First-Principles Assessment of the Structure and Stability of 15 Intrinsic Point Defects in Zinc-Blende Indium Arsenide

 ,
, 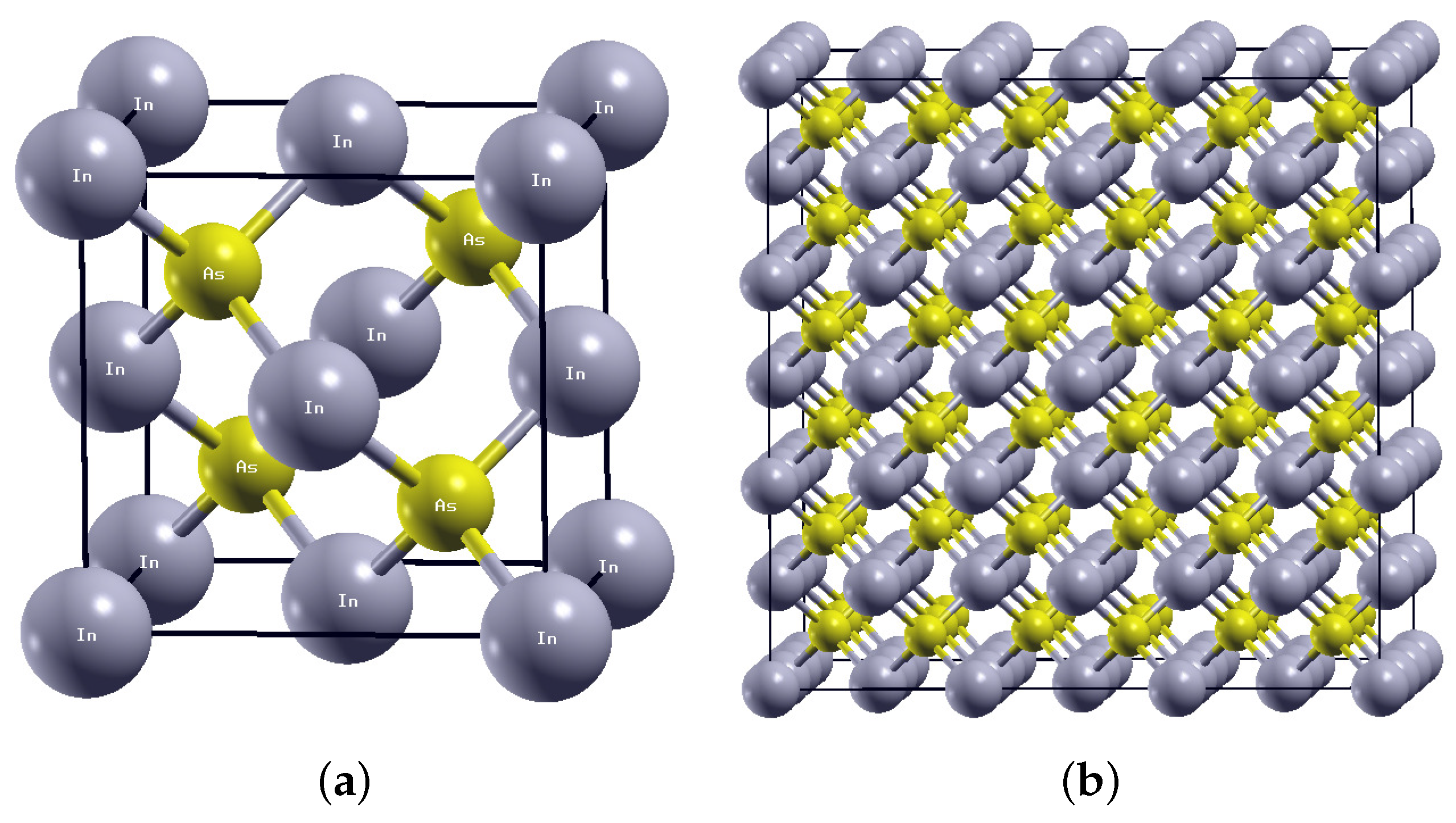{kind=link}
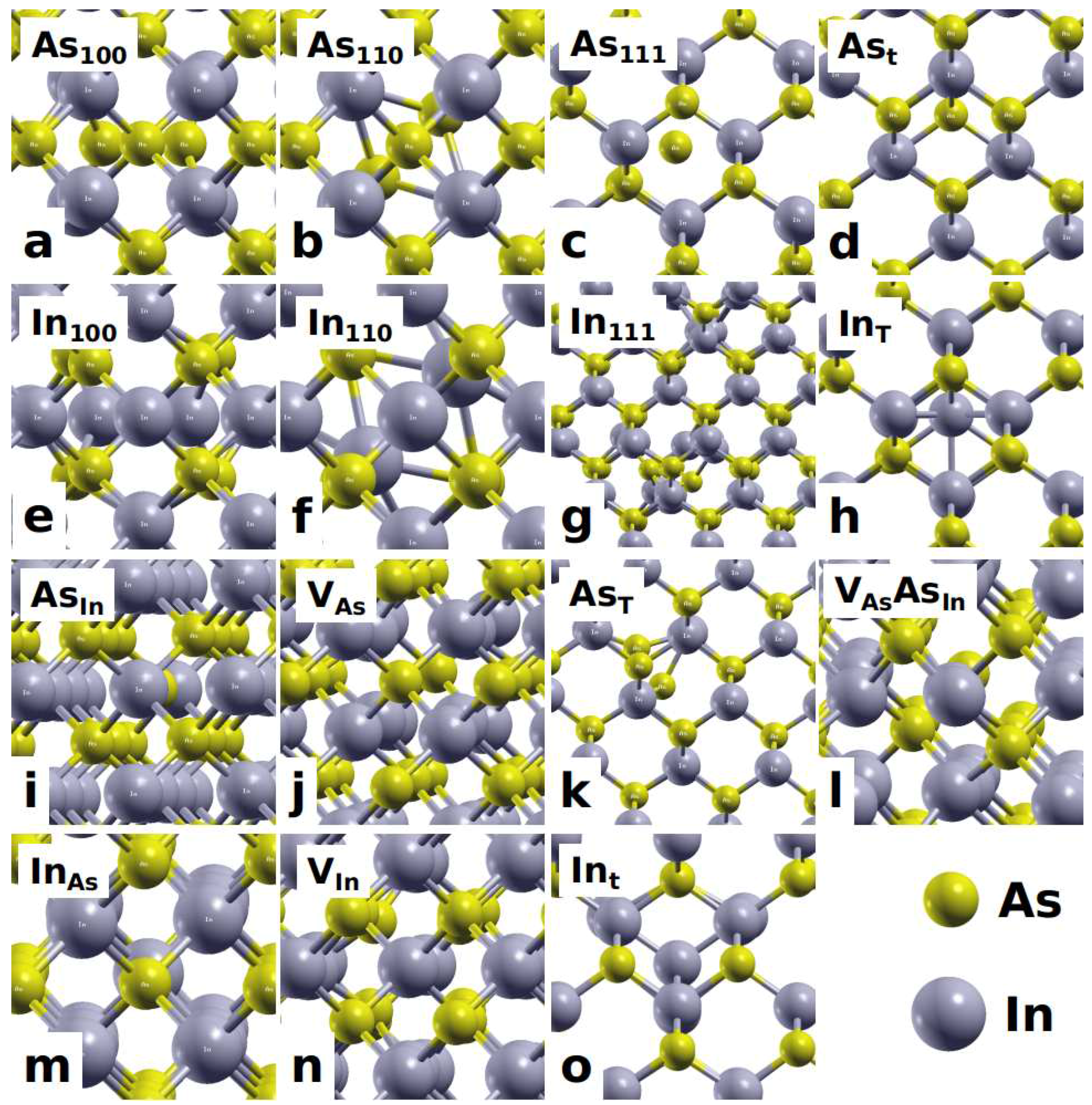{kind=link}
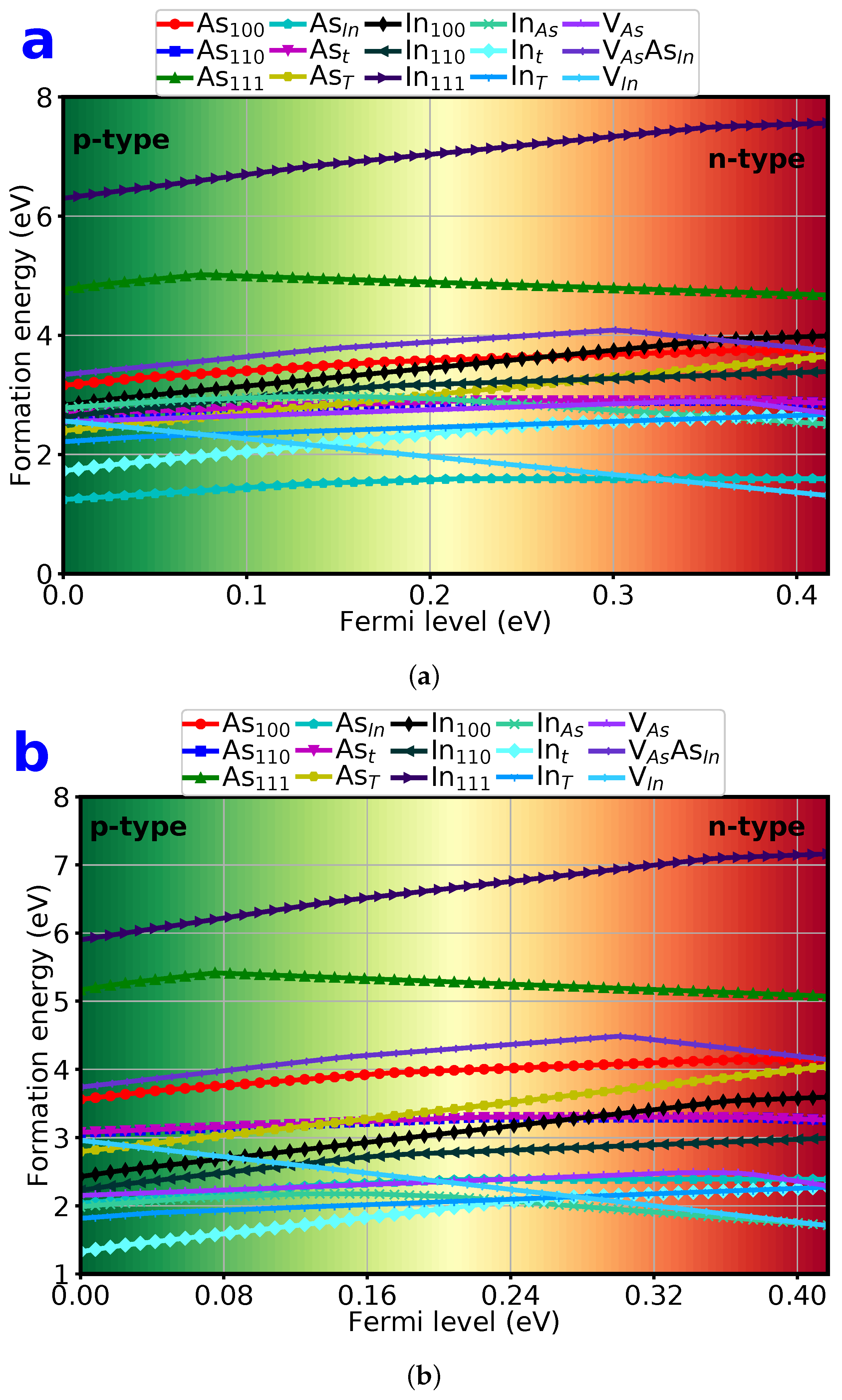{kind=link}
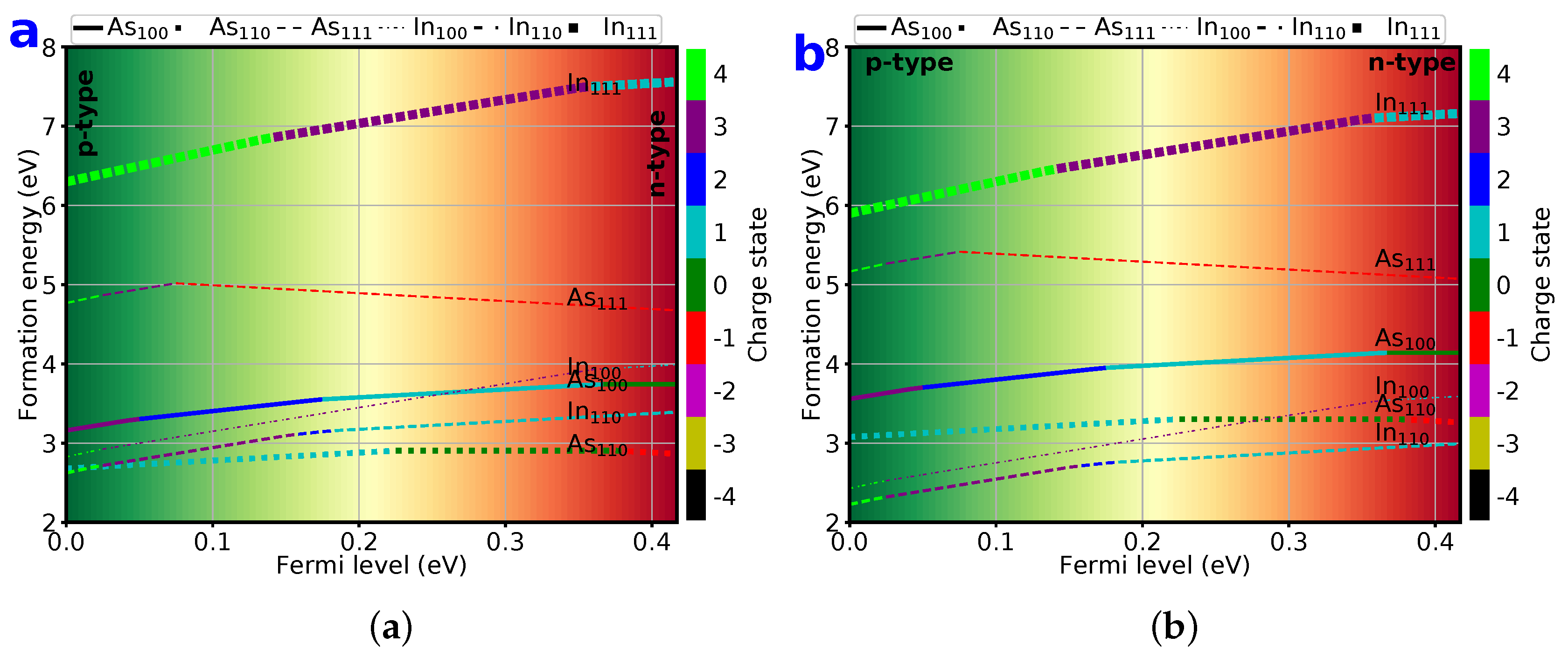{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Defect Formation Energy
2.2. Details of Density Functional Theory Calculations
3. Results and Analysis
3.1. Atomic Structures of Intrinsic Defects
3.2. Defect Formation Energies
3.3. Formation Energy of Dumbbell Interstitials
3.4. Formation Energy of Tetrahedral Interstitials
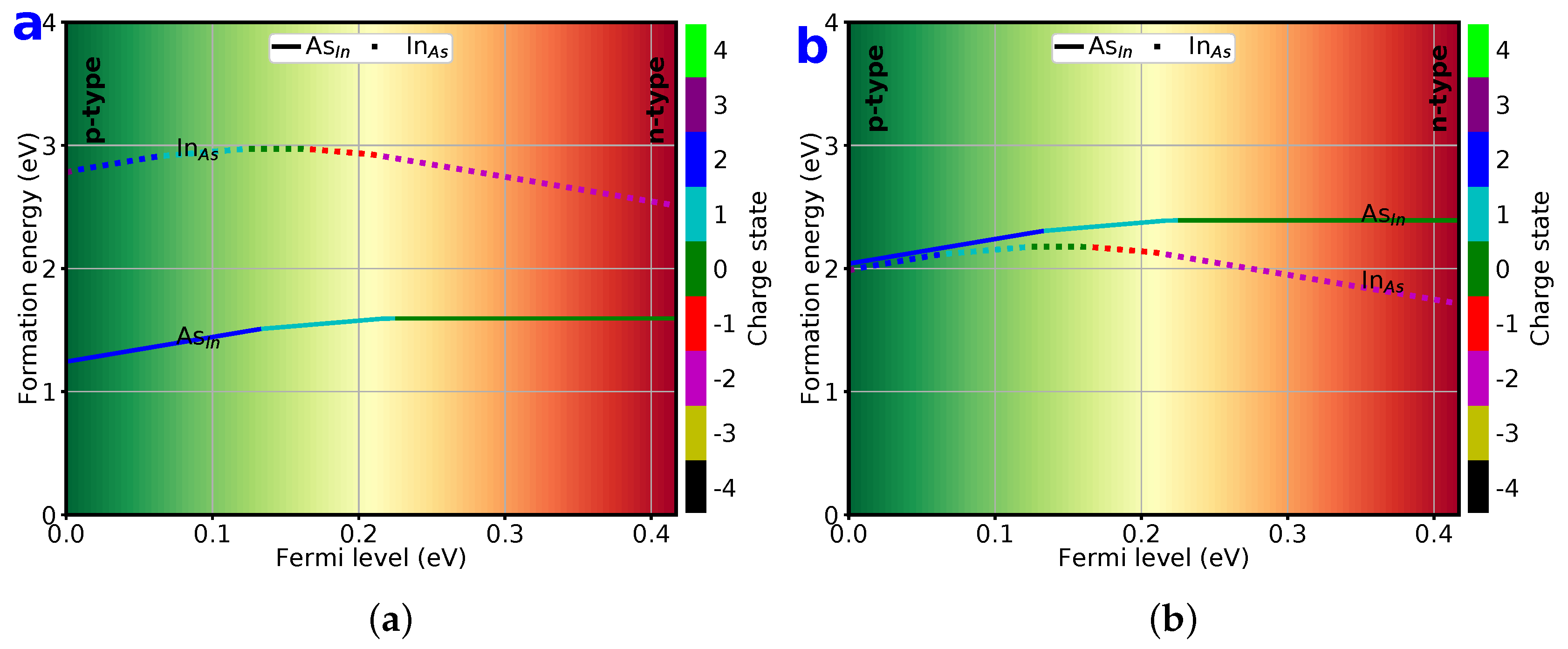
3.5. Formation Energy of Substitutionals
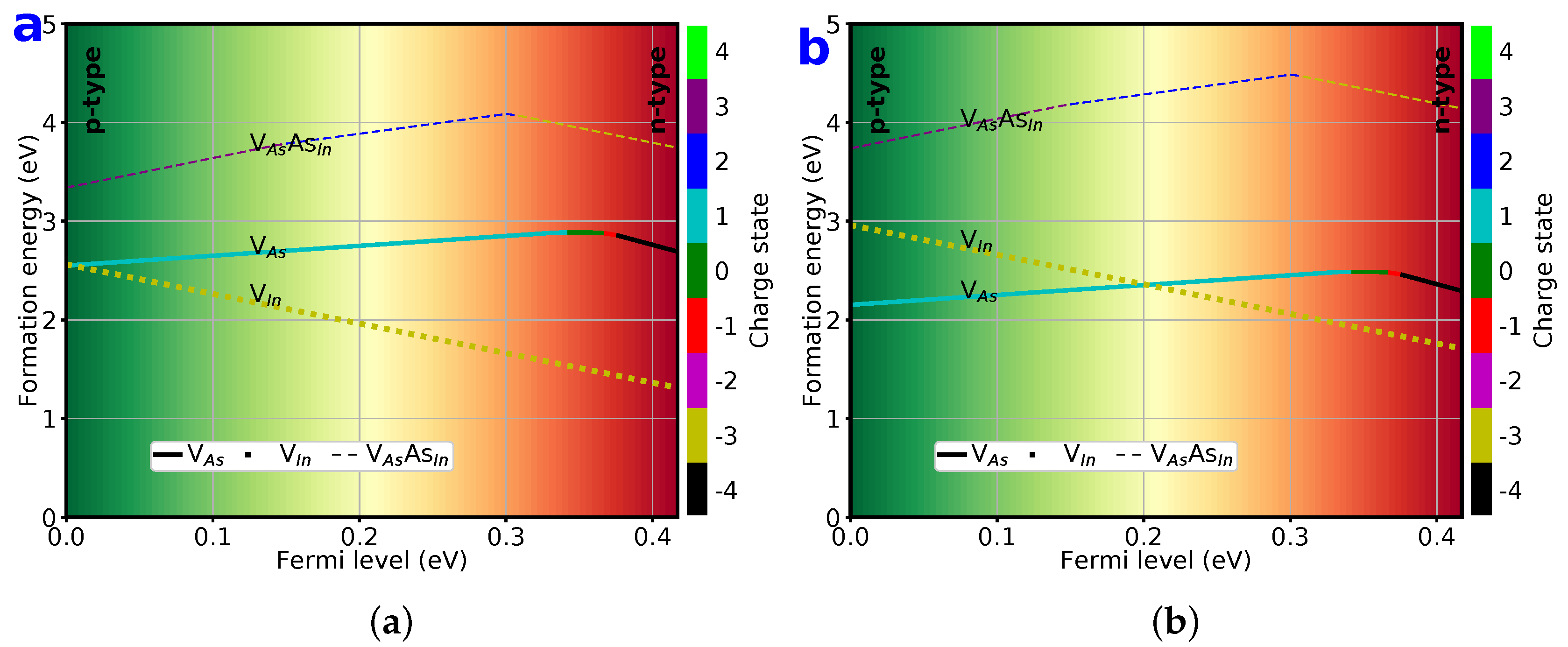
3.6. Formation Energy of Vacancies
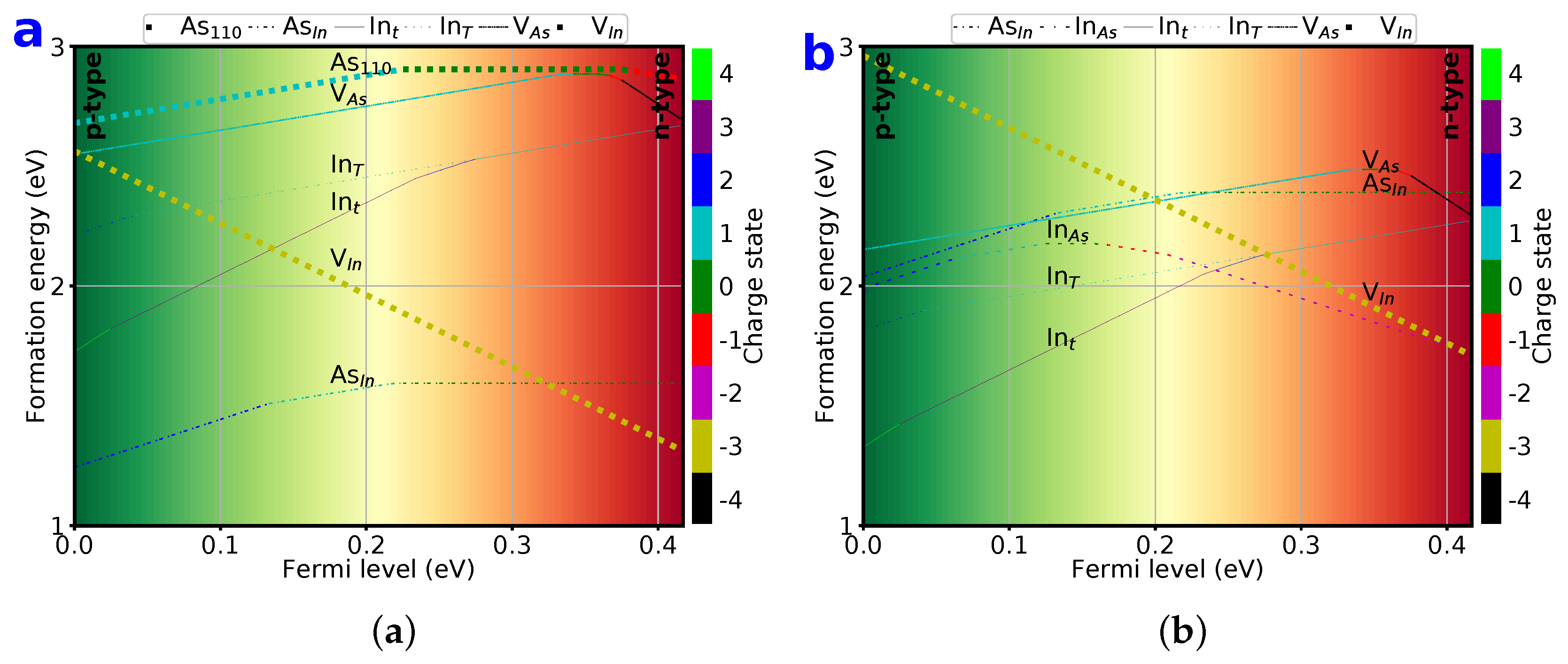
3.7. Lowest 6 Formation Energy of Point Defects
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
References
- Razeghi, M. High-power laser-diodes based on ingaasp alloys. Nature 1994, 369, 631–633. [Google Scholar] [CrossRef]
- Kim, S.-W.; Sujith, S.; Lee, B.Y. InAsxSb1-x alloy nanocrystals for use in the near infrared. Chem. Commun. 2006, 4811–4813. [Google Scholar] [CrossRef]
- Marshall, A.R.J.; Tan, C.H.; Steer, M.J.; David, J.P.R. Electron dominated impact ionization and avalanche gain characteristics in InAs photodiodes. Appl. Phys. Lett. 2008, 93, 111107. [Google Scholar] [CrossRef]
