. English. 1995, 34, 1973–1995. [Google Scholar] [CrossRef]
- Fenton, H.J.H. Oxidation of tartaric acid in presence of iron. J. Chem. Soc. Trans. 1894, 65, 899–910. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; Van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Boerrigter, P.M.; Te Velde, G.; Baerends, J.E. Three-dimensional numerical integration for electronic structure calculations. Int. J. Quantum Chem. 1988, 33, 87–113. [Google Scholar] [CrossRef]
- Fonseca Guerra, C.; Snijders, J.G.; Te Velde, G.; Baerends, E.J. Towards an order- N DFT method. Theor. Chem. Acc. 1998, 99, 391–403. [Google Scholar] [CrossRef]
- Schreckenbach, G.; Ziegler, T. The calculation of NMR shielding tensors based on density functional theory and the frozen-core approximation. Int. J. Quantum Chem. 1996, 60, 753–766. [Google Scholar] [CrossRef]
- Versluis, L.; Ziegler, T. The determination of molecular structures by density functional theory. The evaluation of analytical energy gradients by numerical integration. J. Chem. Phys. 1988, 88, 322. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Ehlers, A.; Baerends, E.-J. Geometry optimizations in the zero order regular approximation for relativistic effects. J. Chem. Phys. 1999, 110, 8943. [Google Scholar] [CrossRef] [Green Version]
- Head, J.D.; Zerner, M.C. A Broyden-Fletcher-Goldfarb-Shanno optimization procedure for molecular geometries. Chem. Phys. Lett. 1985, 122, 264–270. [Google Scholar] [CrossRef]
- Fan, L.; Ziegler, T. Nonlocal density functional theory as a practical tool in calculations on transition states and activation energies. Applications to elementary reaction steps in organic chemistry. J. Am. Chem. Soc. 1992, 114, 10890–10897. [Google Scholar] [CrossRef]
- Fan, L.; Ziegler, T. Application of density functional theory to infrared absorption intensity calculations on main group molecules. J. Chem. Phys. 1992, 96, 9005. [Google Scholar] [CrossRef]
- Bérces, A.; Dickson, R.M.; Fan, L.; Jacobsen, H.; Swerhone, D.; Ziegler, T. An implementation of the coupled perturbed Kohn-Sham equations: Perturbation due to nuclear displacements. Comput. Phys. Commun. 1997, 100, 247–262. [Google Scholar] [CrossRef]
- Jacobsen, H.; Bérces, A.; Swerhone, D.P.; Ziegler, T. Analytic second derivatives of molecular energies: A density functional implementation. Comput. Phys. Commun. 1997, 100, 263–276. [Google Scholar] [CrossRef]
