Co-Crystal with Unusual High Z′ and Z′′ Values Derived from Hexamethylenetetramine and 4-fluorophenol (1/1)


Abstract
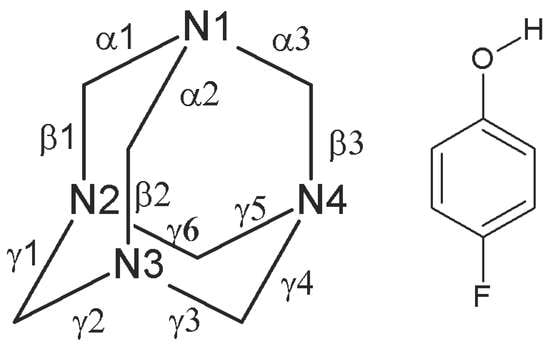
:
1. Introduction
2. Materials and Methods
2.1. Preparation of HMTA-4FP Co-Crystals
2.2. X-ray Analysis
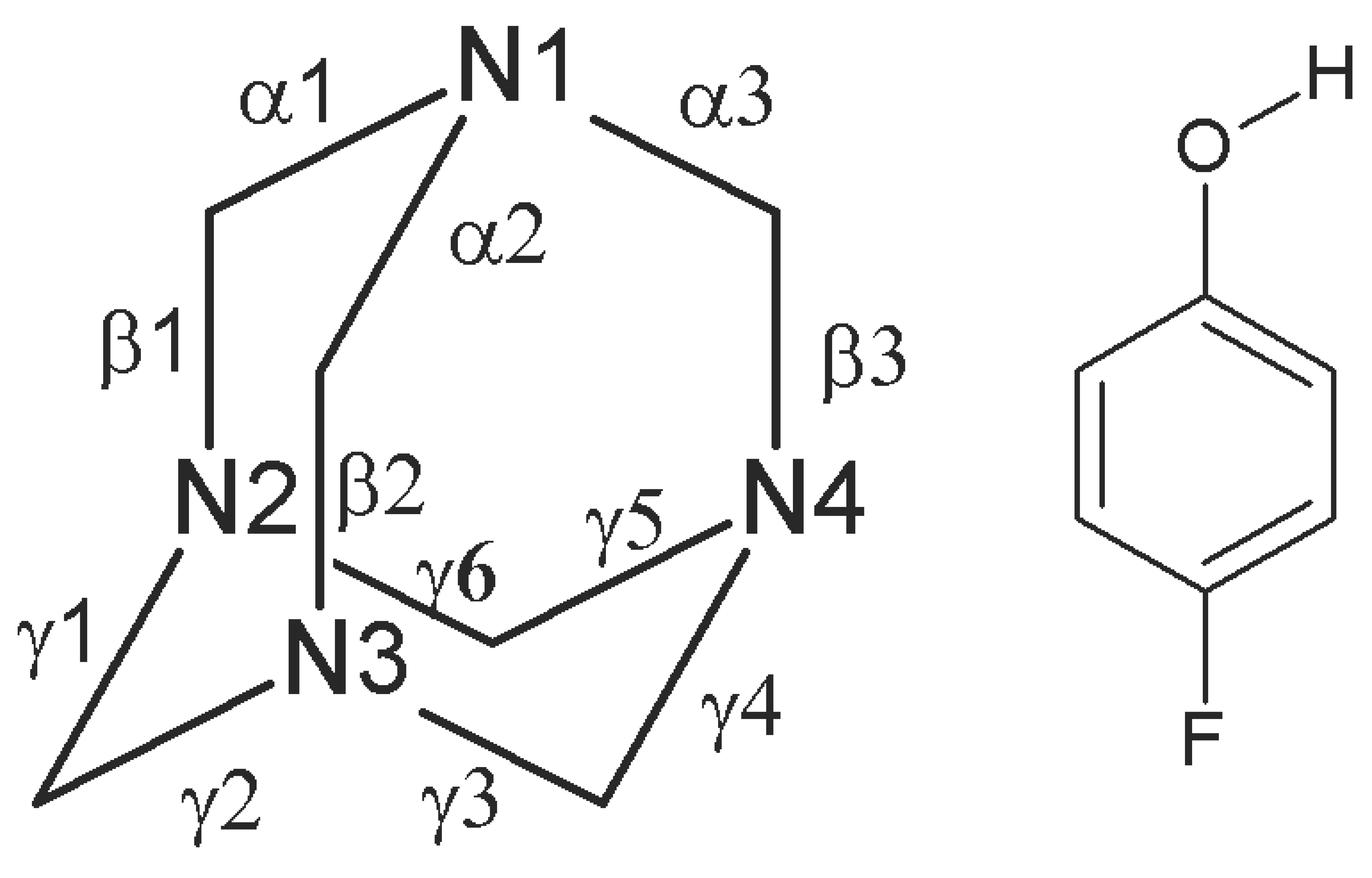
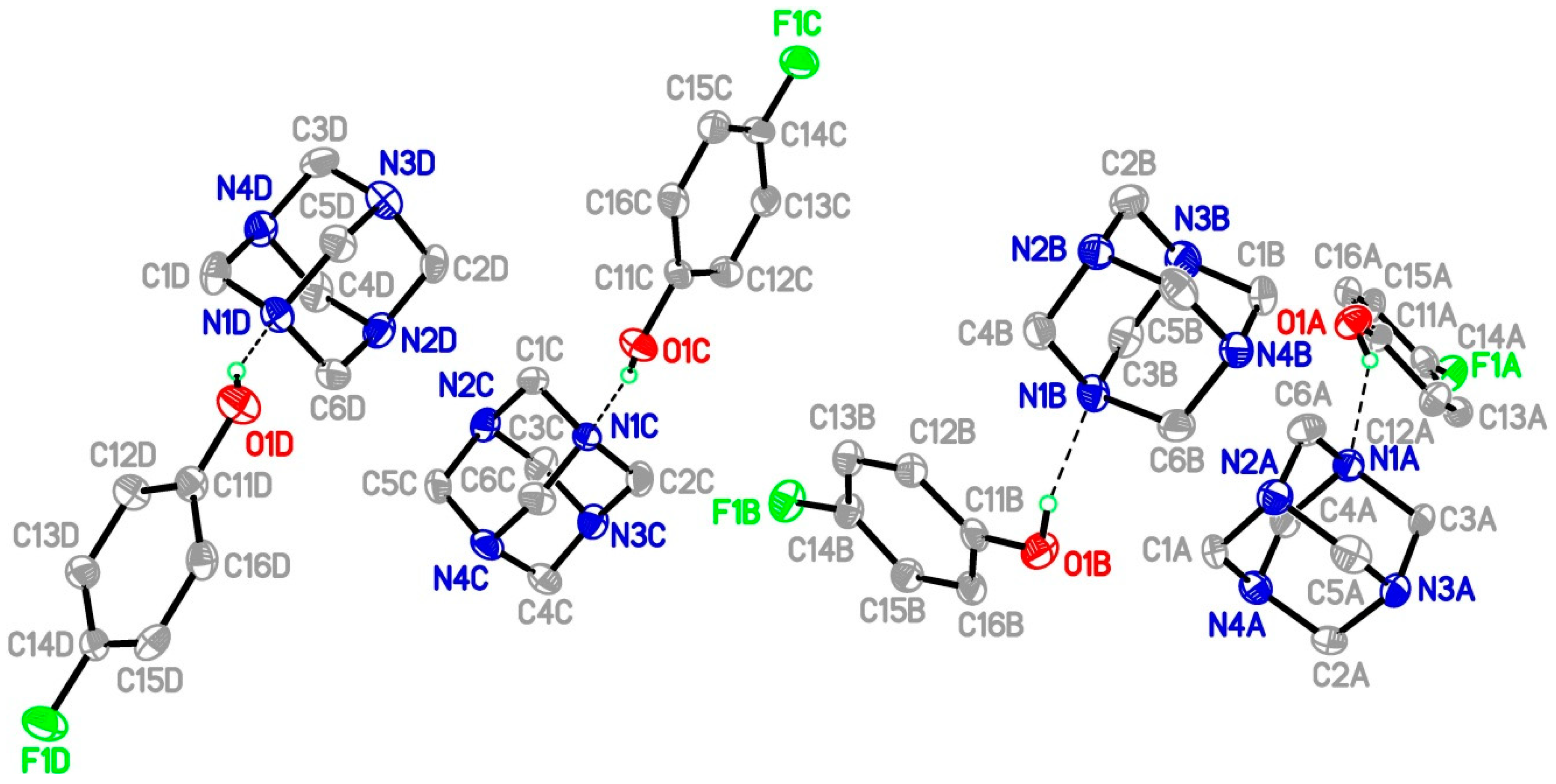
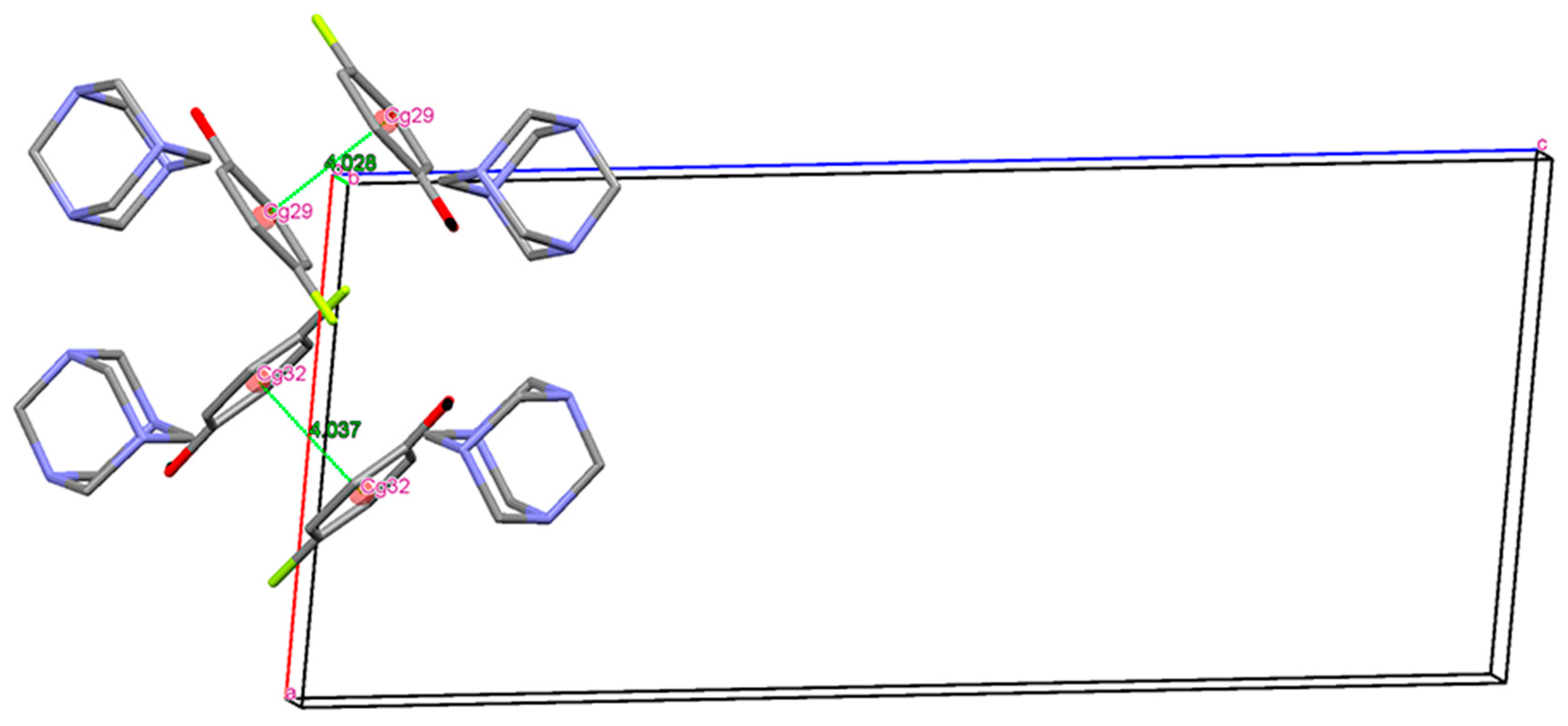
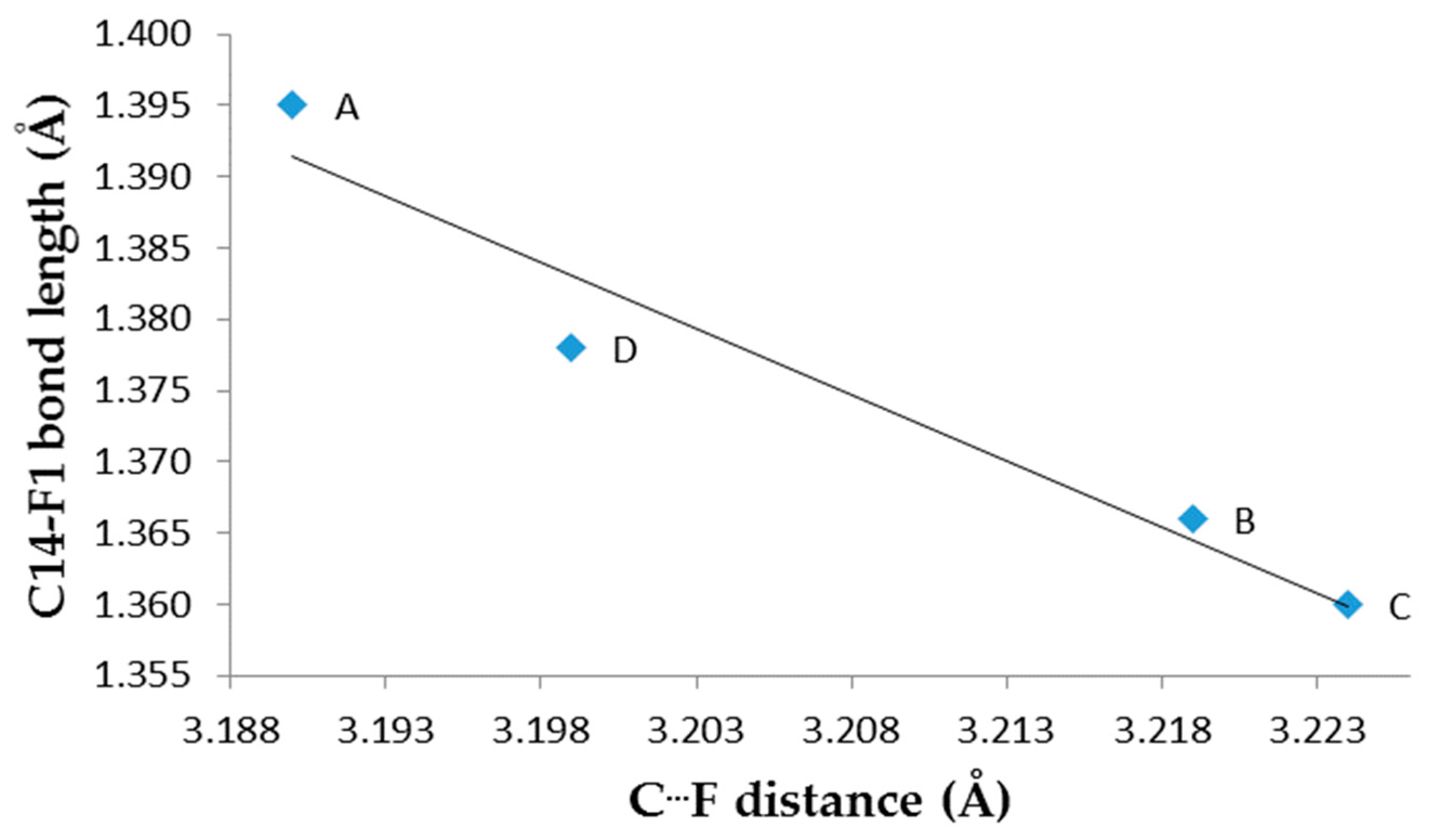
3. Results and Discussion
Single Crystal X-Ray Diffraction
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Majerz, I.; Kwiatkowska, E.; Koll, A. The influence of hydrogen bond formation and the proton transfer on the structure of complexes of phenols with N-methylmorpholine. J. Mol. Struct. 2007, 831, 106–113. [Google Scholar] [CrossRef]
- Jin, S.; Liu, H.; Gao, X.J.; Lin, Z.; Chen, G.; Wang, D. Seven organic salts assembled from hydrogen-bonds of N−H···O, O−H···O, and C−H···O between acidic compounds and bis(benzimidazole). J. Mol. Struct. 2014, 1075, 124–138. [Google Scholar] [CrossRef]
- Vijayalakshmi, S.; Kalyanaraman, S. Non linear optical analyses of hexamine: Phenol co-crystals based on hydrogen bonding: A comparative study. Spectrochim. Acta A 2014, 120, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. B 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Cetina, M.; Benci, K.; Wittine, K.; Mintas, M. Weak C−H···π and C−H···F interactions form higher-order supramolecular structures in cytosine and uracil (Z)-4′-benzamido-2′-butenyl derivatives. Cryst. Growth. Des. 2012, 12, 5262–5270. [Google Scholar] [CrossRef]