- Chelikowsky, J.R.; Cohen, M.L. Nonlocal pseudopotential calculations for the electronic structure of eleven diamond and zinc-blende semiconductors. Phys. Rev. B 1976, 14, 556–582. [Google Scholar] [CrossRef]
- Gorczyca, I.; Christensen, N.E.; Alouani, M. Calculated optical and structural properties of InP under pressure. Phys. Rev. B 1989, 39, 7705–7712. [Google Scholar] [CrossRef]
- Chang, K.J.; Froyen, S.; Cohen, M.L. Pressure coefficients of band gaps in semiconductor. Solid State Commun. 1984, 50, 105–107. [Google Scholar] [CrossRef]
- Del Alamo, J.A. Nanometre-scale electronics with III-V compound semiconductors. Nature 2011, 479, 317–323. [Google Scholar] [CrossRef]
- Jones, K.S.; Lind, A.G.; Hatem, C.; Moffatt, S.; Ridgway, M.C. A Brief Review of Doping Issues in III-V Semiconductors. ECS Trans. 2013, 53, 97–105. [Google Scholar] [CrossRef]
- Dayeh, S.A.; Susac, D.A.; Kavanagh, K.L.; Yu, E.T.; Wang, D. Structural and Room-Temperature Transport Properties of Zinc Blende and Wurtzite InAs Nanowires. Adv. Funct. Mater. 2009, 19, 2102–2108. [Google Scholar] [CrossRef]
- Adachi, S. Properties of Semiconductor Alloys: Group-IV, III-V and II-VI Semiconductors; Wiley Online Library: Hoboken, NJ, USA, 2009. [Google Scholar]