- Alexiadis, A.; Kassinos, S. On the use of the BLYP functional for the DFT calculation of graphite-hydrogen systems. J. Nuc. Mat. 2010, 396, 307–308. [Google Scholar] [CrossRef]
- Yankov, E.P.; Bakalska, R.I.; Horkel, E.; Svatunek, D.; Delchev, V.B. Experimental and theoretical study of the excited-state tautomerism of 6-azauracil in water surroundings. Chem. Phys. 2018, 515, 663–671. [Google Scholar] [CrossRef]
- de Oliveira, A.Z.; Jorge, F.E. Structural, electronic, electrical, and magnetic properties of Rhn (1≤ n ≤13) clusters. Comput. Theo. Chem. 2020, 1177, 112765. [Google Scholar] [CrossRef]
- Delchev, V.B.; Horkel, E.; Svatunek, D. Excited-state photocyclodimerization of 6-azauracil to oxazetidine cyclodimer: A mechanism elucidation in water surroundings. J. Mol. Struc. 2020, 1205, 127571. [Google Scholar] [CrossRef]
- Klamt, A.; Schuurmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 2 1993, 799–805. [Google Scholar] [CrossRef]
- Klamt, A. Conductor-like Screening Model for real solvents: A new approach to the quantitative calculation of solvation phenomena. J. Phys. Chem. 1995, 99, 2224. [Google Scholar] [CrossRef]
- Klamt, A.; Jones, V. Treatment of the outlying charge in continuum solvation models. J. Chem. Phys. 1996, 105, 9972. [Google Scholar] [CrossRef]
- Ye, S.; Neese, F. Accurate Modeling of Spin-State Energetics in Spin-Crossover Systems with Modern Density Functional Theory. Inorg. Chem. 2010, 49, 772–774. [Google Scholar] [CrossRef]
- Boguslawski, K.; Jacob, C.R.; Reiher, M. Can DFT accurately predict spin densities? Analysis of discrepancies in iron nitrosyl complexes. J. Chem. Theory Comput. 2011, 7, 2740–2752. [Google Scholar] [CrossRef]
- Kepp, K.P. Consistent descriptions of metal–ligand bonds and spin-crossover in inorganic chemistry. Coord. Chem. Rev. 2013, 257, 196–209. [Google Scholar] [CrossRef]
- Fouqueau, A.; Mer, S.; Casida, M.E. Comparison of density functionals for energy and structural differences between the high- [5T2g: (t2g)4(eg)2] and low- [1A1g: (t2g)6(eg)0] spin states of the hexaquoferrous cation [Fe(H2O)6]2+. J. Chem. Phys. 2004, 120, 9473–9486. [Google Scholar] [CrossRef] [PubMed]
- Reiher, M.; Salomon, O.; Hess, B.A. Reparameterization of hybrid functionals based on energy differences of states of different multiplicity. Theor. Chem. Acc. 2001, 107, 48–55. [Google Scholar] [CrossRef]