- Hathwar, V.R.; Chopra, D.; Panini, P.; Guru Row, T.N. Revealing the polarizability of organic fluorine in the trifluoromethyl group: Implications in supramolecular chemistry. Cryst. Growth. Des. 2014, 14, 5366–5369. [Google Scholar] [CrossRef]
- Chopra, D. Is organic fluorine really “not” polarizable? Cryst. Growth. Des. 2012, 12, 541–546. [Google Scholar] [CrossRef]
- Chattopadhyay, B.; Hemantha, H.P.; Narendra, N.; Sureshbabu, V.V.; Warren, J.E.; Helliwell, M.; Mukherjee, A.K.; Mukherjee, M. Polymorphism in a symmetrical dipeptidyl urea with Z′ > 1. Cryst. Growth. Des. 2010, 10, 2239–2246. [Google Scholar] [CrossRef]
- Steed, K.M.; Steed, J.W. Packing problems: High Z′ crystal structures and their relationship to co-crystals, inclusion compounds, and polymorphism. Chem. Rev. 2015, 115, 2895–2933. [Google Scholar] [CrossRef] [PubMed]
- Bombicz, P. The way from isostructurality to polymorphism. Where are the borders? The role of supramolecular interaction sand crystal symmetries. Cryst. Rev. 2017, 23, 118–151. [Google Scholar] [CrossRef]
- Van Eijck, B.P.; Kroon, J. Structure predictions allowing more than one molecule in the asymmetric unit. Acta Cryst. B 2000, 56, 535–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, H.R.; Choudhury, A.R. Can C-H···F-C hydrogen bonds alter crystal packing features in the presence of N-H···O-C hydrogen bond? J. Mol. Struct. 2017, 1150, 469–480. [Google Scholar] [CrossRef]