- Park, H.D.; Prokes, S.M.; Cammarata, R.C. Growth of epitaxial InAs nanowires in a simple closed system. Appl. Phys. Lett. 2005, 87, 063110. [Google Scholar] [CrossRef]
- Dayeh, S.A.; Yu, E.T.; Wang, D. III-V nanowire growth mechanism: V/III ratio and temperature effects. Nano Lett. 2007, 7, 2486–2490. [Google Scholar] [CrossRef] [PubMed]
- Dick, K.A.; Deppert, K.; Martensson, T.; Mandl, B.; Samuelson, L.; Seifert, W. Failure of the vapor-liquid-solid mechanism in Au-assisted MOVPE growth of InAs nanowires. Nano Lett. 2005, 5, 761–764. [Google Scholar] [CrossRef] [PubMed]
- Bashouti, M.Y.; Tung, R.T.; Haick, H. Tuning the Electrical Properties of Si Nanowire Field-Effect Transistors by Molecular Engineering. Small 2009, 5, 2761–2769. [Google Scholar] [CrossRef]
- Dayeh, S.A.; Aplin, D.P.R.; Zhou, X.; Yu, P.K.L.; Yu, E.T.; Wang, D. High electron mobility InAs nanowire field-effect transistors. Small 2007, 3, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Dayeh, S.A.; Yu, E.T.; Wang, D. Transport Coefficients of InAs Nanowires as a Function of Diameter. Small 2009, 5, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Park, D.W.; Jeon, S.G.; Lee, C.-R.; Lee, S.J.; Song, J.Y.; Kim, J.O.; Noh, S.K.; Leem, J.-Y.; Kim, J.S. Structural and electrical properties of catalyst-free Si-doped InAs nanowires formed on Si(111). Sci. Rep. 2015, 5, 16652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ning, F.; Tang, L.-M.; Zhang, Y.; Chen, K.-Q. First-principles study of quantum confinement and surface effects on the electronic properties of InAs nanowires. J. Appl. Phys. 2013, 114, 224304. [Google Scholar] [CrossRef]
