
Halogen Bonding and CO-Ligand Blue-Shift in Hybrid Organic—Organometallic Cocrystals [CpFe(CO)2X] (C2I4) (X = Cl, Br)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. General Co-Crystallization Procedure
2.3. X-ray Structure Determinations
2.4. Computations
3. Results
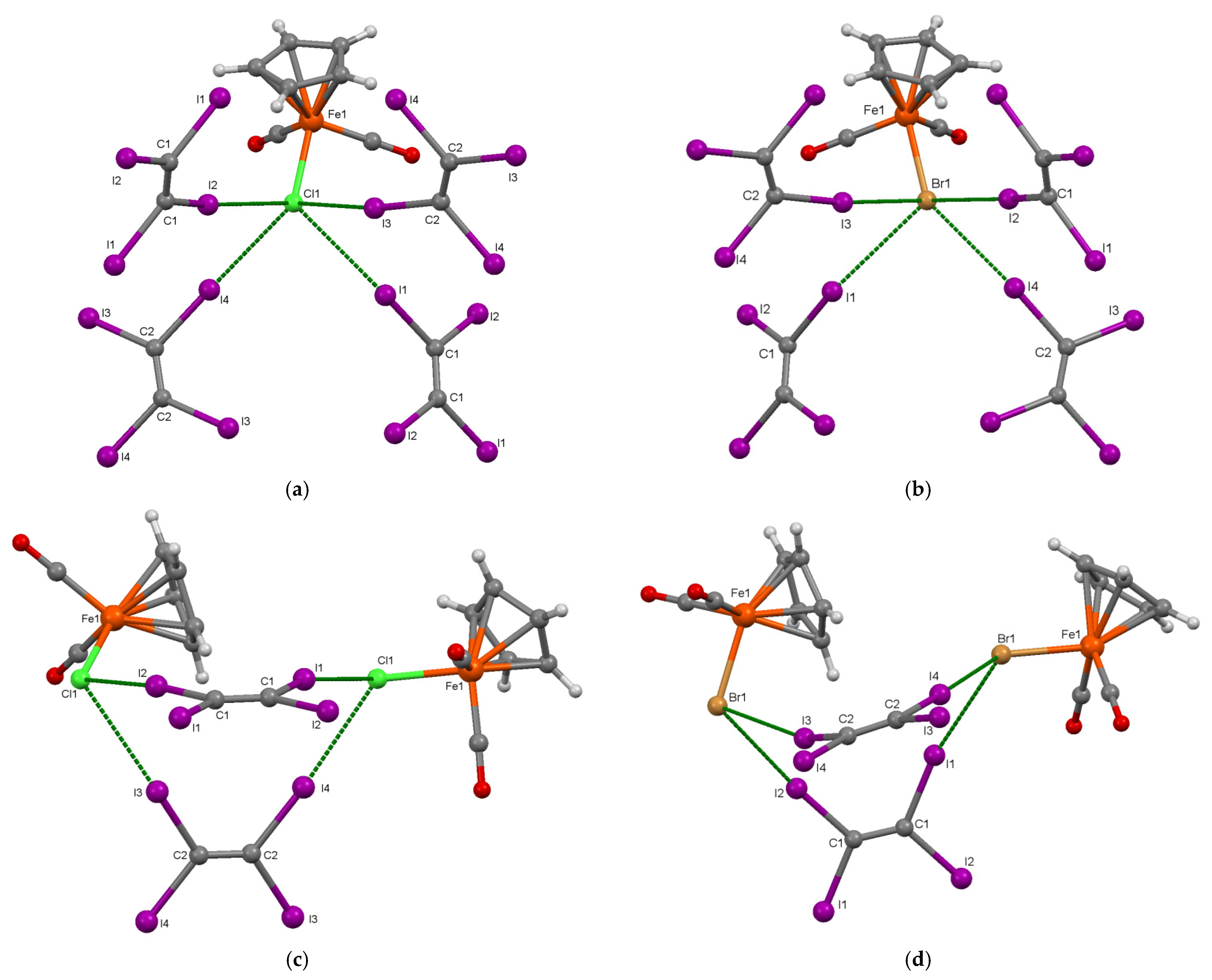
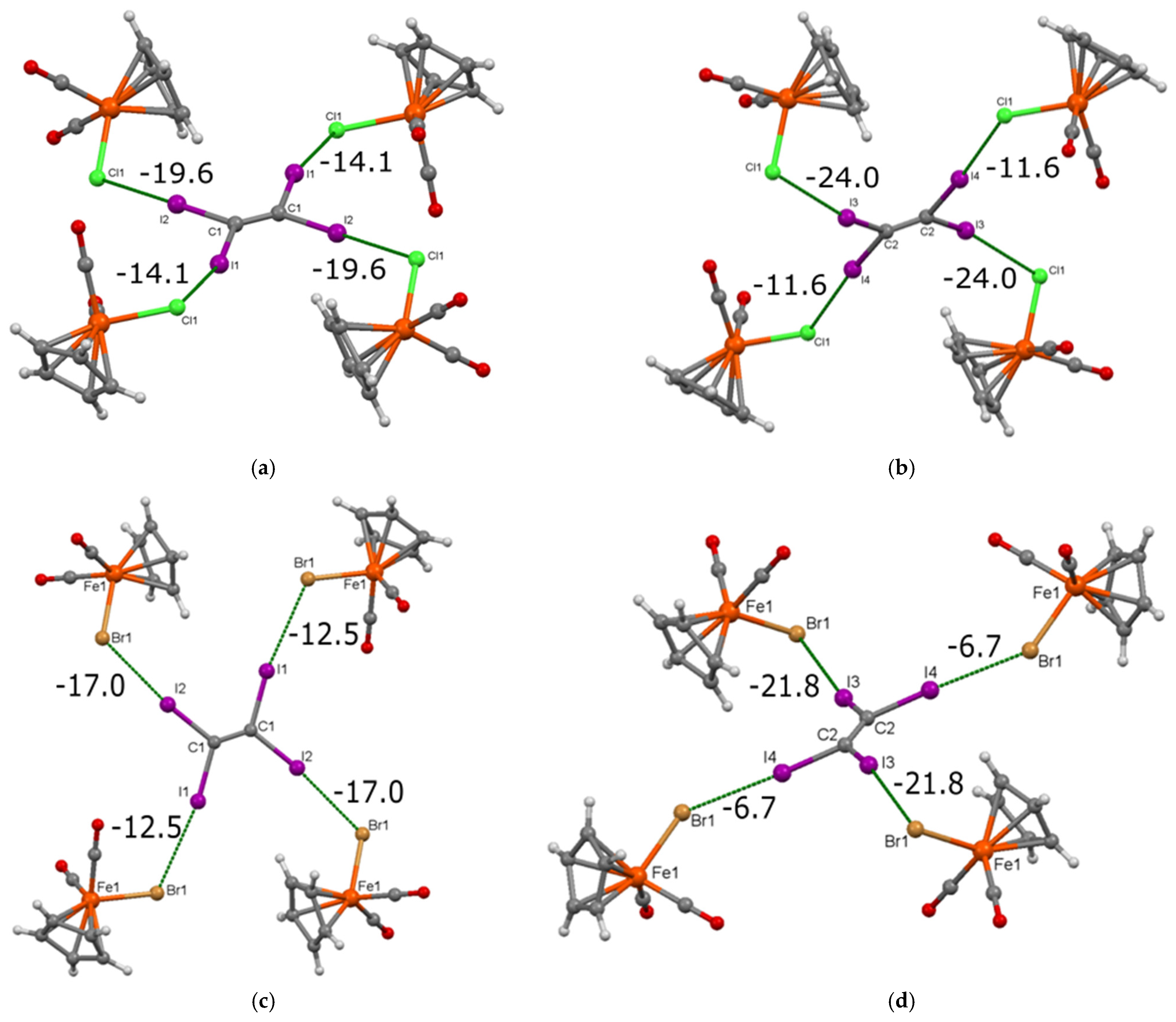
3.1. Preparation and Crystal Structure of [CpFe(CO)2X]·(C2I4) (X = Cl, Br)
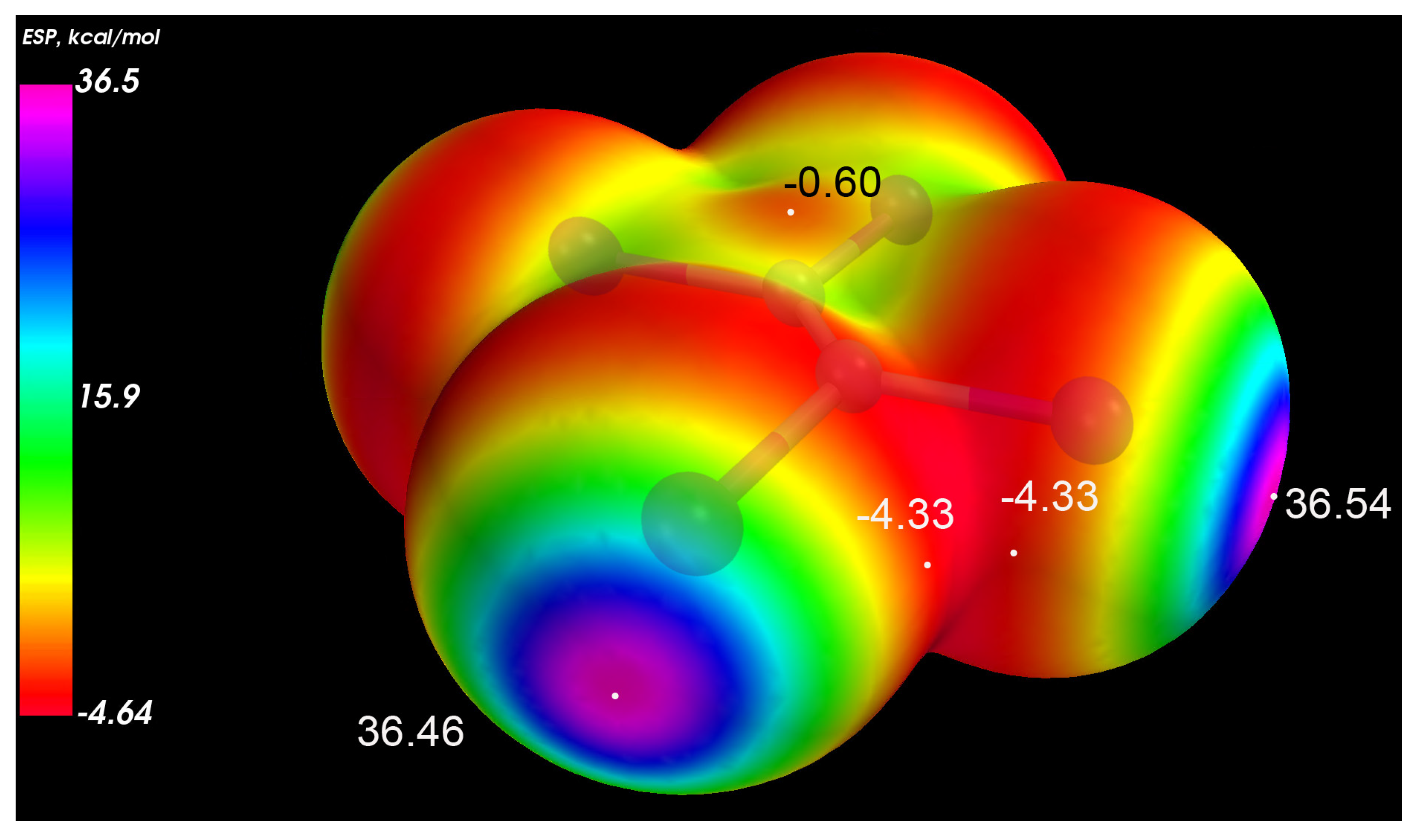
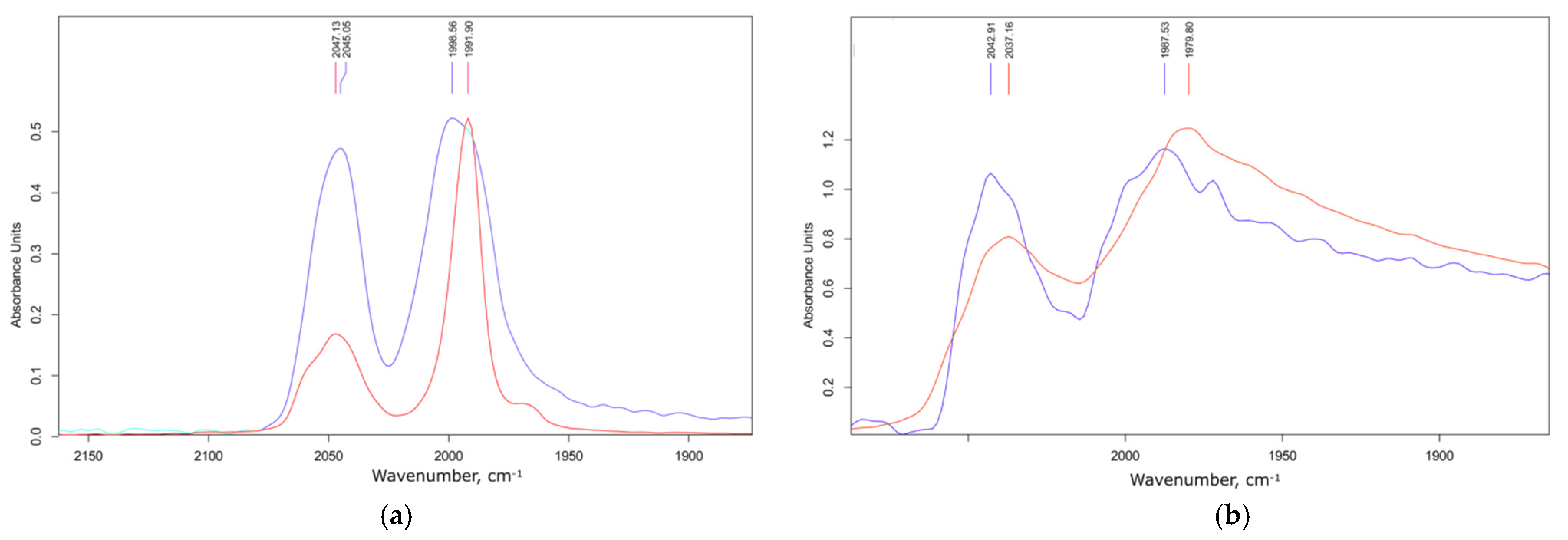
3.2. Electronic Structure and IR Spectroscopy
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, L.P.; Munakata, M.; Kuroda-Sowa, T.; Maekawa, M.; Suenaga, Y.; Kitamori, Y. Silver(I) tetraiodoethylene complexes with twisted olefin moiety. Inorg. Chim. Acta 1999, 290, 251–255. [Google Scholar] [CrossRef]