- Stoe & Cie. X-Area Diffractometer Control Software; Stoe & Cie: Darmstadt, Germany, 2001. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Lemmerer, A. Seven hexamethylenetetramine (HMTA) complexes with mono- and dicarboxylic acids: Analysis of packing modes of HMTA complexes in the literature. Acta Cryst. B 2011, 67, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Terpstra, M.; Craven, B.M.; Stewart, R.F. Hexamethylenetetramine at 298 K: New refinements. Acta Cryst. A 1993, 49, 685–692. [Google Scholar]
- Chandrasekhar, S.; Mukherjee, S. Salts of hexamethylenetetramine with organic acids: Enhanced anomeric interactions with a lowering of molecular symmetry revealed by crystal structures. J. Mol. Struct. 2015, 1082, 188–194. [Google Scholar] [CrossRef]
- Usman, A.; Chantrapromma, S.; Fun, H.K.; Poh, B.L.; Karalai, C. A 1:1 adduct of hexamethylenetetramine and 4-hydroxy-3-methoxybenzaldehyde. Acta Cryst. C 2002, 58, o48–50. [Google Scholar] [CrossRef]
- Cremer, D.; Pople, J.A. General definition of ring puckering coordinates. J. Am. Chem. Soc. 1975, 97, 1354–1355. [Google Scholar] [CrossRef]
- Oswald, D.H.; Allan, D.R.; Day, G.M.; Samuel Motherwell, W.D.; Parsons, S. Realizing predicted crystal structures at extreme conditions: The low-yemperature and high-pressure crystal structures of 2-chlorophenol and 4-fluorophenol. Cryst. Growth. Des. 2005, 53, 1055–1071. [Google Scholar] [CrossRef]