- Alam, K.; Sajjad, R.N. Electronic Properties and Orientation-Dependent Performance of InAs Nanowire Transistors. IEEE Trans. Electron Devices 2010, 57, 2880–2885. [Google Scholar] [CrossRef]
- Dos Santos, C.L.; Piquini, P. Diameter dependence of mechanical, electronic, and structural properties of InAs and InP nanowires: A first-principles study. Phys. Rev. B 2010, 81, 075408. [Google Scholar] [CrossRef]
- Repp, S.; Weber, S.; Erdem, E. Defect Evolution of Nonstoichiometric ZnO Quantum Dots. J. Phys. Chem. C 2016, 120, 25124–25130. [Google Scholar] [CrossRef]
- Dodd, P.E.; Massengill, L.W. Basic mechanisms and modeling of single-event upset in digital microelectronics. IEEE Trans. Nucl. Sci. 2003, 50, 583–602. [Google Scholar] [CrossRef]
- Ziegler, J.F.; Lanford, W.A. Effect of Cosmic Rays on Computer Memories. Science 1979, 206, 776–788. [Google Scholar] [CrossRef] [PubMed]
- Dick, K.A.; Caroff, P.; Bolinsson, J.; Messing, M.E.; Johansson, J.; Deppert, K.; Wallenberg, L.R.; Samuelson, L. Control of III-V nanowire crystal structure by growth parameter tuning. Semicond. Sci. Technol. 2010, 25, 024009. [Google Scholar] [CrossRef]