- Yamamoto, H.M.; Yamaura, J.-I.; Kato, R. Multicomponent Molecular Conductors with Supramolecular Assembly: Iodine-Containing Neutral Molecules as Building Blocks. J. Am. Chem. Soc. 1998, 120, 5905–5913. [Google Scholar] [CrossRef]
- Eliseeva, A.A.; Ivanov, D.M.; Novikov, A.S.; Rozhkov, A.V.; Kornyakov, I.V.; Dubovtsev, A.Y.; Kukushkin, V.Y. Hexaiododiplatinate(II) as a useful supramolecular synthon for halogen bond involving crystal engineering. Dalton Trans. 2020, 49, 356–367. [Google Scholar] [CrossRef]
- Efimenko, Z.M.; Eliseeva, A.A.; Ivanov, D.M.; Galmés, B.; Frontera, A.; Bokach, N.A.; Kukushkin, V.Y. Bifurcated μ2-I·(N,O) Halogen Bonding: The Case of (Nitrosoguanidinate)NiII Cocrystals with Iodine(I)-Based σ-Hole Donors. Cryst. Growth Des. 2020, 21, 588–596. [Google Scholar] [CrossRef]
- Eliseeva, A.A.; Ivanov, D.M.; Rozhkov, A.V.; Ananyev, I.V.; Frontera, A.; Kukushkin, V.Y. Bifurcated Halogen Bonding Involving Two Rhodium(I) Centers as an Integrated σ-Hole Acceptor. JACS Au 2021, 1, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Bailey, R.D.; Hook, L.L.; Watson, R.P.; Hanks, T.W.; Pennington, W.T. Tetraiodoethylene: A supramolecular host for Lewis base donors. Cryst. Eng. 2000, 3, 155–171. [Google Scholar] [CrossRef]
- Torubaev, Y.V.; Skabitskiy, I.V.; Rusina, P.; Pasynskii, A.A.; Rai, D.K.; Singh, A. Organometallic halogen bond acceptors: Directionality, hybrid cocrystal precipitation, and blueshifted CO ligand vibrational band. Cryst. Eng. Comm. 2018, 20, 2258–2266. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. Software update: The ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 2017, 8, e1327. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868, Erratum in Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 3297. [Google Scholar] [CrossRef] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297. [Google Scholar] [CrossRef] [PubMed]
- Metz, B.; Stoll, H.; Dolg, M. Small-core multiconfiguration-Dirac–Hartree–Fock-adjusted pseudopotentials for post-d main group elements: Application to PbH and PbO. J. Chem. Phys. 2000, 113, 2563–2569. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Lenthe, E.v.; Baerends, E.J.; Snijders, J.G. Relativistic regular two-component Hamiltonians. J. Chem. Phys. 1993, 99, 4597–4610. [Google Scholar] [CrossRef]
- van Wüllen, C. Molecular density functional calculations in the regular relativistic approximation: Method, application to coinage metal diatomics, hydrides, fluorides and chlorides, and comparison with first-order relativistic calculations. J. Chem. Phys. 1998, 109, 392–399. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Izsák, R.; Neese, F. An overlap fitted chain of spheres exchange method. J. Chem. Phys. 2011, 135, 144105. [Google Scholar] [CrossRef]
- Pantazis, D.A.; Neese, F. All-Electron Scalar Relativistic Basis Sets for the Lanthanides. J. Chem. Theory Comput. 2009, 5, 2229–2238. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Anisimov, A. VisMap. Available online: https://github.com/aaan1s/VisMap (accessed on 3 December 2021).