- Lawson Daku, L.M.; Vargas, A.; Hauser, A.; Fouqueau, A.; Casida, M.E. Assessment of Density Functionals for the High-Spin/Low-Spin Energy Difference in the Low-Spin Iron(II) Tris(2,2′-bipyridine) Complex. Chem. Phys. Chem. 2005, 6, 1393–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera, A.M.; Staples, R.J.; Kryatov, S.V.; Nazarenko, A.Y.; Rybak-Akimova, E.V. Nickel(II) and copper(II) complexes with pyridine-containing macrocycles bearing an aminopropyl pendant arm: Synthesis, characterization, and modifications of the pendant amino group. Dalton Trans. 2003, 846–856. [Google Scholar] [CrossRef]
- Shiren, K.; Ogo, S.; Fujinami, S.; Hayashi, H.; Suzuki, M.; Uehara, A.; Watanabe, Y.; Moro-oka, Y. Synthesis, structures, and properties of bis(μ-oxo)nickel(III) and bis(μ-superoxo)nickel(II) complexes: An unusual conversion of a NiIII2(μ-O)2 core into a NiII2(μ-OO)2 core by H2O2 and oxygenation of ligand. J. Am. Chem. Soc. 2000, 122, 254–262. [Google Scholar] [CrossRef]
- Sankaralingam, M.; Balamurugan, M.; Palaniandavar, M.; Vadivelu, P.; Suresh, C.H. Nickel(II) complexes of pentadentate N5 ligands as catalysts for alkane hydroxylation by using m-CPBA as oxidant: A combined experimental and computational study. Chem.-A Eur. J. 2014, 20, 11346–11361. [Google Scholar] [CrossRef]
- Sandhiya, L.; Zipse, H. O-O Bond Homolysis in Hydrogen Peroxide. J. Comput. Chem. 2017, 38, 2186–2192. [Google Scholar] [CrossRef]
- Scott, S.L.; Wen-Jang, C.; Bakac, A.; Espenson, J.H. Spectroscopic Parameters, Electrode Potentials, Acid Ionization Constants, and Electron Exchange Rates of the 2,2′-Azinobis(3-ethylbenzothiazoline-6-sulfonate) Radicals and Ions. J. Phys. Chem. 1993, 97, 6710–6714. [Google Scholar] [CrossRef]
- Miessler, G.L.; Fischer, P.J.; Tarr, D.A. Inorganic Chemistry, 5th ed.; Pearson Education, Inc.: Upper Saddle River, NJ, USA, 2014; pp. 185–188. [Google Scholar]
- Ilyasov, I.R.; Beloborodov, V.L.; Selivanova, I.A.; Terekhov, R.P. ABTS/PP Decolorization Assay of Antioxidant Capacity Reaction Pathways. Int. J. Mol. Sci. 2020, 21, 1131. [Google Scholar] [CrossRef] [Green Version]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
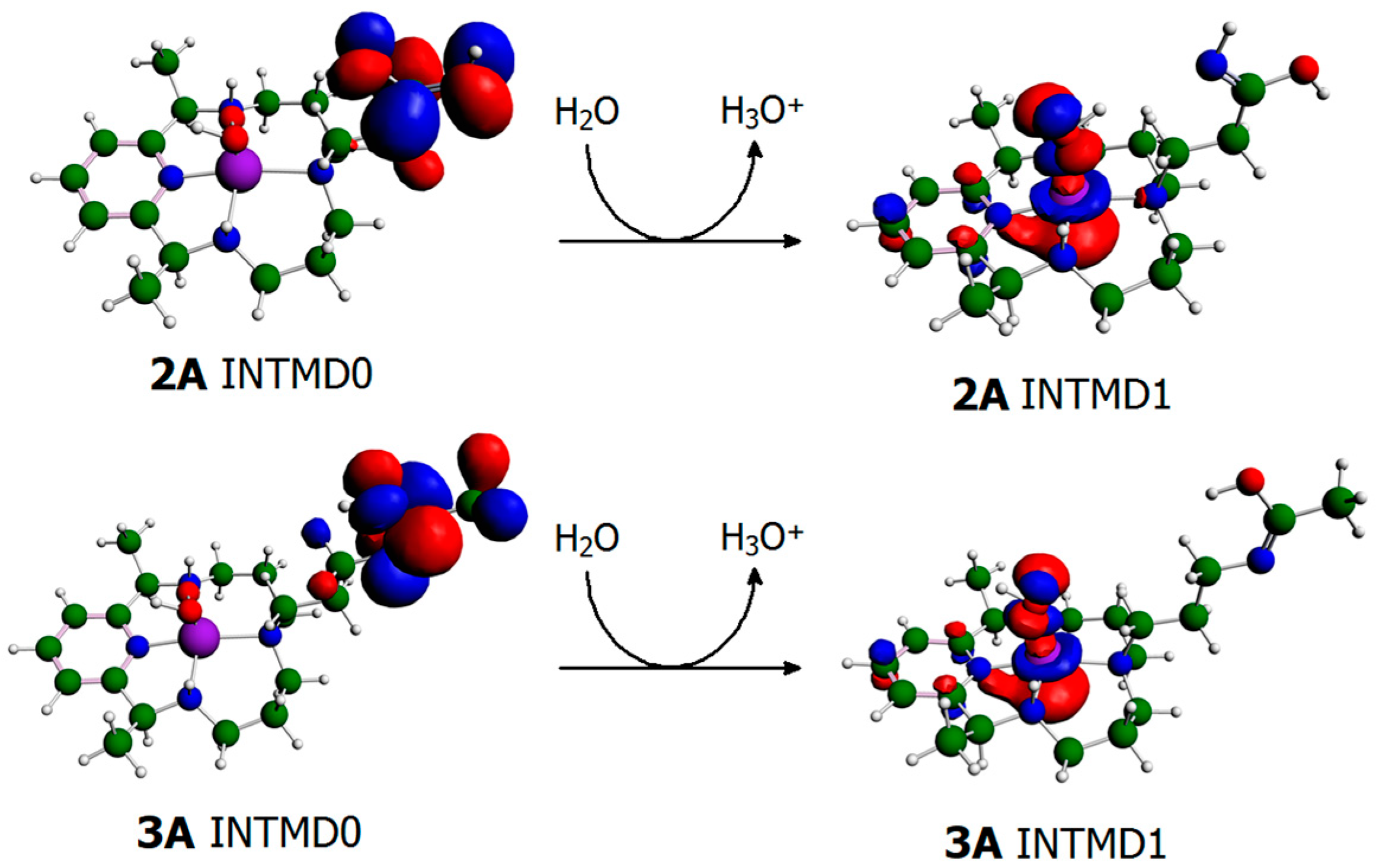{kind=link}
{kind=link}
| XC Functional | HF Exchange Admixture | Bond Energy Splitting (ELS-EHS) |
|---|---|---|
| BLYP | 0 | −0.9 |
| B3LYP* | 0.15 | 7.7 |
| B3LYP | 0.20 | 11.5 |
| Catalyst | U (kcal mol−1) | H (kcal mol−1) | S (cal mol−1 K−1) | G (kcal mol−1) |
|---|---|---|---|---|
| 1ALS | −6462.93 | −6462.34 | 149.98 | −6507.05 |
| 1BLS | −6551.99 | −6551.40 | 154.40 | −6597.43 |
| 1BHS | −6560.72 | −6560.12 | 154.90 | −6606.31 |