- Kim, Y.-S.; Hummer, K.; Kresse, G. Accurate band structures and effective masses for InP, InAs, and InSb using hybrid functionals. Phys. Rev. B 2009, 80, 035203. [Google Scholar] [CrossRef]
- Reveil, M.; Huang, H.-L.; Chen, H.-T.; Liu, J.; Thompson, M.O.; Clancy, P. Ab Initio Studies of the Diffusion of Intrinsic Defects and Silicon Dopants in Bulk InAs. Langmuir 2017, 33, 11484–11489. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lukose, B.; Thompson, M.O.; Clancy, P. Ab initio modeling of vacancies, antisites, and Si dopants in ordered InGaAs. J. Appl. Phys. 2017, 121, 045106. [Google Scholar] [CrossRef]
- Northrup, J.E.; Zhang, S.B. Dopant and defect energetics - Si in GaAs. Phys. Rev. B 1993, 47, 6791–6794. [Google Scholar] [CrossRef]
- Lee, C.-W.; Lukose, B.; Thompson, M.O.; Clancy, P. Energetics of neutral Si dopants in InGaAs: An ab initio and semiempirical Tersoff model study. Phys. Rev. B 2015, 91, 094108. [Google Scholar] [CrossRef]
- Lee, S.G.; Chang, K.J. Energetics and hydrogen passivation of carbon-related defects in InAs and In0.5Ga0.5As. Phys. Rev. B 1996, 53, 9784–9790. [Google Scholar] [CrossRef]
- Freysoldt, C.; Grabowski, B.; Hickel, T.; Neugebauer, J.; Kresse, G.; Janotti, A.; Van de Walle, C.G. First-principles calculations for point defects in solids. Rev. Mod. Phys. 2014, 86, 253–305. [Google Scholar] [CrossRef]