- Balula, S.S.; Coelho, A.C.; Braga, S.S.; Hazell, A.; Valente, A.A.; Pillinger, M.; Seixas, J.D.; Romão, C.C.; Gonçalves, I.S. Influence of Cyclodextrins on Catalytic Olefin Epoxidation with Metal–Carbonyl Compounds. Crystal Structure of the TRIMEB Complex with CpFe(CO)2Cl. Organometallics 2007, 26, 6857–6863. [Google Scholar] [CrossRef]
- Dahl, T.; Hassel, O. A Close Relationship between the Crystal Structure of an Acceptor and that of in Addition Compound. Acta Chem. Scand. 1966, 20, 2009. [Google Scholar] [CrossRef] [Green Version]
- Dahl, T.; Hassel, O. Relationship between Crystal Structures of Tetraiodoethylene and its 1:1 Pyrazine Compound. Acta Chem. Scand. 1968, 22, 715. [Google Scholar] [CrossRef]
- Dahl, T.; Hassel, O. Crystal Structures of Tetrabromoethylene and of Pyrazine 1:1 of Tetrabromo-resp Tetraiodoethylene. Acta Chem. Scand. 1968, 22, 8. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Parthasarathy, R. The nature of halogen—Halogen interactions: Are short halogen contacts due to specific attractive forces or due to close packing of nonspherical atoms? J. Am. Chem. Soc. 1989, 111, 8725–8726. [Google Scholar] [CrossRef]
- Torubaev, Y.; Skabitsky, I.; Lyssenko, K.A. Stages of Kitaigorodsky Aufbau Principle Detached in the Cocrystals of Cp2MX2 (M = Ti, Zr; X = Cl, Br, I) with σ- and π-Hole Donors. Cryst. Growth Des. 2021, 22, 1244–1252. [Google Scholar] [CrossRef]
- Awwadi, F.F.; Willett, R.D.; Peterson, K.A.; Twamley, B. The Nature of Halogen·Halide Synthons: Theoretical and Crystallographic Studies. J. Phys. Chem. A 2007, 111, 2319–2328. [Google Scholar] [CrossRef]
- Freytag, M.; Jones, P.G.; Ahrens, B.; Fischer, A.K. Hydrogen bonding and halogen–halogen interactions in 4-halopyridinium halides. New J. Chem. 1999, 23, 1137–1139. [Google Scholar] [CrossRef]
- Frey, M.; Jones, P.G. Secondary Bonding Interactions in Some Halopyridinium and Dihalopyridinium Halides. Z. Für Nat. B 2001, 56, 889–896. [Google Scholar] [CrossRef]
- Fotović, L.; Stilinović, V. Halogenide anions as halogen and hydrogen bond acceptors in iodopyridinium halogenides. Cryst. Eng. Comm. 2020, 22, 4039–4046. [Google Scholar] [CrossRef]
- Fotović, L.; Bedeković, N.; Stilinović, V. Evaluation of Halogenopyridinium Cations as Halogen Bond Donors. Cryst. Growth Des. 2021, 21, 6889–6901. [Google Scholar] [CrossRef]
- Fotović, L.; Bedeković, N.; Stilinović, V. Isostructural Halogen Exchange and Halogen Bonds: The Case of N-(4-Halogenobenzyl)-3-halogenopyridinium Halogenides. Cryst. Growth Des. 2022, 22, 1333–1344. [Google Scholar] [CrossRef] [PubMed]
- Aakeröy, C.B.; Baldrighi, M.; Desper, J.; Metrangolo, P.; Resnati, G. Supramolecular Hierarchy among Halogen-Bond Donors. Chem. Eur. J. 2013, 19, 16240–16247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bistoni, G.; Rampino, S.; Scafuri, N.; Ciancaleoni, G.; Zuccaccia, D.; Belpassi, L.; Tarantelli, F. How pi back-donation quantitatively controls the CO stretching response in classical and non-classical metal carbonyl complexes. Chem. Sci. 2016, 7, 1174–1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torubaev, Y.; Skabitskiy, I.; Shapovalov, S.; Tikhonova, O.; Popova, A. Halogen Bonding and CO-Ligand Blue-Shift in Hybrid Organic—Organometallic Cocrystals [CpFe(CO)2X] (C2I4) (X = Cl, Br). Crystals 2022, 12, 412. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12030412
Torubaev Y, Skabitskiy I, Shapovalov S, Tikhonova O, Popova A. Halogen Bonding and CO-Ligand Blue-Shift in Hybrid Organic—Organometallic Cocrystals [CpFe(CO)2X] (C2I4) (X = Cl, Br). Crystals. 2022; 12(3):412. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12030412
Chicago/Turabian StyleTorubaev, Yury, Ivan Skabitskiy, Sergey Shapovalov, Olga Tikhonova, and Anna Popova. 2022. "Halogen Bonding and CO-Ligand Blue-Shift in Hybrid Organic—Organometallic Cocrystals [CpFe(CO)2X] (C2I4) (X = Cl, Br)" Crystals 12, no. 3: 412. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst12030412