| 2ALS | −6577.25 | −6576.66 | 146.71 | −6620.40 |
| 2BLS | −6647.97 | −6647.38 | 153.69 | −6693.20 |
| 2BHS | −6661.41 | −6660.82 | 155.29 | −6707.12 |
| 3ALS | −7260.42 | −7259.83 | 154.60 | −7305.93 |
| 3BLS | −7320.64 | −7320.04 | 161.11 | −7368.08 |
| 3BHS | −7332.76 | −7332.16 | 163.90 | −7381.03 |
| 4LS | −5637.29 | −5636.70 | 143.20 | −5679.39 |
| 5ALS | −6395.05 | −6394.46 | 148.87 | −6438.84 |
| 5BLS | −6544.61 | −6544.02 | 153.36 | −6589.74 |
| 5BHS | −6546.03 | −6545.44 | 147.94 | −6589.55 |
| 6ALS | −7781.61 | −7781.02 | 170.97 | −7831.99 |
| 6BLS | −7938.21 | −7937.62 | 167.01 | −7987.41 |
| Atoms | 1A [1B] | 2A [2B] | 3A [3B] | 4 | 5A [5B] | 6A [6B] |
|---|---|---|---|---|---|---|
| Bond Length (Å) | ||||||
| Ni–N(1) | 1.888 [2.023] | 1.887 [2.017] | 1.892 [2.054] | 1.881 | 1.886 [2.037] | 1.887 [1.901] |
| Ni–N(2) | 2.009 [2.157] | 2.006 [2.170] | 2.018 [2.190] | 2.005 | 2.010 [2.201] | 2.006 [2.026] |
| Ni–N(3) | 2.054 [2.123] | 2.053 [2.137] | 2.056 [2.178] | 2.050 | 2.099 [2.134] | 2.091 [2.070] |
| Ni–N(4) | 2.009 [2.157] | 2.005 [2.170] | 2.011 [2.190] | 2.005 | 2.010 [2.201] | 2.003 [2.024] |
| Ni–Laxial | - [1.979] | - [1.993] | - [2.048] | - | - [2.103] | - [-] |
| N(1)–N(3) | 3.941 [4.117] | 3.940 [4.116] | 3.949 [4.101] | 3.930 | 3.985 [4.120] | 3.978 [3.970] |
| N(2)–N(4) | 3.978 [4.253] | 3.972 [4.278] | 3.989 [4.300] | 3.973 | 3.983 [4.329] | 3.973 [4.008] |
| Bond Angle (°) | ||||||
| N(1)–Ni–N(2) | 83.48 [80.36] | 83.57 [80.33] | 83.34 [79.25] | 83.69 | 83.49 [79.77] | 83.49 [83.28] |
| N(1)–Ni–N(3) | 179.08 [166.29] | 178.71 [164.42] | 179.38 [151.37] | 178.68 | 179.98 [162.02] | 179.62 [178.55] |
| N(1)–Ni–N(4) | 83.48 [80.36] | 83.54 [80.33] | 83.55 [79.25] | 83.69 | 83.49 [79.77] | 83.58 [83.26] |
| N(2)–Ni–N(3) | 96.44 [99.55] | 96.42 [99.15] | 96.42 [98.54] | 96.21 | 96.51 [99.16] | 96.49 [96.60] |
| N(2)–Ni–N(4) | 163.63 [160.66] | 163.97 [160.62] | 163.89 [158.20] | 164.49 | 164.38 [159.22] | 164.56 [163.64] |
| N(3)–Ni–N(4) | 96.44 [99.55] | 96.25 [99.15] | 96.59 [98.54] | 96.21 | 96.51 [99.16] | 96.38 [96.62] |
| pKa | 3.03 a | 11.36 a | 11.30 a | - | 6.75 b | 9.36 a |
| Group | Complex | Spin State | Deprotonation of H2O2 | Binding of HOO− to Ni | |
|---|---|---|---|---|---|
| Gas Phase | Aqueous | ||||
| G1 | 1A, 4, 5A, 6A | 1A, 2A, 3A, 4, 5A, 6A | Low-Spin | No | Yes |
| G2 | 2A, 3A, 6B | 6B | Low-Spin | Yes | Yes |
| G3 | 1B, 5B | 1B, 2B, 3B, 5B | High-Spin Low-Spin | No Yes | No Yes |