- Leslie, M.; Gillan, N.J. The energy and elastic dipole tensor of defects in ionic crystals calculated by the supercell method. J. Phys. C Solid State Phys. 1985, 18, 973. [Google Scholar] [CrossRef]
- Schultz, P.A. Charged Local Defects in Extended Systems. Phys. Rev. Lett. 2000, 84, 1942–1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makov, G.; Payne, M.C. Periodic boundary conditions in ab initio calculations. Phys. Rev. B 1995, 51, 4014–4022. [Google Scholar] [CrossRef]
- Van de Walle, C.G.; Neugebauer, J. First-principles calculations for defects and impurities: Applications to III-nitrides. J. Appl. Phys. 2004, 95, 3851–3879. [Google Scholar] [CrossRef]
- Freysoldt, C.; Neugebauer, J.; Van de Walle, C.G. Fully Ab Initio Finite-Size Corrections for Charged-Defect Supercell Calculations. Phys. Rev. Lett. 2009, 102, 016402. [Google Scholar] [CrossRef] [PubMed]
- Lany, S.; Zunger, A. Assessment of correction methods for the band-gap problem and for finite-size effects in supercell defect calculations: Case studies for ZnO and GaAs. Phys. Rev. B 2008, 78, 235104. [Google Scholar] [CrossRef]
- Komsa, H.-P.; Rantala, T.T.; Pasquarello, A. Finite-size supercell correction schemes for charged defect calculations. Phys. Rev. B 2012, 86, 045112. [Google Scholar] [CrossRef]
- Castleton, C.W.M.; Höglund, A.; Mirbt, S. Managing the supercell approximation for charged defects in semiconductors: Finite-size scaling, charge correction factors, the band-gap problem, and the ab initio dielectric constant. Phys. Rev. B 2006, 73, 035215. [Google Scholar] [CrossRef]