- Zierkiewicz, W.; Michalska, D.; Czarnik-Matusewicz, B.; Rospenk, M. Molecular structure and infrared spectra of 4-fluorophenol: A combined theoretical and spectroscopic study. J. Phys. Chem. A 2003, 107, 4547–4554. [Google Scholar] [CrossRef]
- Dalvit, C.; Invernizzi, C.; Vulpetti, A. Fluorine as a hydrogen-bond acceptor: Experimental evidence and computational calculations. Chem. Eur. J. 2014, 20, 11058–11068. [Google Scholar] [CrossRef] [PubMed]
- Gamekkanda, J.C.; Sinha, A.S.; Desper, J.; Daković, M.; Aakeröy, C.B. Competition between hydrogen bonds and halogen bonds: A structural study. New J. Chem. 2018, 42, 10539–10547. [Google Scholar] [CrossRef]
- Desiraju, G.R. The C−H···O Hydrogen bond in crystals: What is it? Acc. Chem. Res. 1991, 24, 290–296. [Google Scholar] [CrossRef]
- Steiner, T. C−H···O hydrogen bonding in crystals. Cryst. Rev. 2003, 9, 177–228. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Desiraju, G.R. C−H···F Hydrogen bonds in solid solutions of benzoic acid and 4-fluorobenzoic acid. Cryst. Growth. Des. 2018, 18, 3607–3615. [Google Scholar] [CrossRef]
- Hu, Y.; Hu, H.; Li, Y.; Chen, R.; Yang, Y.; Wang, L. Supramolecular assemblies of tetrafluoroterephthalic acid and N-heterocycles via various strong hydrogen bonds and weak C−H···F interactions: Synthons cooperation, robust motifs and structural diversity. J. Mol. Struct. 2016, 1122, 256–267. [Google Scholar] [CrossRef]
- Horiguchi, M.; Okuhara, S.; Shimano, E.; Fujimoto, D.; Takahashi, H.; Tsue, H.; Tamura, R. Control of the mode of polymorphic transition inducing preferential enrichment by modifying the molecular structure or adding seed crystals: Significant influence of CH/F hydrogen bonds. Cryst. Growth. Des. 2008, 8, 540–548. [Google Scholar] [CrossRef]