| G4 | 2B, 3B | - | High-Spin Low-Spin | Yes Yes | No Yes |
| Catalyst | Homolysis | Heterolysis | ||||
|---|---|---|---|---|---|---|
| High-Spin (S = 3/2) | Low-Spin (S = 1/2) | High-Spin (S = 1) | ||||
| Ni | O | Ni | O | Ni | O | |
| 1A/1B | 1.5123 [1.5421] | 1.0991 [1.0394] | 0.1737 [0.7512] | 1.0821 [−0.0319] | 0.6587 [0.6955] | 1.3137 [1.2724] |
| 2A/2B | 1.5097 [1.5406] | 1.1041 [1.0416] | 0.1726 [0.7044] | 1.0887 [0.0094] | 0.6559 [0.6922] | 1.3183 [1.2761] |
| 3A/3B | 1.5158 [1.5434] | 1.0903 [1.0353] | 1.1987 [1.1716] | −0.5527 [−0.5365] | 0.8605 [0.7324] | 1.2500 [1.2461] |
| 4 | 1.5239 [1.5540] | 1.0890 [1.0281] | 0.1850 [0.7622] | 1.0756 [−0.0504] | 0.6571 [0.6916] | 1.3165 [1.2775] |
| 5A/5B | 1.5276 [1.5457] | 1.0736 [1.0294] | 0.3890 [0.4053] | 0.6143 [0.5946] | 0.6822 [0.7125] | 1.3076 [1.2471] |
| 6A/6B | 1.5268 [1.5468] | 1.0775 [1.0318] | 1.1828 [0.2419] | −0.5288 [1.0087] | 0.6854 [0.7080] | 1.2774 [1.2515] |
| Catalyst | TS2 | INTMD2 | ||||
|---|---|---|---|---|---|---|
| ΔE (kcal mol−1) | vi (cm−1) | r (Å) | ΔE (kcal mol−1) | r (Å) | ||
| O1–O2 | O1–O2 | Ni–O1 | Ni–O1 | |||
| 1A/1B | +27.7 | −142 | 2.116 | 1.987 | −26.5 | 1.910 |
| 2A/2B | +27.0 | −105 | 2.109 | 2.000 | −25.7 | 1.910 |
| 3A/3B | +26.2 | −291 | 2.119 | 1.985 | −27.0 | 1.885 |
| 4 | +25.9 | −100 | 2.121 | 1.973 | −28.3 | 1.917 |
| 5A/5B | +27.2 | −242 | 2.124 | 1.973 | −21.6 | 1.878 |
| 6A/6B | +27.5 | −210 | 2.122 | 1.994 | −22.5 | 1.891 |
| H2O2 | +41.8 | −254 | 2.100 | - | +53.0 a | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taping, J.J.E.; Billones, J.B.; Organo, V.G. Influence of Varying Functionalization on the Peroxidase Activity of Nickel(II)–Pyridine Macrocycle Catalysts: Mechanistic Insights from Density Functional Theory. Computation 2020, 8, 52. https://0-doi-org.brum.beds.ac.uk/10.3390/computation8020052
Taping JJE, Billones JB, Organo VG. Influence of Varying Functionalization on the Peroxidase Activity of Nickel(II)–Pyridine Macrocycle Catalysts: Mechanistic Insights from Density Functional Theory. Computation. 2020; 8(2):52. https://0-doi-org.brum.beds.ac.uk/10.3390/computation8020052
Chicago/Turabian StyleTaping, Jerwin Jay E., Junie B. Billones, and Voltaire G. Organo. 2020. "Influence of Varying Functionalization on the Peroxidase Activity of Nickel(II)–Pyridine Macrocycle Catalysts: Mechanistic Insights from Density Functional Theory" Computation 8, no. 2: 52. https://0-doi-org.brum.beds.ac.uk/10.3390/computation8020052