- Taylor, S.E.; Bruneval, F. Understanding and correcting the spurious interactions in charged supercells. Phys. Rev. B 2011, 84, 075155. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Furthuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.O.; Gunnarsson, O. The density functional formalism, its applications and prospects. Rev. Mod. Phys. 1989, 61, 689–746. [Google Scholar] [CrossRef]
- Bechstedt, F.; Belabbes, A. Structure, energetics, and electronic states of III-V compound polytypes. J. Phys. Condens. Matter 2013, 25, 273201. [Google Scholar] [CrossRef] [PubMed]
- Tahini, H.A.; Chroneos, A.; Murphy, S.T.; Schwingenschlögl, U.; Grimes, R.W. Vacancies and defect levels in III–V semiconductors. J. Appl. Phys. 2013, 114, 063517. [Google Scholar] [CrossRef] [Green Version]
- Madelung, O.; Schulz, M.; Weiss, H. (Eds.) Semiconductors, Physics of Group IV Elements and III-V Compounds; Springer-Verlag: New York, NY, USA, 1982; Volumn 17. [Google Scholar]
- Shu, H.; Liang, P.; Wang, L.; Chen, X.; Lu, W. Tailoring electronic properties of InAs nanowires by surface functionalization. J. Appl. Phys. 2011, 110, 103713. [Google Scholar] [CrossRef]
- Thompson, A.G.; Rowe, J.E.; Rubenste, M. Preparation and Optical Properties of InAs1-xPx Alloys. J. Appl. Phys. 1969, 40, 3280–3288. [Google Scholar] [CrossRef]