- Rivera, A.; Rojas, J.J.; Ríos-Motta, J.; Bolte, M. Crystal structure and C−H···F hydrogen bonding in the fluorinated bis-benzoxazine: 3,3′-(ethane-1,2-diyl)bis(6-fluoro-3,4-dihydro-2H-1,3-benzoxazine). Acta Cryst. E 2016, 72, 1509–1511. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Cryst. D 2009, 65, 148–155. [Google Scholar] [CrossRef]
- Bondi, A. Van der Waals volumes and radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CCDC No. | 1949826 |
|---|---|
| Empirical formula | C6H12N4·C6H5FO |
| Formula weight | 252.29 |
| Temperature | 173(2) K |
| Crystal system | triclinic |
| Space group | P1 |
| Unit cell dimensions | a = 10.1873(10) Å, α = 96.068(7)° |
| b = 10.2048(9) Å, β = 96.058(8)° | |
| c = 23.554(2) Å, γ = 90.179(8)° | |
| Volume | 2421.1(4)Å3 |
| Z | 8 |
| Calculated density | 1.384 g/cm3 |
| Absorption coefficient | 0.10 mm−1 |
| F(000) | 1072 |
| Crystal size mm3 | 0.28 × 0.27 × 0.27 |
| θmin/θmax/o | 1.8/25.0 |
| Limiting indices | −12 ≤ h ≤ 12, −12 ≤ k ≤ 12, −28 ≤ l ≤ 28 |
| Reflections collected | 8402 |
| Independent reflections | 8402 |
| Refinement method | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 8402/0/657 |
| Goodness of fit on F2 | 1.941 |
| R1/wR2[I > 2σ(I)] | 0.1076/0.2724 |
| R1/wR2(all data) | 0.1442/0.2931 |
| Largest diff. peak and hole/e.Å−1 | 0.580 /−0.482 |