- Dreyer, C.E.; Alkauskas, A.; Lyons, J.L.; Janotti, A.; Van de Walle, C.G. First-Principles Calculations of Point Defects for Quantum Technologies. Annu. Rev. Mater. Res. 2018, 48, 1–26. [Google Scholar] [CrossRef]
- Höglund, A.; Castleton, C.W.M.; Göthelid, M.; Johansson, B.; Mirbt, S. Point defects on the (110) surfaces of InP, InAs, and InSb: A comparison with bulk. Phys. Rev. B 2006, 74, 075332. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, Q.; Chen, N.; Huang, D.; Heller, E.R.; Cardimona, D.A.; Gao, F. First-Principles Assessment of the Structure and Stability of 15 Intrinsic Point Defects in Zinc-Blende Indium Arsenide. Crystals 2019, 9, 48. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst9010048
Peng Q, Chen N, Huang D, Heller ER, Cardimona DA, Gao F. First-Principles Assessment of the Structure and Stability of 15 Intrinsic Point Defects in Zinc-Blende Indium Arsenide. Crystals. 2019; 9(1):48. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst9010048
Chicago/Turabian StylePeng, Qing, Nanjun Chen, Danhong Huang, Eric R. Heller, David A. Cardimona, and Fei Gao. 2019. "First-Principles Assessment of the Structure and Stability of 15 Intrinsic Point Defects in Zinc-Blende Indium Arsenide" Crystals 9, no. 1: 48. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst9010048