| Parameter | A | B | C | D | HMTA | 4-FP |
|---|---|---|---|---|---|---|
| α1 | 1.486(10) | 1.500(10) | 1.498(9) | 1.472(10) | 1.462(5) a | |
| α2 | 1.476(9) | 1.462(10) | 1.504(10) | 1.504(10) | ||
| α3 | 1.493(10) | 1.482(9) | 1.442(10) | 1.468(10) | ||
| β1 | 1.445(10) | 1.460(10) | 1.466(11) | 1.481(10) | ||
| β2 | 1.456(10) | 1.469(11) | 1.443(11) | 1.431(10) | ||
| β3 | 1.446(10) | 1.452(10) | 1.494(10) | 1.472(11) | ||
| γ1 | 1.482(10) | 1.470(10) | 1.484(10) | 1.468(10) | ||
| γ2 | 1.469(11) | 1.469(10) | 1.466(10) | 1.467(10) | ||
| γ3 | 1.475(11) | 1.467(10) | 1.427(11) | 1.478(10) | ||
| γ4 | 1.469(10) | 1.467(10) | 1.460(11) | 1.489(11) | ||
| γ5 | 1.463(11) | 1.473(10) | 1.443(10) | 1.493(11) | ||
| γ6 | 1.472(11) | 1.458(10) | 1.465(10) | 1.448(10) | ||
| F1—C14 | 1.395(8) | 1.366(8) | 1.360(8) | 1.378(8) | 1.361 b; 1.3406 c | |
| O1—C11 | 1.372(9) | 1.372(9) | 1.359(9) | 1.358(9) | 1.380 b; 1.3651 c | |
| C13—C14 | 1.383(10) | 1.378(10) | 1.385(10) | 1.361(10) | 1.365, 1.368 b; 1.3864, 1.3890 c | |
| C14—C15 | 1.350(10) | 1.385(10) | 1.366(10) | 1.379(10) | 1.365, 1.368 b; 1.3864, 1.3890 c | |
| C12—C13 | 1.417(10) | 1.387(10) | 1.393(10) | 1.398(10) | 1.379 b; 1.3958 c | |
| C15—C16 | 1.400(11) | 1.401(11) | 1.394(11) | 1.385(11) | 1.379 b; 1.3925 c |
| D-H···A | d(D-H) | d(H∙∙∙A) | d(D∙∙∙A) | <(DHA) |
|---|---|---|---|---|
| O(1A)-H(1A)∙∙∙N(1A) | 0.84 | 1.98 | 2.741 (8) | 151 |
| O(1B)-H(1B)∙∙∙N(1B) | 0.84 | 1.92 | 2.740 (8) | 166 |
| O(1C)-H(1C)∙∙∙N(1C) | 0.84 | 1.93 | 2.756 (8) | 169 |
| O(1D)-H(1D)∙∙∙N(1D) | 0.84 | 1.94 | 2.739 (8) | 158 |
| C(5B)-H(5B1)∙∙∙O(1C)#1 | 0.99 | 2.54 | 3.505(10) | 166 |
| C(5A)-H(5A1)∙∙∙O(1D)#1 | 0.99 | 2.56 | 3.521(10) | 164 |
| C(4C)-H(4C1)∙∙∙O(1B)#2 | 0.99 | 2.59 | 3.567(11) | 168 |
| C(3D)-H(3D2)∙∙∙O(1A)#3 | 0.99 | 2.56 | 3.529(10) | 167 |
| C(1D)-H(1D1)∙∙∙F(1A) #4 | 0.99 | 2.41 | 3.200(10) | 136 |
| C(3A)-H(3A2)∙∙∙F(1D) #5 | 0.99 | 2.40 | 3.199(9) | 137 |
| C(12D)-H(12D)∙∙∙F(1A) #6 | 0.95 | 2.55 | 3.190(8) | 125 |
| C(12C)-H(2C2)∙∙∙F(1B) | 0.99 | 2.45 | 3.219(9) | 134 |
| C(4B)-H(4B2)∙∙∙F(1C) #7 | 0.99 | 2.45 | 3.224(9) | 135 |
| C(4A)-H(4A1)∙∙∙N(2C) #6 | 0.99 | 2.62 | 3.589(10) | 165 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rivera, A.; Sadat-Bernal, J.; Ríos-Motta, J.; Bolte, M. Co-Crystal with Unusual High Z′ and Z′′ Values Derived from Hexamethylenetetramine and 4-fluorophenol (1/1). Crystals 2019, 9, 520. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst9100520
Rivera A, Sadat-Bernal J, Ríos-Motta J, Bolte M. Co-Crystal with Unusual High Z′ and Z′′ Values Derived from Hexamethylenetetramine and 4-fluorophenol (1/1). Crystals. 2019; 9(10):520. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst9100520
Chicago/Turabian StyleRivera, Augusto, John Sadat-Bernal, Jaime Ríos-Motta, and Michael Bolte. 2019. "Co-Crystal with Unusual High Z′ and Z′′ Values Derived from Hexamethylenetetramine and 4-fluorophenol (1/1)" Crystals 9, no. 10: 520. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst9100520